

Abstract
Sequential cleavages of the β-amyloid precursor protein cleaving enzyme 1 (BACE1) by β-secretase and γ-secretase generate the amyloid β-peptides, believed to be responsible of synaptic dysfunction and neuronal cell death in Alzheimer's disease (AD). Levels of BACE1 are increased in vulnerable regions of the AD brain, but the underlying mechanism is unknown. Here we show that oxidative stress (OS) stimulates BACE1 expression by a mechanism requiring γ-secretase activity involving the c-jun N-terminal kinase (JNK)/c-jun pathway. BACE1 levels are increased in response to OS in normal cells, but not in cells lacking presenilins or amyloid precursor protein. Moreover, BACE1 is induced in association with OS in the brains of mice subjected to cerebral ischaemia/reperfusion. The OS-induced BACE1 expression correlates with an activation of JNK and c-jun, but is absent in cultured cells or mice lacking JNK. Our findings suggest a mechanism by which OS induces BACE1 transcription, thereby promoting production of pathological levels of amyloid β in AD.
Keywords: Alzheimer's Disease, BACE1, JNK pathway, oxidative stress, γ-secretase
The central pathological event in Alzheimer's disease (AD) is an accumulation of amyloid β (Aβ)-peptide in the brain, the product of two sequential endoproteolytic cleavages of the β-amyloid precursor protein (APP), a transmembrane type 1 protein. The β-secretase (β-site amyloid precursor protein cleaving enzyme 1, BACE1) cleaves the ectodomain of APP, producing an APP C-terminal fragment. This is further cleaved within the transmembrane domain by a γ-secretase, resulting in the release of Aβ peptides, with two C-terminal variants, at residue 40 (Aβ40) or at residue 42 (Aβ42) (Selkoe 2001). Presenilin 1 (PS1) is the catalytic subunit of the γ-secretase. Altered activities of both γ-secretase and BACE1 are involved in the pathogenesis of AD. PS1 gene mutations linked with early-onset familial AD increase the production of Aβ42, which aggregates and accumulates faster than Aβ40 (Lemere et al. 1996; Citron et al. 1997). The expression and the activity of PS1 and BACE1 are elevated in the brain of late-onset sporadic AD patients (Fukumoto et al. 2002; Holsinger et al. 2002; Yang et al. 2003; Matsui et al. 2007). The mechanisms responsible for the increased PS1 expression as well as of BACE1 activity in sporadic AD are unknown.
We and others have shown that the expression and activity of BACE1 is increased by oxidants and by the lipid peroxidation product 4-hydroxynonenal (HNE) (Tamagno et al. 2002, 2005; Kao et al. 2004; Tong et al. 2005), and that there is a significant correlation of BACE1 activity with oxidative markers in sporadic AD brain tissue (Borghi et al. 2007). Oxidative stress (OS) increases during normal ageing and is believed to be an early event in AD pathology (Nunomura et al. 2001; Cutler et al. 2004), which may contribute to membrane damage, cytoskeletal alterations and cell death (Perry et al. 2000a,b). OS and Aβ production are proportionally linked to each other because Aβ induces OS in vivo, and in vitro (Hensley et al. 1994; Mark et al. 1997; Murakami et al. 2005; Tabner et al. 2005), and OS increases the production of Aβ (Paola et al. 2000; Tamagno et al. 2005; Tong et al. 2005).
In the present study, we first found that OS increase the γ-secretase activity. We then demonstrated that the increased expression of BACE1 induced by OS is regulated by the γ-secretase activity, which in turn is mediated by the c-jun N-terminal kinase (JNK)/c-jun pathway.
Materials and methods
Cell culture, treatments and transfection
SK-N-BE neuroblastoma cells and wild-type, PS1/PS2-deficient, APP deficient and JNK-deficient mouse embryonic fibroblasts (MEFs) were generated and cultured as described previously (Herreman et al. 1999, 2003; Heber et al. 2000; Leissring et al. 2002; Tamagno et al. 2003). SK-N-BE cells and MEFs were left for 16 h in serum-free medium before any treatments. Cells were incubated for 1, 3 and 6 h with HNE (Calbiochem, Darmstadt, Germany) at a concentration of 5 μmol/L, or H2O2 (Sigma Chemical Company, St. Louis, MO, USA) at a concentration of 20 μmol/L. APPko–MEFs were incubated with 1 μmol/L Aβ 1–40 and Aβ 1–42 (Bachem, Weil am, Rhein, Germany) for 1 h. The Aβ peptides were dissolved in water at 1 mg/mL and then immediately added to the cells to avoid aggregation. Five microlitres of the solutions were applied to Formvar-coated grids, negatively stained with 5% uranyl acetate, and observed under a Philips CM10 transmission electron microscope at 80 kV. Actinomycin D (Sigma Chemical Company) was used at 1 μmol/L concentration 30 min before pro-oxidants. The BACE1 inhibitor IV was added at the concentration of 15 nmol/L.
The γ-secretase inhibitor L685,458 (Bachem) was added (at a final concentration of 1 μmol/L) to SK-N-BE cells, 8 h before treatments with pro-oxidants. The cell permeable JNK-inhibitor peptide (JIP)/JIP-1-HIV-TAT peptide (Phoenix Pharmaceuticals, Karlsruhe, Germany) was injected intraperitoneally to mice at a dose of 0.3 mg/kg 30 min before ischaemic injury or sham operation. Fusion constructs were amplified by PCR and cloned into pcDNA3, as described previously (Passer et al. 1999a,b; Cao and Sudhof 2001). Transient transfection of cDNAs into cells was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions.
Animals and surgery
BalbC mice (Harlan Italy, San Pietro al Natisone, Udine, Italy) weighing 20 g were housed in a controlled environment at 25 ± 2°C with alternating 12 h light and dark cycles. They were provided with Piccioni pellet diet (no. 48, Gessate Milanese, Milano, Italy) and water ad libitum. All mice were acclimatized in our animal facility for at least 1 week before experiments. Animal care was in compliance with Italian regulations on the protection of animals used for experimental and other scientific purposes (n. 86/609/Comunita Economica Europea) as well as with the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health.
Animals were anaesthetized through intraperitoneal injection (20 mg/kg) of Zoletil 100 (Laboratoires Virbac, Carros, France). Anaesthetized mice were placed onto a thermostatically controlled heating pad, a rectal temperature probe was inserted and body temperature was monitored and maintained at 37°C. Half of the animals were intraperitoneally injected with the cell permeable JIP/JIP-1-HIV-TATpeptide (Phoenix Pharmaceuticals) at a dose of 0.3 mg/kg, 30 min before ischaemic injury or sham operation.
To provoke transient middle cerebral artery occlusion (MCAO), a monofilament surgical suture was inserted into the exposed external carotid artery, advanced into the internal carotid artery and wedged into the cerebral arterial circle to obstruct the origin of the middle cerebral artery (Huang et al. 1994). Ischaemic injury was maintained for 30 min. During ischaemia the animals were monitored for body temperature, respiration pattern, loss of righting reflex and unresponsiveness, corneal reflexes and fixed and dilated pupils. Reperfusion was administered for 1, 3 and 6 h. At the end of the reperfusion, the anaesthetized mice were killed by decapitation. After decapitation, brains were rapidly dissected at 0°C, removed and transferred to an appropriate ice-chilled homogenizing medium consisting of 50 mmol/L Tris, pH 7.6, 5 mmol/L EDTA, 150 mmol/L NaCl and protease inhibitors for biochemical assays.
Tissue and cell extracts
Cytosolic and nuclear extracts of mouse brain tissues were prepared by the Meldrum et al.(1997). Briefly, cerebral cortical tissues were homogenized at 10% (w/v) in a Potter Elvehjem homogenizer (Wheaton, Millville, NJ, USA) using a homogenization buffer containing 20 mmol/L HEPES, pH 7.9, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 1% Nonylphenyl-polyethylene glycol, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 0.5 mmol/L phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin and 2.5 μg/mL leupeptin. Homogenates were centrifuged at 1000 g for 5 min at 4°C. Supernatants were removed and centrifuged at 105 000 g at 4°C for 40 min to obtain the cytosolic fraction, The pelleted nuclei were resuspended in extraction buffer containing 20 mmol/L HEPES, pH 7.9, 1.5 mmol/L MgCl2, 300 mmol/L NaCl, 0.2 mmol/L EDTA, 20% glycerol, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 0.5 mmol/L phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin and 2.5 μg/mL leupeptin. The suspensions were incubated on ice for 30 min for high-salt extraction followed by centrifugation at 15 000 g for 20 min at 4°C. The resulting supernatants containing nuclear proteins were carefully removed and protein content was measured using a commercially available assay (Bio-Rad, Segrate, Italy). Preparation of cell lysates and nuclear extracts were performed as described previously (Tamagno et al. 2002, 2005). To determine the γ-secretase activity and the protein levels of full-length N-cadherin, membrane fractions were isolated as described by Kim et al. (2005).
Oxidative stress determinations
Reactive oxygen species (ROS) were measured in cytosolic fractions using the probe 2′,7′dichloro-fluorescin diacetate. It is a stable, non-fluorescent molecule that is hydrolyzed by intracellular esterases to non-fluorescent 2′,7′-dichlorofluorescein, which is rapidly oxidized, in the presence of peroxides, to a highly fluorescent adduct which is measured fluorimetrically (Ravindranath 1994). Antioxidant levels in the cytosolic fractions were evaluated in terms of GSSG/GSH ratio, by the Owens and Belcher (1965) method. A mixture was directly prepared in a cuvette: 0.05 mmol/L Na phosphate buffer, pH 7.0; 1 mmol/L EDTA, pH 7.0; and 10 mmol/L 5-5′-Dithiobis-(2-nitrobenzoic acid) plus an aliquot of the sample. GSH content was evaluated after 2 min at 412 nm and expressed as μg/mg protein. Suitable volumes of diluted GSH reductase and of NADPH were then added to evaluate the total GSH level. The ratio between GSSG and GSH content is considered a measure of antioxidant status.
Antibodies and immunoblot analysis
The following antibodies were used: polyclonal anti-BACE1 antibody (Chemicon, Temecula, CA, USA); polyclonal anti-PS1, anti-PS2, anti-pJNK, anti-JNK, anti-pc-jun, anti-c-jun antibodies (Cell Signalling Technology); monoclonal β-actin and polyclonal N-cadherin antibodies (Sigma Chemical Company, Beverly, MA, USA); monoclonal 2C11, that recognizes the APP N-terminus (Boehringer, Mannheim, Germany). Lysates, nuclear and membrane fraction extracts were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis on 9.3% (pJNK, JNK, pc-jun, c-jun, BACE1 and β-actin) or 7.5% (full-length APP, N-cadherin) acrylamide gels using the mini-PROTEAN II electrophoresis cell (Bio-Rad). Proteins were transferred onto nitrocellulose membranes (Hybond-C extra Amersham Life Science, Arlington Heights, IL, USA). Non-specific binding was blocked with 50 g/L non-fat dry milk in 50 mmol/L Tris–HCl, pH 7.4, containing 200 mmol/L NaCl and 0.5 mmol/L Tween-20 (Tris-buffered saline Tween). The blots were incubated with different primary antibodies, followed by incubation with peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulins in Tris-buffered saline Tween containing 20 g/L non-fat dry milk. Reactions were developed with an enhanced chemiluminescence system according to the manufacturer's protocol (Amersham-Pharmacia Biotech Italia, Cologno Monzese, Italy).
Enzyme-linked immunosorbent assay
The levels of Aβx-40 and Aβx-42 were measured by sandwich ELISA method following the manufacturer's instructions (IBL, Gunma, Japan). Samples were analysed following the manufacturer's instructions. The Aβ concentration was detected using a Benchmark Microplate Reader and evaluated by ‘microplate manager’ v. 5.1 software (Bio-Rad). ELISA analysis of all samples was performed in two different experiments.
BACE1 activity
The activity of BACE1 was determined using a commercially available secretase kit from R&D Systems (Wiesbaden, Germany) according to the manufacturer's protocol, as previously described (Tamagno et al. 2006). To confirm the specificity of BACE1 cleavage a BACE1 cell permeable inhibitor IV (Calbiochem) was added together with the fluorescent substrate. Data were expressed as a percentage of the activity level of control cells.
Analysis of gene expression
For the quantitative SYBR Green (2× iQ YBR Green PCR Super Mix; Bio-Rad Laboratories) real-time PCR, 40 ng of cDNA was used per reaction. Primer sequences, designed with PRIMER 3 software, were:
Murine BACE1: 5′-GCATGATCATTGGTGGTATC-3′ and 5′-CCATCTTGAGATCTTGACCA-3′.
Murine β actin: 5′-AGCTATGAGCTGCCTGACGGC-3′ and 5′-CATGGATGCCACAGGATTCCA-3′.
Human BACE1: 5′-CATTGGAGGTATCGACCACTCGCT-3′ and 5′-CCACAGTCTTCCATGTCCAAGGTG-3′.
Human β actin: 5′-GGCACTCTTCCAGCCTTCCTTC-3′ and 5′-GCGGATGTCCACGTCACACTTCA-3′.
Murine PS1: 5′-TGCGGCCA TCATGATCAGTGTC-3′ and 5′-ATAAGCCAGGCGTGGATGAC-3′.
Murine presenilin enhancer-2 (PEN-2): 5′-GTTGAACCTGTG CCGGAAGTAC-3′ and 5′-TGTAGGCTGGGGCGAGGAAC-3′.
Human PS1: 5′-TAATAAGCCAGGCATGGATGAC-3′ and 5′-CATGGCCCTGGTGTTTATCAAG-3′.
Human PEN2: 5′-GGAGAAATTGAACCTGTGCCGG-3′ and 5′-CCTCTCGGAAGAACCAGAAGATG-3′.
Quantitative PCR was performed on a real-time iCycler sequence detector instrument (Bio-Rad Laboratories). After 3 min of initial denaturation, the amplification profile included 30 s denaturation at 95°C and extension at 72°C. Primer annealing was carried out for 30 s at 60°C. The results were obtained with the comparative Ct method using the arithmetic formula 2−ΔΔCt. Samples obtained from a least three independent experiments were used to calculate the means and SD.
RNA interference
Neuroblastoma cells were transfected with short hairpin RNA (shRNA) directed against human PS1 and PS2 or non-silencing shRNA, with Arrest-In transfection reagent, according to the manufacturer's instructions. The following sequences were used. PS1: NM_000021 : TRCN000061738 PS2: NM_000447 : 2HS_93093. The Arrest-In transfection reagent and non-silencing shRNA and shRNA sequences for PS1 and PS2 were purchased from Open Biosystems (Huntsville, AL, USA). The cells were harvested 48 h after shRNA transfection for sample preparation.
Luciferase reporter gene assay
Neuroblastoma cells were plated onto 96-well plate, in triplicate, and co-transected with APP-Gal4, a fusion protein of APP695 and the binding and activating domains of Gal4, pG5BLuc and promoter cytomegalo virus Renilla. As positive control of the induction of the γ-secretase activity, constructs coding for the wild-type PS1 and BACE1 were transfected together with the luciferase triplet plasmids. After 24 h cells were incubated for 3 h with 5 μmol/L HNE or with 20 μmol/L H2O2 (all in pyruvate and serum-free medium). Following incubation with pro-oxidant agents, cells were lysed according to the manufacturer's protocol (Promega, Italia, Milano, Italy) and Luciferase and Renilla signals were acquired with a Luminometer (Perkin-Elmer, Wallac MicroBeta TriLux, Waltham, MA, USA) (Minopoli et al. 2006).
In vitro peptide cleavage assay for γ-secretase activity measurement
To evaluate γ-secretase activity, membrane enriched fractions were isolated as described by Kim et al. (2005). Briefly, brains were homogenized in a hypotonic buffer containing 10 mmol/L Tris–HCl (pH 7.4), 1 mmol/L EGTA and 1 mmol/L EDTA. To extract the dissolved proteins from the crude membranes, the supernatants were centrifuged at 12 000 g for 20 min. The pellets were dissolved in 300–500 μL of a hypotonic buffer containing 0.2% CHAPS (Sigma Chemical Company) at 4°C for 30 min. The supernatants were collected after centrifugation. To measure the enzyme activity, 20 μg proteins were incubated with 20 μmol/L of a fluorescent conjugated peptide substrate [NMA-GGVVIATVK (DNP)-DRDRDR-NH2, Calbiochem] at 37°C for 2 h (Kim et al. 2005). The degree of substrate cleavage was measured by the emitted fluorescence using a reader (Perkin-Elmer LS-55) with an excitation wavelength of 355 nm and an emission wavelength of 440 nm. To confirm the specificity of γ-secretase cleavage the L685,458 inhibitor was added together with the fluorescent substrate.
Activator protein 1 transcription factor determination
The activity of protein 1 (AP-1) was determined using a commercially available kit (Active Motif, Rixensart, Belgium), with a 96-well plate on which an oligonucleotide that contains a TPA (12.0-Tetradecanoyl phorbol 13-acetate) response element (5′-TGAGTCA-3′) was immobilized. AP-1 contained in nuclear extracts specifically binds to this oligonucleotide. The primary antibodies used in the kit recognize accessible epitopes on c-fos, fos-b, fos related antigen-1, fos related antigen-2, c-jun, jun-b, jun-d proteins upon DNA binding.
Statistical analysis
Statistical analysis was performed using the unpaired Student's t-test or ANOVA, followed by the Bonferroni post hoc test.
Results
Oxidative stress up-regulates PS1/PEN-2 expression and γ-secretase activity
We previously showed that oxidant agents and HNE up-regulate BACE1 expression and activity, as well as the production of Aβ in differentiated NT2 cells (Tamagno et al. 2005). To test the effect of the same agents on the γ-secretase, we determined the expression levels of PS1 and PEN-2 and also the activity of the γ-secretase on APP.
Real-time PCR in SK-N-BE neuroblastoma cells revealed that treatment with HNE or H2O2 induced a 1.5- to twofold increase of PS1 and PEN-2 mRNA levels, when compared with untreated cells (Fig. 1a and b). Pre-treatment of cells with actinomycin D blocked the over-expression of PS1 (Fig. 1a) and PEN-2 (Fig. 1b) induced by the stress agents.
Fig. 1.

Oxidative stress increases presenilin (PS) 1/PEN2 expression and the γ-secretase activity. (a) PS1 and (b) PEN-2 mRNA levels were significantly increased, as revealed by real-time PCR, after treatment of SK-N-BE neuroblastoma cells with 4-hydroxynonenal (HNE) or H2O2. Pre-treatment of cells with 1 μmol/L actinomycin D was able to completely rescue the increase of PS1 and PEN2 expression. (c) A significant increase of γ-secretase activity on the amyloid precursor protein (APP)-Gal4 fusion protein, as shown by the luciferase reporter assay, occurs after treatment of SK-N-BE neuroblastoma cells with oxidative agents. Values are expressed as arbitrary units of luminescence. (d) and (e) Incubation of SK-N-BE cells with HNE and H2O2 resulted in a significant increase of intracellular and secreted Aβx-40 and Aβx-42. Experiments were performed in triplicate; *p < 0.05 versus control.
We evaluated the γ-secretase activity on APP using an APP-Gal4 luciferase reporter assay, after exposure of SK-N-BE neuroblastoma cells to the same agents (Fig. 1c). The level of luciferase gene expression in SK-N-BE cells treated with HNE and H2O2 increased markedly after a 3 h incubation with HNE and H2O2, by 50% and 120%, respectively (Fig. 1c). A control value was obtained, based on data from transfection with an empty vector (pcDNA3), or from untransfected cells, and gave the same results (data not shown). As a positive control, the expression of wild-type PS1 resulted in a 50% increase of luciferase gene expression (Fig. 1c). To evaluate the effect of the increased β- and γ-secretase activities in SK-N-BE neuroblastoma cells treated with HNE and H2O2, we quantified the concentrations of Aβx-40 and Aβx-42 in cell media by ELISA assay. The data reported in Fig. 1(d and e) indicate that HNE and H2O2 treatments lead to a significant increase of 40% and 90% in the steady-state concentrations of intracellular Aβx-40 and Aβx-42 respectively (Fig. 1d). Secreted levels of Aβx-40 and Aβx-42 were increased of 20% and 100% respectively (Fig. 1e). Thus, OS resulted in a higher increase of Aβx-42 when compared with that of Aβx-40, that is more intense for secreted than intracellular Aβ levels, as shown by the comparison of Aβ42/40 ratios (intracellular ratio Aβ42/40: 0.38 ± 0.02 control vs. 0.50 ± 0.05 OS p < 0.05; secreted ratio Aβ42/40: 0.10 ± 0.008 control vs. 0.24 ± 0.007 OS p < 0.02).
The γ-secretase cleavage regulates the activation of BACE1 induced by oxidative stress
The H2O2 and HNE induce an increased expression and activity of BACE1 and PS1/PEN-2, we therefore investigated whether the induced activation of BACE1 requires the γ-secretase and APP processing. It was found that HNE and H2O2 induced significant increases in the levels of BACE1 mRNA, protein and enzyme activity in SK-N-BE human neuroblastoma cells, as expected (Fig. 2a–c). Treatment with cell permeable inhibitor IV (Calbiochem) inhibited BACE1 and blocked the increase of activity induced by HNE and H2O2 (Fig. 2c). The anti-BACE1 antibody recognized a 75 kDa band as seen in the gel that corresponded to the mature, N-glycosylated, form of the protein. A lower 70 kDa band was observed that corresponded to pro-BACE1, a precursor of the mature BACE1 (Capell et al. 2000) (Fig. 2b).
Fig. 2.
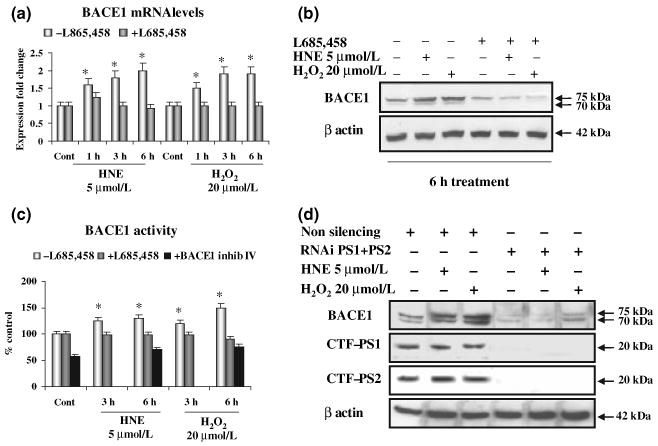
Inhibition of γ-secretase decreases β-site amyloid precursor protein cleaving enzyme 1 (BACE1) mRNA and protein levels, and its protease activity. (a–c) In SK-N-BE cells, inhibition of γ-secretase with the transition analogue L685,458 prevented the increase of BACE1 mRNA level (a), protein level (b) and enzyme activity (c) of BACE1, that are induced by 4-hydroxynonenal (HNE) and H2O2 in control cells. Addition of BACE1 inhibitor IV halved the basal activity of BACE1 and completely blocked its significant increase induced by oxidative stress (OS) (c). (d) In SK-N-BE cells, simultaneous knock-down of PS1/2 by RNA interference reduced the basal level of BACE1 protein levels and prevented the increase of BACE1 by HNE and H2O2. Non-silencing shRNA-treated cells exhibited BACE1 levels similar to those of wild-type cells. Experiments were performed in triplicate; *p < 0.05.
Pre-incubation of cells with L685,458, an inhibitor of the γ-secretase, completely prevented the HNE- or H2O2-induced increase of BACE1 mRNA, protein and activity levels (Fig. 2a–c). To complement this result, we silenced PS1 and PS2 expression using a double-stranded RNA-mediated interference in SK-N-BE cells. Accordingly, we observed that the inhibition of the production of PS prevented the increase of BACE1 protein levels induced by H2O2 or HNE (Fig. 2d).
To confirm that the γ-secretase cleavage is required to induce the up-regulation of BACE1 we used MEFs from PS1/PS2 double knock-out (PSdko) mice (Herreman et al. 1999, 2003). Treatment with HNE and H2O2 induced a 100–150% increase of BACE1 mRNA, protein and activity levels in wild-type MEFs, as expected (Fig. 3a–c). Instead, HNE or H2O2 failed to induce any changes of BACE1 mRNA, protein or activity levels in PSdko-MEFs, when the same experimental conditions were applied (Fig. 3a–c). To confirm that the activation of BACE1 requires the PS, we re-introduced PS1/PS2 into PSdko-MEFs by transfection of PSdko-MEFs with PS1 and PS2 constructs. The re-expression of PS1/PS2 in PSdko-MEFs restored the increase of BACE1 protein levels induced by HNE and H2O2 to the same level of that observed in the wild-type MEFs (Fig. 3d).
Fig. 3.
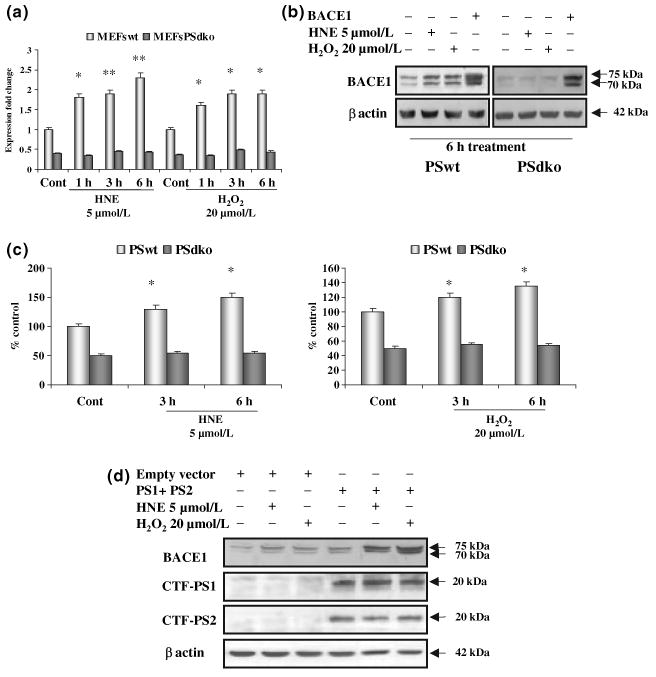
Presenilins are needed for oxidative stress dependent up-regulation of β-site amyloid precursor protein cleaving enzyme 1 (BACE1) expression and activity. (a–c) Pro-oxidative agents increased BACE1 transcription (a), protein levels (b) and activity (c) in WT-MEFs (PSwt), but not in MEFs lacking PS1 and PS2 (PSdko). (d) Transfection of PSdko-MEFs with PS1 and PS2 increased the basal level of BACE1 and restored the capacity of pro-oxidative agents to induce BACE1, as seen by western blot analysis. Experiments were performed in triplicate; *p < 0.05.
The γ-secretase cleavage regulates the expression of BACE1 in the brain of mice subjected to cerebral ischaemia/reperfusion
To confirm that γ-secretase functions as a positive regulator of BACE1 activity in response to OS in vivo, we used a well-established animal model, in which OS is induced in mice by transient MCAO followed by reperfusion (Huang et al. 1994; Cho et al. 2007).
Mice that had undergone ischaemic injury, followed by 1 or 3 h of reperfusion, exhibited a 30% or 90% increase in ROS in the brain, respectively, as shown in Fig. 4a. The increased production of ROS was accompanied by a significant depletion of reduced GSH content, as indicated by the increased ratio of GSSG/GSH (Fig. 4b).
Fig. 4.
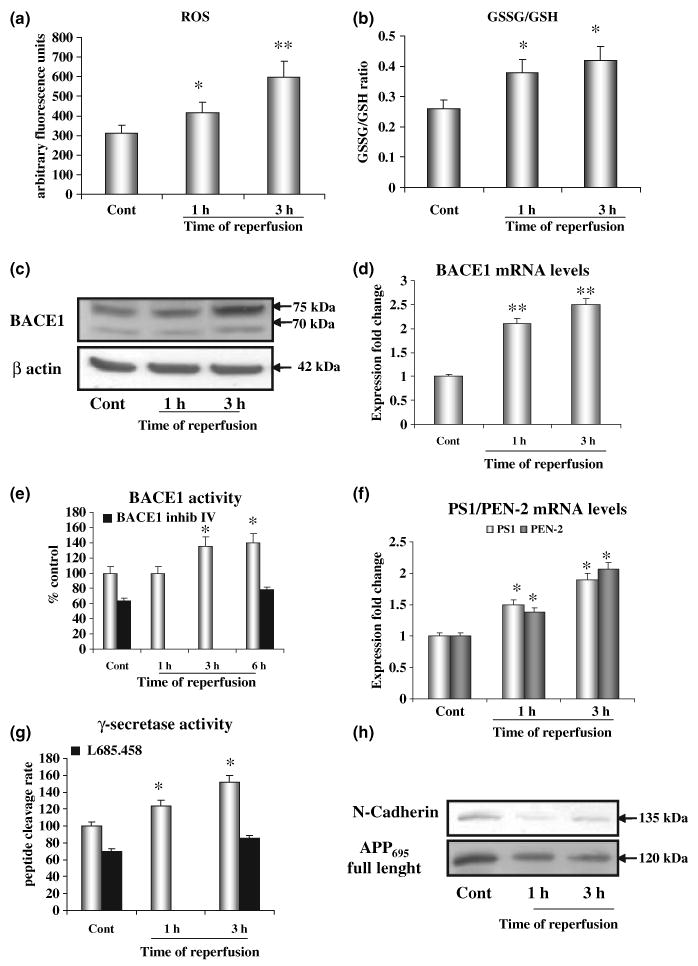
Cerebral ischaemia/reperfusion induces oxidative stress and β-site amyloid precursor protein cleaving enzyme 1 (BACE1) expression and γ-secretase activity in mice brain. (a and b) Mice subjected to ischaemia and 1 or 3 h of reperfusion exhibit a significant increase in the production of reactive oxygen species (a) as well as a significant depletion in GSH content evaluated as a significant increase in the GSSG/GSH ratio (b). (c–e) Compared with sham-operated animals, BACE1 protein levels (c), mRNA levels (d) and enzyme activity (e) are significantly increased in brains of mice subjected to a stroke. Addition of BACE1 inhibitor IV halved the basal activity of BACE1 and completely blocked the increase induced by oxidative stress (OS) (e). (f) PS1 and PEN-2 transcription is increased in the ischaemic cortex after 1 and 3 h of reperfusion. (g) Increase in the activity of γ-secretase is seen in ischaemic-reperfused mice, as shown by the in vitro peptide cleavage assay, within 1 h and up to 3 h of reperfusion. Addition of L685,458 halved the basal activity of γ-secretase and completely blocked the increase induced by OS (g). (h) At 1 and 3 h of reperfusion, a significant decrease in protein levels of full-length N-cadherin and amyloid precursor protein (APP), substrates of γ-secretase, was also observed. Equal protein loading of membrane enriched fractions was controlled by staining the membrane with Ponceau Red (data not shown). There were six mice per group: sham operated (cont), or ischaemia followed by reperfusion for 1, 3 or 6 h; *p < 0.05; **p < 0.02.
We next measured BACE1 mRNA, protein and activity levels. The protein levels of BACE1 were significantly increased during reperfusion, ranging from a 50% increase compared with sham-operated animals after 1 h of reperfusion to 150% after 3 h (Fig. 4c). At 1 and 3 h of reperfusion a 2- to 2.5-fold increase of BACE1 mRNA was observed (Fig. 4d) and BACE1 activity was found to be significantly increased after 3 and 6 h of reperfusion, when compared with sham-operated animals (Fig. 4e). The use of a specific inhibitor of BACE1 halved the basal activity of BACE1 and completely blocked the increase induced by oxidation (Fig. 4e).
The γ-secretase activity was increased in the brain in response to ischaemia/reperfusion. After 1–3 h of reperfusion, brain tissue showed a 100% increase in the PS1 and PEN-2 mRNA (Fig. 4f). The activity of the γ-secretase cleavage of APP was tested using a specific peptide cleavage assay (Kim et al. 2005), as well as using a measurement of the levels of full-length N-cadherin and of full-length APP, as substrates of the γ-secretase (Marambaud et al. 2003; Uemura et al. 2006). At 1–3 h of reperfusion there was a significant increase in the fluorescence intensity read-out (40–50%), which is directly proportional to the γ-secretase activity (Fig. 4g), while a strong decrease in the amounts of full-length N-cadherin and APP were observed (Fig. 4h). Equal protein loading of membrane enriched fractions was ensured by monitoring the staining of the membranes with Ponceau Red (data not shown).
The JNK/c-jun pathway mediates the OS-dependent up-regulation of γ-secretase and BACE1
The signalling pathways potentially involved in the OS-dependent up-regulation of BACE1 mediated by the γ-secretase cleavage were next investigated. The c-jun N-terminal kinase (JNK) signalling pathway is a potential candidate, as it can be activated by both Aβ and OS (Wang et al. 2004; Yao et al. 2005). First, we tested if the JNK pathway was activated in the OS models used in this study. HNE or H2O2 induced a robust activation of JNK and c-jun in SK-N-BE neuroblastoma cells, as shown by increased levels of phospho-JNK and phospho-c-jun (Fig. 5a). Similarly, brains of ischaemic/reperfused mice showed a significant increase of JNK and c-jun phosphorylation, the levels increasing by 100% after 1 h of reperfusion, to 120% after 3 h (Fig. 5a). To confirm that there was an activation of the JNK pathway in our experimental models we evaluated the activation of the transcription factor AP-1. It is composed of a mixture of heterodimeric complexes of proteins derived from the fos and jun families. Phosphorylation of AP-1 family members by kinases is required for transactivation activity. In the case of c-jun, the activation domain is regulated to a large extent by the JNK (Karin 1995). Three hours incubation of SK-N-BE cells with HNE or H2O2 and 1–3 h of reperfusion of ischaemic brains resulted in a significant activation of AP-1 (Fig. 5b). To ascertain the role of the JNK/AP-1 pathway in the OS-dependent up-regulation of the γ-secretase and BACE1, we used MEFs deficient for JNK (JNKko-MEFs). Treatment of cells with HNE or H2O2 resulted in a 90–100% increase in BACE1 mRNA in WT-MEFs over 1–6 h incubation periods. By contrast, there was no BACE1 mRNA increase induced by HNE or H2O2 in JNKko–MEFs (Fig. 5c). PS1 mRNA was also increased in WT-MEFs, but not in JNKko-MEFs, upon treatment with HNE or H2O2 (Fig. 5d). The same result was observed for PEN-2 (data not shown).
Fig. 5.
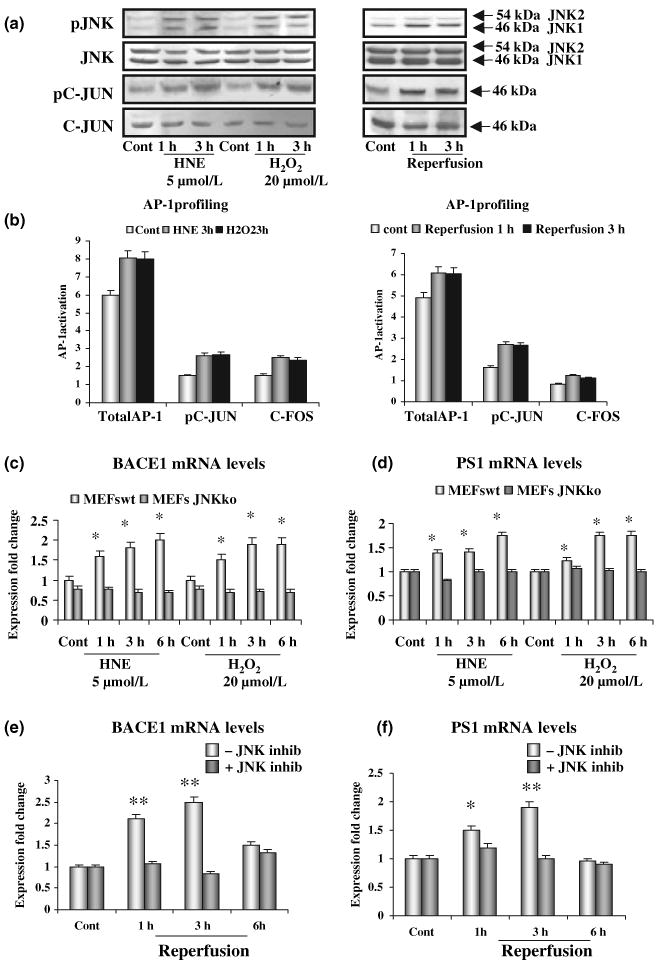
The c-jun N-terminal kinase (JNK)/activator protein 1 (AP-1) signalling pathway mediates oxidative stress (OS)-induced increases in β-site amyloid precursor protein cleaving enzyme 1 (BACE1) and PS1 expression. (a) Significant increase in phosphorylation of JNK and c-jun after treatment of SK-N-BE neuroblastoma cells with HNE or H202, and after 1 and 3 h of reperfusion, in brains of ischaemic mice. (b) Activation of AP-1 transcription factor, together with pc-jun and c-fos, follows OS SK-N-BE cells and mouse models of OS. (c) BACE1 transcription, measured by real-time PCR, is not affected by oxidative stimuli in JNKko–MEFs. (d) Levels of PS1 mRNA are not changed by oxidative stimuli in JNKko–MEFs. (e and f) Pre-treatment of mice with the JNK inhibitory peptide prevents the increase of BACE1 and PS1 transcription observed in ischaemic/reperfused mice; *p < 0.05, **p < 0.02.
To check the activation of the JNK/AP-1 pathway in the transient MCAO-reperfusion model, a cell permeable selective JIP (JIP-1-HIV-TAT peptide), able to abrogate the activation of JNK, without affecting its phosphorylation level (Borsello et al. 2003), was injected intraperitoneally 30 min prior to the MCAO.
Accordingly, the 2- to 2.5-fold increase of BACE1 mRNA after 1–3 h of reperfusion was also completely prevented by pre-treatment with the JNK inhibitor peptide (Fig. 5e). Similarly, the levels of PS1 mRNA also showed a 1.5- to twofold increase after 1–3 h of reperfusion and again the increase was prevented by pre-treatment with the JNK inhibitor peptide (Fig. 5f). The inhibitor peptide also prevented the increase of PEN-2 (data not shown).
Role of the γ-secretase cleavage-derived APP derivatives on the up-regulation of BACE1
As APP is the substrate of the γ-secretase, we next investigated whether the presence of APP was essential for obtaining the up-regulation of BACE1. For this experiment we used MEFs lacking APP695 (APPko) (Heber et al. 2000; Leissring et al. 2002). Neither HNE nor H2O2 induced any change in BACE1 mRNA and protein levels in APPko–MEFs, in contrast to the result obtained with wild-type MEFs (Fig. 6a). The re-introduction of APP into APPko–MEFs by transfection with the APP695 construct restored the increase of BACE1 protein levels to the same levels observed in the WT-MEFs (Fig. 6a).
Fig. 6.
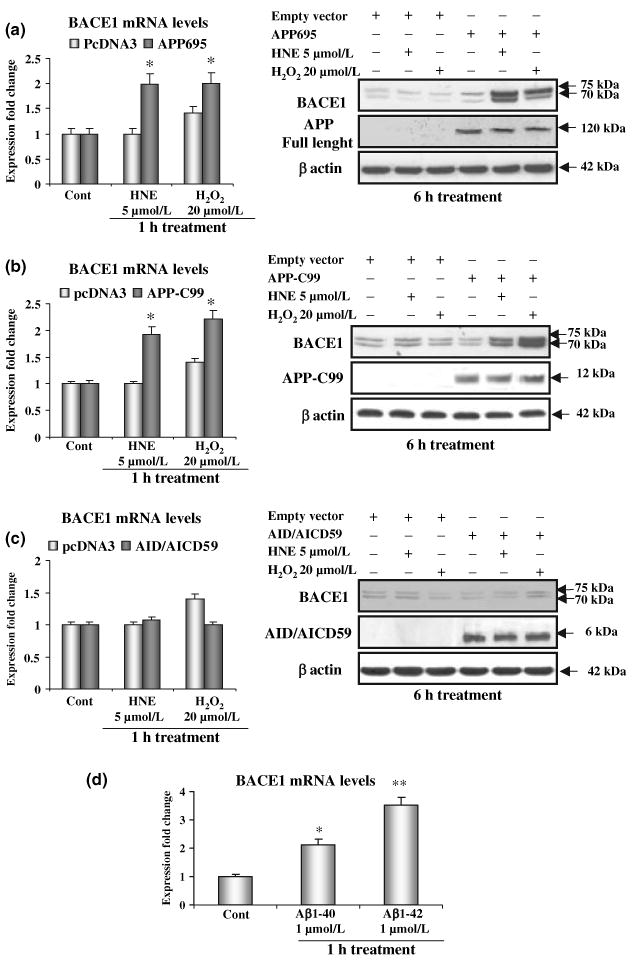
Role of amyloid precursor protein (APP) derivatives on the up-regulation of β-site amyloid precursor protein cleaving enzyme 1 (BACE1). (a) Transfection with APP695, significantly increases BACE1 expression and protein levels in APPko–mouse embryonic fibroblasts (MEFs). (b) Transfection with APP-C99, expressing the APP C-terminal domain resulting from the β-secretase cleavage restores the up-regulation of BACE1 in APPko-MEFs. (c) Transfection with APP intracellular domain (AID/AICD) 59, the APP derivative released by the γ-secretase cleavage at position 40 of the Aβ domain, has no effect on BACE1 expression and protein levels in APPko-MEFs. (d) Incubation with freshly prepared Aβ 1–42 induced a 3.5-fold increase in BACE1 mRNA in APPko–MEFs. Incubation with freshly prepared Aβ 1-40 induced a twofold increase in BACE1 mRNA in APPko–MEFs; *p < 0.05, **p < 0.02.
The activation of BACE1 depends on both the activity of the γ-secretase and the presence of APP, we therefore explored whether the APPko phenotype can be rescued by transfecting the cells with APP–Carboxy terminal fragments, or by treating them with Aβ peptides. We first transfected APPko–MEFs with APP-C99, the APP derivative resulting from the β-cleavage, both in normal conditions or under OS (Fig. 6b). An increase of BACE1 mRNA by 100–150% and of protein levels by 100–200%, were observed upon transfection with APP-C99 (Fig. 6b). We then looked at the APP intracellular domain (AID/AICD) as a potential player of the positive feedback on APP processing. To accomplish this APPko–MEFs were transfected with an AID/AICD construct (AID/AICD59) which expresses the APP C-terminal fragment of 59 amino acid residues, the latter resulting from the γ-secretase cleavage at position 40 of the Aβ domain. The expression of AID/AICD59 did not modify BACE1 mRNA and protein levels (Fig. 6c). This result is consistent with the Aβ peptides, resulting from γ-secretase cleavage of APP derivatives, being the signalling molecules in the loop. We incubated APPko–MEFs with Aβ 1–40 and Aβ 1–42 synthetic peptides, at a concentration of 1 μmol/L. The state of the Aβ preparations was examined by electron microscopy and no fibrillar or globular structures were detected (data not shown). APPko–MEFs showed a 3.5-fold increase of BACE1 mRNA after 1 h treatment with Aβ 1–42, whereas incubation with Aβ 1–40 produced a twofold increase in BACE1 expression (Fig. 6d).
Discussion
We found that both in cultured cells and in vivo, the up-regulation of BACE1 induced by OS is mediated by γ-secretase activity, and also that it requires the activation of the JNK/c-jun pathway. The results have three implications for the pathogenesis of sporadic AD. First, they suggest that OS, as effect of ageing, can increase the expression of both PS1/PEN2 and BACE1, thereby enhancing Aβ production. The activity of the γ-secretase is modified by PS1 mutations, as well as by molecules that interact directly with PS1, generally inhibiting the function of the γ-secretase complex (Beher et al. 2004; Matsuda et al. 2005; Cai et al. 2006; Heneka and O'banion 2007). OS is the only known factor able to augment the γ-secretase cleavage by increasing the expression of PS1, the catalytic subunit of the endoprotease. Secondly, our data reveal the existence of a positive feedback loop in which increased γ-secretase activity results in up-regulation of BACE1 expression, which is mediated in part by the release of Aβ peptides that act as signalling molecules (Tamagno et al., in preparation). Moreover, the effect of the up-regulation of BACE1 is not only the increased production of total Aβ, but also an augmented production of N terminally truncated Aβ species (Lee et al. 2003), which exert higher rates of aggregation and toxicity than the full-length peptides (Piccini et al. 2005). Thirdly, we showed that the activation of the positive feed-forward loop between the β- and the γ-secretase requires the JNK/c-jun signalling pathway. The JNK/c-jun signalling cascade, which is activated in the AD brain (Zhu et al. 2003a,b Lagalwar et al. 2006), responds to cell stress and mediates apoptosis (Kanzawa et al. 2006; Pugazhenthi et al. 2006; Zhang et al. 2007). The extracellular regulated kinases, that promote cell survival, are also activated in AD brain. Of note, extracellular regulated kinases inhibit the γ-secretase activity (Kim et al. 2005) and decrease the expression of BACE1 (Tamagno et al., submitted). Thus, the positive or negative cellular responses to OS parallel the activities of the β- and the γ-secretases that generate APP.
The sources and nature of OS in brain regions vulnerable in AD have not been established, but are likely to include processes involved in normal ageing. During normal ageing, and to a greater extent in AD, levels of protein oxidation and lipid peroxidation are increased (Butterfield and Kanski 2001; Cutler et al. 2004). Hydroxyl radicals, a ROS generated from H2O2, are potent inducers of membrane lipid peroxidation and the generation of HNE, a neurotoxic lipid peroxidation product, is associated with Aβ pathology in AD (Mark et al. 1997; Sayre et al. 1997). OS and HNE have been shown to induce activation of JNK/c-jun in neurons, which at high enough levels can trigger apoptosis (Camandola et al. 2000; Tamagno et al. 2003). Our findings suggest that the activation of JNK/c-jun mediates the increased expression of PS1 and BACE1 in response to OS and HNE. The latter mechanism is consistent with previous findings suggesting that c-jun can induce the expression of PS1 (Pastorcic and Das 2002) and BACE1 (Sambamurti et al. 2004).
Oxidative stress produces several effects that may contribute to synaptic function and cell death in AD (Mattson 2004; Bayer et al. 2006; Nunomura et al. 2006). We and other groups observed that OS induces in vitro an increased production of Aβ (Paola et al. 2000; Tamagno et al. 2005; Tong et al. 2005), and in this report we show a possible mechanism of this effect. OS correlates with ageing, the major risk factor of sporadic AD. This study presents evidence that OS contributes to Aβ accumulation. Aβ, in turn, induces OS and HNE production resulting in JNK/c-jun activation and increased levels of β- and γ-secretases, which further enhances Aβ production.
Acknowledgments
We thank Drs B. De Strooper and U. Mueller for providing PS- and APP-deficient MEF cells. We thank Dr L. D'Adamio for providing fusion constructs. We are grateful to Drs Pistoia and Pagnan for their help in analysing BACE1 activity. This work was supported by the Italian Ministry of Health and Regione Piemonte, De MARI and Telethon Foundations and by the Intramural Research Program of the National Institute on Aging of the NIH. Support was also provided by the National Institutes of Health (AG024028 to XWZ) and Philip Morris USA Inc. and Philip Morris International (MAS). [Correction added after online publication (17 December 2007): Additional sentence added at the end of Acknowledgements section]
Abbreviations used
- AD
Alzheimer's disease
- AID/AICD
amyloid precursor protein intracellular domain
- AP-1
activator protein 1
- APP
amyloid precursor protein
- Aβ
β amyloid
- BACE1
β-site amyloid precursor protein cleaving enzyme 1
- HNE
4-hydroxynonenal
- JIP
c-jun N-terminal kinase-inhibitory peptide
- JNK
c-jun N-terminal kinase
- MCAO
middle cerebral artery occlusion
- MEFs
mouse embryonic fibroblasts
- OS
oxidative stress
- PEN-2
presenilin enhancer-2
- PS
presenilin
- PSdko
PS1/PS2 double knock-out
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
References
- Bayer TA, Schafer S, Breyhan H, Wirths O, Treiber C, Multhaup G. A vicious cycle: role of oxidative stress, intraneuronal Abeta and Cu in Alzheimer's disease. Clin Neuropathol. 2006;25:163–171. [PubMed] [Google Scholar]
- Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcer I, Shearman MS. Selected non-steroidal anti-inflammatory drugs and their derivatives target gamma-secretase at a novel site. Evidence for an allosteric mechanism. J Biol Chem. 2004;279:43419–53426. doi: 10.1074/jbc.M404937200. [DOI] [PubMed] [Google Scholar]
- Borghi R, Patriarca S, Traverso N, Piccini A, Storace D, Garuti A, Cirmena G, Odetti P, Tabaton M. The increased activity of BACE1 correlates with oxidative stress in Alzheimer's disease. Neurobiol Aging. 2007;28:1009–1014. doi: 10.1016/j.neurobiolaging.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PGH, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nature Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Kanski J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech Ageing Dev. 2001;122:945–962. doi: 10.1016/s0047-6374(01)00249-4. [DOI] [PubMed] [Google Scholar]
- Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, Foster DA, Sisodia SS, Gorelick FS, Greengard P. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci USA. 2006;103:1941–1946. doi: 10.1073/pnas.0510708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal increases AP-1-binding activity through caspase activation in neurons. J Neurochem. 2000;74:159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Capell A, Steiner H, Willem M, Kaiser H, Meyer C, Walter J, Lammich S, Multhaup G, Haass C. Maturation and pro-peptide cleavage of beta secretase. J Biol Chem. 2000;275:30849–30854. doi: 10.1074/jbc.M003202200. [DOI] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nature Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizzary MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, Von Kretzchmar H, Von Hoch C, Sisodia S, Tremml P. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, O'banion MK. Inflammatory processes in Alzheimer's disease. J Immunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Van Gassen G, Benthair M, Nyabi O, Crassaert K, Muller U, Annaert W, De Strooper B. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA. 1999;96:11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Hartman D, Annaert W, et al. Gamma-secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J Cell Sci. 2003;116:1127–1136. doi: 10.1242/jcs.00292. [DOI] [PubMed] [Google Scholar]
- Holsinger RMD, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor β-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Kanzawa T, Iwado E, Aoki H, Iwamaru A, Hollingsworth EF, Sawaya R, Kondo S, Kondo Y. Ionizing radiation induces apoptosis and inhibits neuronal differentiation in rat neural stem cells via the c-jun NH2-terminal kinase (JNK) pathway. Oncogene. 2006;25:3638–3648. doi: 10.1038/sj.onc.1209414. [DOI] [PubMed] [Google Scholar]
- Kao SC, Krichevsky AM, Kosik KS, Tsai LH. BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem. 2004;279:1942–1949. doi: 10.1074/jbc.M309219200. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kim SK, Park HJ, Hong HS, Baik EJ, Jung MW, Mook-Jung I. ERK 1/2 is an endogenous negative regulator of the γ-secretase activity. FASEB J. 2005;20:512–514. doi: 10.1096/fj.05-4055fje. [DOI] [PubMed] [Google Scholar]
- Lagalwar S, Guillozet-Bongaarts AL, Berry RW, Binder LI. Formation of phosphor-SAPK/JNK granules in the hippocampus is an early event in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:455–464. doi: 10.1097/01.jnen.0000229236.98124.d8. [DOI] [PubMed] [Google Scholar]
- Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VM. Secretion and intracellular generated of truncated Abeta in beta-site amyloid-beta precursors protein-cleaving enzyme expressing human neurons. J Biol Chem. 2003;278:4458–4466. doi: 10.1074/jbc.M210105200. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Murphy MP, Mead TR, et al. A physiologic signaling role for the γ-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002;99:4697–4702. doi: 10.1073/pnas.072033799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Lopera F, Kosik KS, et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta42 deposition and severe cerebellar pathology. Nature Med. 1996;2:1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Giliberto L, Matsuda Y, Davies P, McGowan E, Pickford F, Ghiso J, Frangione B, D'Adamio L. The familial BRI2 gene binds the Alzheimer gene amyloid-beta precursor protein and inhibits amyloid-beta production. J Biol Chem. 2005;280:28912–28916. doi: 10.1074/jbc.C500217200. [DOI] [PubMed] [Google Scholar]
- Matsui T, Ingelsson M, Fukumoto H, Ramasamy K, Kowa H, Frosch MP, Irizarry MC, Hyman BT. Expression of APP pathway mRNAs and proteins in Alzheimer's disease. Brain Res. 2007;1161:116–123. doi: 10.1016/j.brainres.2007.05.050. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum DR, Shenkar R, Sheridan BC, Cain BS, Abraham E, Harken AH. Hemorrhage activates myocardial NFkappaB and increases TNF-alpha in the hearth. J Mol Cell Cardiol. 1997;29:2849–2854. doi: 10.1006/jmcc.1997.0506. [DOI] [PubMed] [Google Scholar]
- Minopoli G, Stante M, Napolitano F, et al. Essential roles for Fe65, Alzheimer's amyloid precursor binding protein, in the cellular response to DNA damage. J Biol Chem. 2006;282:831–835. doi: 10.1074/jbc.C600276200. [DOI] [PubMed] [Google Scholar]
- Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, Shirasawa T. Formation and stabilization model of the 42-mer Abeta radical: implications for the long-lasting oxidative stress in Alzheimer's disease. J Am Chem Soc. 2005;127:15168–15174. doi: 10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Castellani DJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- Owens CW, Belcher RV. A colorimetric micro-method for the determination of glutathione. Biochem J. 1965;94:705–711. doi: 10.1042/bj0940705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paola D, Domenicotti C, Nitti M, et al. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun. 2000;268:642–646. doi: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- Passer BJ, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D'Adamio L. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer's amyloid beta protein precursor. J Alzheimer Dis. 1999a;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- Passer BJ, Pellegrini L, Vito P, Ganjei JK, D'Adamio L. Interaction of Alzheimer's presenilin-1 and presenilin-2 with Bcl-X(L). A potential role in modulating the threshold of cell death. J Biol Chem. 1999b;274:24007–24013. doi: 10.1074/jbc.274.34.24007. [DOI] [PubMed] [Google Scholar]
- Pastorcic M, Das HK. Activation of transcription of the human presenilin 1 gene by 12-O-tetradecanoylphorbol 13-acetate. Eur J Biochem. 2002;269:5956–5962. doi: 10.1046/j.1432-1033.2002.03320.x. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA. Oxidative damage in Alzheimer's disease: the metabolic dimension. Int J Dev Neurosci. 2000a;18:417–421. doi: 10.1016/s0736-5748(00)00006-x. [DOI] [PubMed] [Google Scholar]
- Perry G, Raina AK, Nunomura A, Wataya T, Sayre LM, Smith MA. How important is oxidative damage? Lessons from Alzheimer's disease. Free Rad Biol Med. 2000b;28:831–834. doi: 10.1016/s0891-5849(00)00158-1. [DOI] [PubMed] [Google Scholar]
- Piccini A, Russo C, Gliozzi A, et al. Beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280:34186–34192. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Phansalkar K, Audesirk G, West A, Cabell L. Differential regulation of c-jun and CREB by acrolein and 4-hydroxynonenal. Free Rad Biol Med. 2006;40:21–34. doi: 10.1016/j.freeradbiomed.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Ravindranath V. Animal models and molecular markers for cerebral ischemia-reperfusion injury in brain. Methods Enzymol. 1994;233:610–619. doi: 10.1016/s0076-6879(94)33064-6. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004;18:1034–1036. doi: 10.1096/fj.03-1378fje. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Tabner BJ, El-Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y, Cooper LJ, Fullwood NJ, Allsop D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer's disease and familial British dementia. J Biol Chem. 2005;280:35789–35792. doi: 10.1074/jbc.C500238200. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, Obbili A, et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Parola M, Guglielmotto M, Santoro G, Bardini P, Marra L, Tabaton M, Danni O. Multiple signaling events in amyloid β-induced, oxidative stress-dependent neuronal apoptosis. Free Rad Biol Med. 2003;35:45–58. doi: 10.1016/s0891-5849(03)00244-2. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Parola M, Bardini P, et al. β-Site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M. The various aggregation states of beta-amyloid 1-42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Rad Biol Med. 2006;41:202–212. doi: 10.1016/j.freeradbiomed.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Tong Y, Zhou W, Fung V, Christensen MA, Qing H, Sun H, Sun X, Song W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. Neural Transm. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Takaaki N, Minomiya H, Sugimoto H, Kinoshita A, Shimohama S. Chatacterization of sequential N-Cadherin cleavage by ADAM10 and PS1. Neurosci Lett. 2006;402:278–283. doi: 10.1016/j.neulet.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LB, Lindholm R, Yan R, et al. Elevated β-secretase expression and activity detected in sporadic Alzheimer disease. Nature Med. 2003;9:3. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Yao M, Nguyen TV, Pike CJ. Beta-amyloid-induced neuronal apoptosis involves c-jun N-terminal kinase-dependent downregulation of Bcl-w. J Neurosci. 2005;25:1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Wu DN, Han D, Zhang GY. Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signalling in ischemic brain injury. FEBS Lett. 2007;581:495–505. doi: 10.1016/j.febslet.2006.12.055. [DOI] [PubMed] [Google Scholar]
- Zhu X, Ogawa O, Wang Y, Perry G, Smith MA. JKK1, an upstream activator of JNK/SAPK, is activated in Alzheimer's disease. J Neurochem. 2003a;85:87–93. doi: 10.1046/j.1471-4159.2003.01645.x. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Lee HG, Nunomura A, Tabaton M, Petersen RB, Perry G, Smith MA. Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxid Redox Signal. 2003b;5:571–576. doi: 10.1089/152308603770310220. [DOI] [PubMed] [Google Scholar]