

Abstract
The role of ryanodine-sensitive intracellular Ca2+ stores present in nonmuscular cells is not yet completely understood. Here we examine the physiological parameters determining the dynamics of caffeine-induced Ca2+ release in individual fura-2–loaded sympathetic neurons. Two ryanodine-sensitive release components were distinguished: an early, transient release (TR) and a delayed, persistent release (PR). The TR component shows refractoriness, depends on the filling status of the store, and requires caffeine concentrations ≥10 mM. Furthermore, it is selectively suppressed by tetracaine and intracellular BAPTA, which interfere with Ca2+-mediated feedback loops, suggesting that it constitutes a Ca2+-induced Ca2+-release phenomenon. The dynamics of release is markedly affected when Sr2+ substitutes for Ca2+, indicating that Sr2+ release may operate with lower feedback gain than Ca2+ release. Our data indicate that when the initial release occurs at an adequately fast rate, Ca2+ triggers further release, producing a regenerative response, which is interrupted by depletion of releasable Ca2+ and Ca2+-dependent inactivation. A compartmentalized linear diffusion model can reproduce caffeine responses: When the Ca2+ reservoir is full, the rapid initial Ca2+ rise determines a faster occupation of the ryanodine receptor Ca2+ activation site giving rise to a regenerative release. With the store only partially loaded, the slower initial Ca2+ rise allows the inactivating site of the release channel to become occupied nearly as quickly as the activating site, thereby suppressing the initial fast release. The PR component is less dependent on the store's Ca2+ content. This study suggests that transmembrane Ca2+ influx in rat sympathetic neurons does not evoke widespread amplification by CICR because of its inability to raise [Ca2+] near the Ca2+ release channels sufficiently fast to overcome their Ca2+-dependent inactivation. Conversely, caffeine-induced Ca2+ release can undergo considerable amplification especially when Ca2+ stores are full. We propose that the primary function of ryanodine-sensitive stores in neurons and perhaps in other nonmuscular cells, is to emphasize subcellular Ca2+ gradients resulting from agonist-induced intracellular release. The amplification gain is dependent both on the agonist concentration and on the filling status of intracellular Ca2+ stores.
Keywords: CICR, fura-2, ryanodine, calcium release, calcium signaling
introduction
Ca2+-induced Ca2+ release (CICR)1 is crucial for excitation-contraction coupling in mammalian cardiac muscle. According to a widely accepted scheme, the initial depolarization of the sarcolemmal membrane triggers Ca2+ influx by opening voltage-gated Ca2+ channels. This Ca2+ influx activates ryanodine receptor/Ca2+ release channels (RyR) on the sarcoplasmic reticulum, from which Ca2+ is then released into the cytosol, producing the large Ca2+ transients required to initiate contraction (Wier, 1990). Experiments in rat cardiac myocytes have shown that voltage-gated Ca2+ influx is the most efficient stimulus to trigger Ca2+ release (Sham et al., 1995). A recent model proposes that single L-type Ca2+ channels in the membrane of t-tubules are functionally coupled and in close proximity to small clusters or RyRs in the sarcoplasmic reticulum, which together constitute a Ca2+ release unit (Stern, 1992; Cannell et al., 1995).
The molecular elements responsible for CICR (voltage-gated Ca2+ channels and ryanodine receptor channels) are also present in vertebrate neurons (reviewed by Kostyuk and Verkhratsky, 1994; Kuba, 1994; Verkhratsky and Shmigol, 1996). This leads to the notion that Ca2+ mobilization from intracellular stores mediated by RyR channels may constitute an important pathway for Ca2+ signaling (McPherson and Campbell, 1993). Nonetheless, several studies indicate that under physiological conditions, neuronal calcium transients originated by Ca2+ influx are not amplified to a large extent by CICR. In the best cases, only a modest augmentation and prolongation of the calcium transient over that which would be produced by the calcium influx alone has been observed (Thayer et al., 1988; Friel and Tsien, 1992; Nohmi et al., 1992; Thayer and Miller, 1990; Ivanenko et al., 1993; Usachev et al., 1993; Kostyuk and Verkhratsky, 1994; Kuba, 1994; Shmigol et al., 1995). Moreover, prolonged depolarizations are often needed to raise [Ca2+] enough to initiate CICR (Llano et al., 1994; Verkhratsky and Shmigol, 1996). This behavior is characteristic of low feedback loop gain models of CICR (Stern, 1992).
If nerve cells are not optimized for widespread amplification of Ca2+ transients resulting from voltage-gated Ca2+ influx, what is the role of their ryanodine-sensitive Ca2+ stores? The answer to this question requires a better understanding of the functional organization of ryanodine receptors and neuronal caffeine-sensitive stores. For instance, we need to establish if released Ca2+, by interacting with regulatory sites on the RyR channel, can participate in feedback loops that may promote or prevent further release. A related question is whether or not neighboring Ca2+ release channels and calcium stores are diffusionally coupled. Finally, we need to discern to what extent calcium depletion of intracellular reservoirs control the release process.
The goal of this study was to analyze the mechanisms that determine the dynamics of Ca2+ release from ryanodine-sensitive intracellular Ca2+ stores in rat sympathetic neurons after its activation by a rapid caffeine application. Since it is likely that endogenous agonists act similarly to caffeine (Hua et al., 1994), the conclusions of our study should be applicable to Ca2+ release initiated by physiological phenomena as well (Pozzan et al., 1994).
This paper presents unequivocal evidence of CICR phenomena when trigger Ca2+ is released at an adequately fast rate from intracellular stores. Our data also suggest that the regenerative release is interrupted by a combination of rapid depletion of releasable Ca2+ and Ca2+-dependent inactivation of release. The most salient features of caffeine-induced Ca2+ mobilization described in this paper could be accounted for with a compartmentalized linear diffusion model which contains a caffeine-sensitive intracellular Ca2+ store capable of releasing Ca2+ in response to changes in cytosolic [Ca2+], separated from the plasma membrane by a diffusional space containing fixed Ca2+ buffers.
materials and methods
Tissue Culture of Rat Sympathetic Neurons
Cultures of superior cervical ganglion neurons were prepared as follows: 10-d-old rats of either sex were used. Ganglia were removed under ether anesthesia and aseptic conditions. They were cleaned and chopped, and then incubated in Ca2+ and Mg2+-free Hank's medium (Sigma Chemical Co., St. Louis, MO) with 1 mg/ml trypsin (Worthington Biochemical Corp., Freehold, NJ) and 2 mg/ml DNAse I (Sigma Chemical Co.) for 30 min at 37°C. After digestion, trypsin was inactivated by dilution in DMEM medium (Gibco BRL, Gaithersburg, MD) containing 10% FCS and 1 mg/ml trypsin inhibitor (Sigma Chemical Co.). The tissue was then incubated in Hank's medium with 2 mg/ml collagenase (Worthington Biochemical Corp.) and 2 mg/ml DNAse I for 30 min at 37°C. Tissue fragments were then triturated with a fire-polished siliconized Pasteur pipette. Cell suspension was centrifuged at 800 rpm for 10 min, washed twice in Hank's medium (Gibco BRL) and resuspended in fresh DMEM (Gibco BRL) supplemented with 10% FCS (Gibco BRL). Cells were seeded on poly-lysine-treated No. 1 glass coverslips (1 × 105cells per well) and maintained for up to 3 wk supplemented with 20 ng/ml of 7S nerve growth factor (Sigma) at 37°C in a humidified atmosphere of 95% air and 5% CO2. Medium was changed three times per week. Most experiments were carried out with cells that had been in culture from 1 to 5 d.
Measurements of Intracellular Ca2+ and Sr2+ Concentrations
Methods are described in detail elsewhere (Hernández-Cruz et al., 1995). Briefly, a coverslip containing sympathetic neurons was transferred to a recording chamber (Mod. RC-25; Warner Instruments, Hamden, CT) on an inverted microscope (Nikon Diaphot TMD; Nikon Corp., Tokyo, Japan). Cells were loaded with fura-2 by incubation with the acetoxymethyl (AM) ester form of the dye (fura-2/AM; Molecular Probes, Eugene, OR), at a final concentration of 1 μM, with no dispersing agents added. Cells were allowed to load for 30–45 min at room temperature and then rinsed continuously for another 15 min before the beginning of the experiments.
Ca2+ or Sr2+ levels were determined by recording pairs of images using alternating illumination with 340- and 380-nm excitation. Dual wavelength excitation was provided by two nitrogen pulsed lasers (3-ns pulse duration), one emitting at its natural wavelength of 337 nm and the second one dye-tuned at 380 nm. These lasers were triggered alternatively at frequencies ranging from 2 to 15 Hz under computer control. (Biolase Imaging System, Newton MA). Background images at 340- and 380-nm illumination were obtained from an area of the coverslip free of cells. These images were stored separately and used for on-line background subtraction. We found that a correction for cell autofluorescence was not essential for calibration purposes. The key elements of the fluorescence Ca2+ imaging system were a high numerical aperture UV objective (Nikon UV-F 100X, 1.3 N.A.), an intensified charge coupled device camera (c2400-87; Hamamatsu, Bridgewater, NJ), and the Biolase Imaging System running under their FL-2 software. The system allows real-time simultaneous acquisition of fluorescence measurements from multiple areas of interest placed on individual cells or within a single cell. All Ca2+ determinations in this study were obtained from entire cells. Ca2+ or Sr2+ concentrations were calculated from fluorescence measurements at 340- and 380-nm excitation wavelengths using the formula:
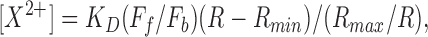 |
where the calculated dissociation constant (K D) for fura-2 is 300 nM for Ca2+ and 7.6 μM for Sr2+ (Kwan and Putney, 1990), F f/F b is the ratio of fluorescence values for X 2+-free/X 2+-bound indicator at 380-nm excitation, R is the ratio of fluorescence at 340/380 nm for the unknown [X 2+], and R min, R max are the ratio of fura-2 fluorescences at 340/380 nm of X 2+-free and X 2+-bound fura-2. The values of F f/F b, R min, and R max for Ca2+ were determined by measuring under identical conditions the fluorescence of a glass capillary 100-μm in diameter containing calibration solutions with 50–100 μM fura-2 (pentapotassium salt; Molecular Probes) and known Ca2+ concentrations in the range from 10 nM to 40 μM. Sr2+ values were determined similarly, using solutions containing 100 μM fura-2 and either no added Sr2+ or 1 mM Sr2+. Although in situ calibrations were attempted in initial experiments, we found it very difficult to manipulate [Ca2+] over the required range. Our Ca2+ and Sr2+ measurements, based exclusively on the in vitro calibrations could be underestimated to some extent because of effects of viscosity and dye binding to cytoplasmic constituents (Konishi et al., 1988). In previous experiments, we estimated that fura-2 reached intracellular concentrations between 30 and 50 μM. This was done by comparing fluorescence levels attained in similar cells 10 min after breaking-in with patch pipettes filled with known concentrations of fura-2 pentapotassium (range 25–100 μM). By comparing responses from cells loaded with fura-2 for 30–45 min with those from cells loaded with much less fura-2 (10-min incubation), we concluded that the additional Ca2+ buffer activity introduced by the dye did not affect significantly the magnitude or the time course of Ca2+ transients (unpublished data).
Solutions
Cells were continuously superfused with a recording solution containing (in mM) NaCl 130, KCl 3, CaCl2 2, MgCl2 2, NaHCO3 1, NaH2PO4 0.5, HEPES-Na 5, and glucose 5, pH 7.4. In some experiments CaCl2was replaced equimolarly by SrCl2. Test solutions were pressure-applied (10 psi) via independent puffer pipettes located within 100 μm from the cell under examination. In some experiments, drugs (ryanodine, tetracaine, and the acetoxymethyl ester of 1,2-bis(2-aminophenoxy)ethane-NNN ′N ′-tetraacetic acid [BAPTA-AM]) were applied via a third puffer pipette. Application of test solutions and drugs were separately controlled by solenoid valves of two Picospritzer II devices (General Valve, Fairfield, NJ) governed by an 486/AT computer running pClamp 5.1 (Axon Instruments, Foster City, CA). Control experiments showed that with this procedure, the external medium surrounding the cell is replaced within <100 ms. Most experiments were conducted in cells that had been kept in culture from 1 to 4 d. Experiments were carried out at 22–23°C. Test solutions used were: caffeine 10 or 20 mM, or tetracaine 500 μM, dissolved in normal saline, and a depolarizing solution containing 140 mM KCl, 10 mM HEPES-Na and either 10 mM CaCl2 or 10 mM SrCl2.
Sr2+ Replacement Procedure
To substitute Sr2+ for Ca2+ both externally and internally, a coverslip containing sympathetic neurons was extensively superfused (45 min or longer) with normal external solution containing no Ca2+ (no Ca2+ added plus 500 μM EGTA) and 2 mM SrCl2. During this period, the dish was exposed three times (1-min duration each) to a Sr2+-containing depolarizing solution (140 mM KCl, 2 mM SrCl2, 10 mM HEPES-Na, pH 7.35). In addition, to promote substitution of Ca2+ with Sr2+ in the intracellular stores, cells were bathed three times with saline containing 2 mM Sr2+ and 10 mM caffeine (1-min duration each at 10-min intervals). The Sr2+ replacement procedure had been completed before the beginning of fluorescence measurements.
Mathematical Modeling of Ca2+ Mobilization from Intracellular Stores in Rat Sympathetic Neurons
The model consists of a linear arrangement representing the extracellular space, as well as the cytosol and the endoplasmic reticulum (ER), with boundary conditions located both at the plasma and at the ER membranes. The ER and the plasma membrane are coupled via n diffusional compartments. In the model, Ca2+ diffuses freely between compartments and binds to fixed Ca2+ buffers. Ca2+ fluxes due to diffusion exchange with neighboring compartments and binding-unbinding to fixed buffers were computed by integrating Fick's law difference equations and mass action law differential equations, respectively. The boundary condition representing the plasma membrane allows Ca2+ influx through voltage-gated Ca2+ channels and Ca2+ extrusion via Ca2+ pumps. Similar Ca2+ pumps are located at the boundary condition representing the membrane of the ER compartment, which also contains RyRchannels which allow Ca2+ release into the cytosol. These channels are regulated by two different cytosolic Ca2+ binding sites operating with a first order kinetic scheme, which control activation and inactivation of the channel, respectively, and a lumenal regulatory binding site which controls channel conductance. The degree of activation or inactivation of this pathway is proportional to the fraction of total sites that are occupied with Ca2+. Caffeine-induced stimulation of RyRs was simulated by an abrupt change in the on-rate for Ca2+ binding to the activation sites, with all remaining rate constants unaltered. A more detailed description of the model is included as an appendix.
results
Rationale of Caffeine-induced Ca2+ Release Experiments
Our basic experimental design allows the controlled activation of calcium influx through voltage-gated Ca2+ channels and Ca2+ release from intracellular stores in intact fura-2–loaded neurons (see Hernández-Cruz et al., 1995). Caffeine application provides two main advantages to analyze CICR phenomena in nerve cells with respect to voltage-gated Ca2+ influx. First, since Ca2+is directly released from intracellular stores, [Ca2+] in the microenvironment surrounding RyRs increases at a faster rate than when Ca2+ diffuses from distant sources (i.e., the plasma membrane). Secondly, by increasing Ca2+ affinity for the RyRs activation sites, caffeine promotes CICR (Sitsapesan and Williams, 1990). At the concentration used in these experiments (10 mM), caffeine produces half-maximal activation of release (Akaike and Sadoshima, 1989; Uneyama et al., 1993). This allows both up- and down-regulation of release by [Ca2+] and other modulators (Hernández-Cruz et al., 1995). Previous work has shown that externally applied caffeine enters readily into cells, reaching equilibrium with extracellular concentration in <0.2 s (Hernández-Cruz et al., 1990; O'Neill et al., 1990; Kuba et al., 1990). Thus, the amount of Ca2+ released will depend on three governing factors: (a) the prevalent cytosolic Ca2+ concentration, (b) the number of RyRs opened per time unit by the drug, and (c) the calcium content of the store (Friel and Tsien, 1992; Hernández-Cruz et al., 1995; Shmigol et al, 1996). Changes of cytoplasmic [Ca2+] resulting from Ca2+ fluxes through the ER membrane can directly enhance (or inhibit) further release by interacting with regulatory Ca2+-binding sites on the RyR channels (Bezprozvanny et al., 1991; Györke and Patade, 1994; Meissner, 1994).
Two Kinetic Components of Caffeine-induced Ca2+ Release
Fig. 1 A depicts Ca2+ transients recorded from a fura-2– loaded sympathetic neuron in response to three brief applications of a high-K+ solution (arrows; see materials and methods), followed by several caffeine applications (10 mM, 40 s). Ca2+ transients elicited by high-K+ and caffeine are termed here K+- and C-Ca2+ transients, respectively. Conditioning high-K+ pulses were applied to replete Ca2+ stores before the first use of caffeine. Notice that the first Ca2+ release comprises an early, fast release that decays before the end of the pulse, (transient release component, TR), followed by a slower and more sustained release (persisting release component, PR). When caffeine was applied again 140 s later, the TR component was obliterated, and only the PR component remained. Subsequent caffeine applications only produced smaller PRs with slower onsets. Fig. 1 B, obtained from a similar experiment, shows superimposed responses to the first and second caffeine applications. Both responses initiated with similar latencies, but the second one had a slower rate of rise and did not bring about a TR component. The first derivatives of these [Ca2+]records are shown in the inset (Fig. 1 C). Notice that during the second caffeine response the initial rate of rise was about half of that observed during the first response. The mean amplitudes of TR and PR components of release, measured shortly after the last conditioning high K+ pulse in 38 cells was TR = 1,684.6 ± 140 nM; PR = 326.8 ± 9.6 nM (mean ± SE).
Figure 1.

(A) Ca2+ transients recorded from a fura-2–loaded sympathetic neuron in response to three applications (500 ms each) of a solution containing high K+ and 10 mM Ca2+ (vertical arrows; see materials and methods) and then to five applications of 10 mM caffeine (40 s each) at intervals of 140 s. Notice that only the first caffeine-induced Ca2+ release showed a transient release (TR) followed by a persistent release (PR) component. (B) Comparison, in a different cell, of the kinetics of intracellular Ca2+ release elicited by the first (continuous line) and the second (dotted line) caffeine applications. Only the PR component is elicited by the second application. (C) First derivatives of the upstroke of the [Ca2+]signals shown in B. Numbers next to traces indicate the initial rate of rise. Horizontal arrows indicate resting [Ca2+].
When exposed to caffeine for the first time, many cells show TR and PR components, even in the absence of conditioning high K+ applications (see Fig. 5). The TR component recuperates if caffeine is applied again 10–15 min later (data not shown). This slow recovery is greatly speeded up if one or more conditioning K+-Ca2+ transients are elicited during the interval between caffeine applications. This behavior is consistent with a use-dependent depletion of a slowly replenished caffeine-sensitive Ca2+ store (Thayer et al., 1988; Friel and Tsien, 1992; Usachev et al., 1993).
Figure 5.

Recovery of the TR component in a sympathetic neuron maintained for 2 d in culture. (A) In this experiment, each caffeine application (except the first one) was preceded with a conditioning K+-Ca2+ transient, whose magnitude was varied by changing the duration of the high K+application between 50 ms and 2 s. (B) Relationship between the area under each C-Ca2+ transient (labeled a through l) and the area under the preceding K+-Ca2+ transient. The gradual increase of the area under the caffeine response suggests a correlation between the filling status of the store and the magnitude of the subsequent Ca2+ release. The generation of a TR component coincides with an abrupt area increase (trace k), suggesting an all-or-none regenerative mechanism. Further increasing the area under the conditioning K+-Ca2+ transient did not increase the size of the TR component nor the area under the C-Ca2+ transient (see trace l). Horizontal arrow indicates resting [Ca2+].
Both Kinetic Components of Caffeine-induced Ca2+ Mobilization Result from Intracellular Ca2+ Release
It has been reported that caffeine can directly inhibit K+ currents in sympathetic neurons (Akaike and Sadoshima, 1989; Dryer et al., 1995). Therefore C-Ca2+ transients could result in part from membrane depolarization as well as intracellular Ca2+ release. It also has been shown that caffeine can directly activate Ca2+ permeable channels in smooth muscle cells (Guerrero et al., 1994). The most rigorous test to rule out these potential problems would be to prevent completely and selectively Ca2+ release from caffeine-sensitive stores. The plant alkaloid ryanodine is the most specific Ca2+ release blocker known. Nevertheless, when ryanodine locks the release channel in a subconducting state (Russeau et al., 1987), it can significantly deplete Ca2+ stores, hence increasing intracellular Ca2+ levels by reducing the capacity of the cell to remove calcium (Kuba, 1994). This effect is most apparent when ryanodine (10–20 μM) is applied continuously (see Fig. 8). In preliminary experiments we noticed that 20 μM of ryanodine can inhibit completely and irreversibly intracellular release after applications as brief as 5 s, without seriously affecting calcium homeostasis. Fig. 2 A shows the use-dependent inhibition of caffeine responses 1 and 4 min after a ryanodine application 5 s in duration. Fig. 2, B and C, depict the first derivatives of selected segments of these [Ca2+] records. Ryanodine treatment did not change significantly basal Ca2+ levels (Fig. 2 A), or the magnitude or kinetics of the K+-Ca2+ transients. The time constant of an exponential function fitted to the decay phase of the K+-Ca2+ transients remained between 2.1 and 2.3 s. Also, the peak rate of rise (7.8 μM s−1 in the control response), diminished only slightly after 1 and 4 min of exposure to the drug (7.1 and 6.4 μM s−1, respectively). Nevertheless, the amplitude and rate of rise of the C-Ca2+ transients dropped from 2.8 μM s−1 to 0.74 and 0.08 μM s−1 after 1 and 4 min of ryanodine exposure (Fig. 2 C). Note that at 1 min post ryanodine, caffeine elicits a delayed and partially inhibited TR component, whereas only a reduced PR persists after 4 min. 10 min after ryanodine application, caffeine responses were completely abolished, both in this cell and in five other cells similarly examined (data not shown). Three conclusions can be drawn from these results: (a) brief ryanodine applications can block Ca2+ release without affecting resting Ca2+ levels or the kinetics of K+-Ca2+ transients, (b) both kinetic components of the caffeine induced-Ca2+ mobilization result entirely from intracellular Ca2+ release, and (c) the TR component appears more susceptible to use-dependent inhibition of release by ryanodine than the PR component.2
Figure 8.

Recording of fura-2 fluorescence changes in a sympathetic neuron after undergoing the Sr2+ replacement procedure (see materials and methods). High K+ (1 s; short arrows) and 10 mM caffeine (20 s; long arrows) were applied alternatively with independent puffer pipettes. Near the middle of the recording, 20 μM ryanodine was added to the bath, and 2 min later, alternate stimulation with high-K+ and caffeine restated. Caffeine responses are eliminated in a use-dependent manner after exposure to ryanodine (asterisks). Horizontal arrow indicates resting [Sr2+].
Figure 2.

Use-dependent ryanodine inhibition of caffeine- induced Ca2+ release. (A) Ca2+ transients elicited in a sympathetic neuron following the applications of high-K+ for 0.5 s (vertical arrows) and 10 mM caffeine for 20 s (horizontal bar). Traces shown were obtained before (continuous trace) and after 1 or 4 min (dotted and dashed traces, respectively) of a single application of 20 μM ryanodine for 5 s with a third puffer pipette. (B) First derivatives of the K+-Ca2+ transients indicated. The magnitude and kinetics of these Ca2+ transients were not significantly affected by ryanodine. (C) First derivatives of caffeine responses. The rate of rise fell from 2.8 μM s−1 before to 0.74 and 0.08 μM s−1 after 1 and 4 min of ryanodine application. Caffeine responses were completely abolished 10 min after ryanodine application (not shown). Horizontal arrow indicate resting [Ca2+].
The Effects of Caffeine Pretreatment on the Kinetic Behavior of Subsequent Responses Are Not Due to Caffeine Desensitization
A conceivable explanation for the effects of caffeine pretreatment on the kinetic behavior of subsequent responses is that it produces a desensitization of Ca2+ stores that persist for several minutes after the agonist is removed. To explore this possibility, the effect of pretreating the cell with a brief conditioning caffeine application on a test response was examined. To distinguish between the effects of caffeine per se from those resulting from depletion of the Ca2+ store, we took advantage of the Ca2+-induced inhibition of Ca2+release characteristic of these cells (Hernández-Cruz et al., 1995). Conditioning caffeine applications were initiated at different intervals from a standard K+-Ca2+ transient. Caffeine, applied at intervals shorter than 10 s (when [Ca2+]is still relatively high), is ineffective at releasing Ca2+. When this interval is increased, caffeine becomes progressively more effective. Fig. 3 A shows that when the conditioning caffeine application does not release any Ca2+, a test caffeine response 90 s later displays both TR and PR components (continuous trace). The same is true when only a small Ca2+ release occurs during the conditioning caffeine application (dashed trace). However, when the conditioning caffeine application brings about a substantial Ca2+ release, the TR component of the succeeding test response is completely abolished (dotted trace). This experiment rules out desensitization of the release, and clearly establishes that the diminution of the TR component during the second caffeine application only occurs if the first application produces substantial opening of Ca2+-release channels and/or Ca2+ efflux from the intracellular stores. This inference is further supported by experiments shown in Fig. 3, B and C. First, two identical applications of 10 mM caffeine were given at an interval of 2 min (continuous traces, Fig. 3 B and C). The second response shown in Fig. 3 C displays the usual inhibition of the TR component. The experiment was then repeated, but now the second response was elicited by delivering 20-mM caffeine with a third pipette, instead of 10 mM (dotted trace). This experiment was designed to try to activate more efficiently desensitized Ca2+ stores with a higher dose of caffeine. Although the second response elicited with 20 mM caffeine was slightly larger (Fig. 3 C, arrow) and had a faster rate of rise (Fig. 3 C, inset), it was still unable to trigger a complete TR response. Similar results were obtained in experiments where even higher caffeine concentrations (i.e., 40 mM) were used to elicit the second response (data not shown).
Figure 3.

Obliteration of the TR component is not due to caffeine desensitization. (A) Caffeine 10 mM (5 s) applied at different intervals from a high K+-initiated Ca2+ transient evokes different amounts of Ca2+ release. When caffeine application does not release any Ca2+ (continuous trace), or releases only a small amount (dashed trace), a test caffeine response 90 s later displays both TR and PR components. When the conditioning caffeine application brings about a substantial Ca2+ release (dotted trace), the transient component of the succeeding test response is abolished. (B and C) Two applications of 10 mM caffeine were given at an interval of 2 min. The first response shows transient and sustained components (B, continuous trace), while the second response displays the usual inhibition of the transient component (C, continuous trace). The experiment was repeated, but now the second response was elicited by delivering 20 mM caffeine with a third pipette (B and C, dotted traces). Notice that caffeine, even at this higher concentration, was unable to trigger a TR component during the second response (arrows). (Inset) Comparison of the first derivatives of traces shown in C. Horizontal arrows indicate resting [Ca2+].
The Kinetic Behavior of Caffeine-induced Ca2+ Release Depends upon the Filling Status of Intracellular Stores
As mentioned above, the obliteration of the TR component during subsequent applications of caffeine is a consequence of the opening of Ca2+ release channels and/or Ca2+ liberation from internal stores during the first application. Experiments were conducted to try to decide between these possibilities. We compared the kinetics of release under conditions where presumably an equal number of release channels were opened, but with the Ca2+ store filled up to different degrees. Fig. 4 A shows the gradual recovery of the TR component after the application of one to four small “filler” conditioning K+-Ca2+ transients. Fig. 4 B depicts the first derivative of Ca2+ concentration records shown in Fig. 4 A. The plot in Fig. 4 C illustrates the relationship between the conditioning K+-Ca2+ transients versus the resulting Ca2+ release (open circles) and the magnitude of the peak rate of rise of the C-Ca2+ transients (filled squares). In this figure, as well as in Fig. 5, the areas under the K+- and C-Ca2+ transients are plotted rather than peak Ca2+ concentrations to better represent the magnitude of the Ca2+ increase. Notice that a TR component was not produced in this cell when caffeine was applied without a conditioning K+-Ca2+ transient. It is clear from Fig. 4 C that both the magnitude of the C-Ca2+ transients and their peak rate of rise reach saturation after three to four conditioning K+-Ca2+ transients. These results are expected if the generation of the TR response requires sufficient loading of intracellular Ca2+ reservoirs with limited capacity for Ca2+ storage. Since the supply of additional Ca2+ by transmembrane Ca2+ influx readily reestablishes the transient component of release, a long-lasting “fatigue” of Ca2+ release channels cannot explain the TR abatement phenomena.
Figure 4.

Recovery of the TR component in a sympathetic neuron maintained in culture for 10 days. (A) Influence of 1–4 filler K+-Ca2+ transients (each initiated by 250-ms depolarizations with high K+ solution) on the Ca2+ release produced by application of 10 mM caffeine for 25 s (horizontal bars). (B) First derivative of records shown in A. (C, open circles and left y-axis) Relationship between the total area under the conditioning K+-Ca2+ transients and the area under the resulting Ca2+ release. (Filled squares and right y-axis) Peak rate of rise obtained from the derivatives shown in B. A transient component was absent from this cell when caffeine was applied without a conditioning K+-Ca2+ transient. Notice that both the magnitude and peak rate of rise of caffeine responses reached saturation after 3–4 conditioning high K+applications. Horizontal arrow indicates resting [Ca2+].
A somewhat different behavior often observed in the recovery of the TR component is shown in Fig. 5. Unlike the cell in Fig. 4, this cell displayed a TR response when first challenged with caffeine (Fig. 5 A, trace a). Cells exhibiting this behavior are common during the first few days in culture. Here, the replenishment of the Ca2+ store was carried out by preceding each caffeine application with a single conditioning K+-Ca2+ transient, whose magnitude was regulated by varying the duration of the high K+application. As shown in the plot shown in Fig. 5 B, the area under the C-Ca2+ transient increases gradually as the area under the filler Ca2+ transient increases. This suggests a close correlation between the filling status of the store and the magnitude of the subsequent Ca2+ release. Interestingly, an abrupt increase in the magnitude of the C-Ca2+ transient occurs when the area under the filler Ca2+ transient reaches a critical value. This discontinuity coincides with the generation of the TR component (Fig. 5 A, trace k). These results support the notion that an all-or-none regenerative mechanism underlies the TR component. Further increasing the area under the K+-Ca2+ transient did not increase the size of the TR component nor the area under the C-Ca2+ transient (Fig. 5 A, trace l), as if the release were self-limited or the stores had a finite capability to accumulate Ca2+. Previous studies have shown that Ca2+ stores of intact cardiac myocytes reach a limiting Ca2+ content that cannot be exceeded regardless of the loading stimulation protocol used (Bassani et al., 1995). Feedback inhibition of Ca2+ uptake by Ca2+ load of intracellular stores (Favre et al., 1996) may explain the apparent limiting Ca2+ content. Notice that the response elicited by the initial caffeine application (trace a) is very similar to that produced when the reservoir is full (see traces k and l). This suggests that in resting sympathetic neurons a mechanism exists that ensures that caffeine-sensitive Ca2+ stores remain filled nearly to the limit of their capacity, thereby allowing an expeditious Ca2+ efflux whenever RyRs open, either in response to Ca2+ or other endogenous agonist.3
We also tested whether or not lower concentrations of caffeine were capable of eliciting a TR response provided that Ca2+ stores were replete. Under conditions where 10 mM caffeine invariably gave a TR component, 3 or 5 mM of caffeine was unable to produce it (data not shown). These results indicate that the development of the TR component requires both a high Ca2+ content of intracellular stores and the activation of a sufficient number of Ca2+ release channels. Either of these conditions, occurring separately, do not support a TR response.
Tetracaine and Intracellular BAPTA Suppress the TR Component
Local anesthetics tetracaine and procaine affect excitation-contraction coupling by reducing Ca2+-induced Ca2+ release (Endo, 1985). Thus, if the TR component was the result of regenerative activation by CICR, it should be reduced or even abolished by tetracaine. For these experiments, tetracaine was applied from a third puffer pipette for 30 s (only during the interval between the last conditioning K+-Ca2+ transient and the test caffeine application). This maneuver allowed us to explore the effects of tetracaine on the kinetics of the release mechanism without affecting voltage-gated Ca2+ influx or Ca2+ loading of intracellular stores. Fig. 6 A shows control responses from a sympathetic neuron to the filler high K+ applications and then to 10 mM caffeine. A TR component is clearly observed in response to caffeine. As shown in Fig. 6 B, the use of 500 μM tetracaine before the caffeine application completely suppressed the TR component, with less effect on the PR component. The TR component recovered after 5 min of tetracaine wash-out (see Fig. 6 C). Similar results were obtained in 5 other experiments. In 13 additional experiments, tetracaine reduced the amplitude of both TR and PR release components to 1,177.8 ± 187 and 269 ± 24.1 nM (mean ± SE), respectively. These values are 30 and 20% smaller, respectively, than the control data (see above). In many of these cases, the upstroke of the TR component was delayed with respect to their control response after exposure to tetracaine (see Fig. 6 D). This delay (measured as the time interval between the peaks of the derivatives of the Ca2+ transients, see Fig. 6 E), ranged form 1.5 to 7.48 s (4.5 ± 0.63 s; mean ± SE; n = 13), and probably results from tetracaine not being present long enough to abolish the positive feedback mechanism that underlies the TR component.
Figure 6.

Effects of tetracaine on the kinetics of caffeine- induced Ca2+ release. (A) Responses of a sympathetic neuron to three 500-ms applications of high K+ (vertical arrows), and then to a 20-s application of 10 mM caffeine (horizontal bar). Both TR and PR components occur in response to caffeine. (B) Effect of a 30-s application of 500 μM tetracaine, delivered from a third puffer pipette before the caffeine application. Tetracaine suppressed the TR component, affecting less the PR component. (C) Recovery of the transient release component after 5 min of tetracaine wash-out. (D) Example of an experiment similar to that illustrated in B, but where tetracaine reduced the TR component and delayed its onset (only caffeine responses are shown). Continuous trace : control response; dotted trace : response after tetracaine application. (E) First derivatives of C-Ca2+ transients shown in D. Horizontal arrows indicate resting [Ca2+].
The tetracaine sensitivity of the TR component supports the notion that it may constitute a regenerative response fueled by CICR. This presumption was corroborated by experiments where the kinetics of caffeine-induced release were examined before and after the cells had been incubated with the acetoxymethyl ester form of the fast calcium buffer BAPTA (10 μM). A combination of BAPTA-AM concentration and incubation time was found such that the time constants of decay of K-Ca2+ transients increased slightly, but their amplitude did not show a drastic reduction (see Fig. 7 C). This condition indicated that intracellular BAPTA had reached sufficient concentration to began competing with Ca2+ extrusion mechanisms, but not enough to significantly compete with fura-2 for Ca2+ binding. As shown in Fig. 7, A and B, after 10 min of incubation, BAPTA had two main effects: (a) it slowed the decay of the filler K+-Ca2+ transients (a monoexponential fit to the decay phase of the third K+-Ca2+ transient gave a time constant of 6.4 s before and 10.6 s after BAPTA), and (b) it eliminated the TR component. Similar results were obtained in 5 additional experiments. In 9 other cases, where the TR component was spared (see an example in Fig. 7 C), the combined amplitude of TR and PR components diminished after incubation with BAPTA from 923.6 ± 91.4 nM to 739.5 ± 91.4 nM (mean ± SE). In addition, TR and PR components after BAPTA treatment were only 500.3 ± 59.6 and 257.7 ± 33 nM, respectively. It is noteworthy that after incubation with BAPTA, the separation between the TR and PR components became much less discernible (Fig. 7 C), as if BAPTA was also impeding Ca2+ binding to inhibitory regulatory sites on the RyRs which may be partially responsible for the decay of the TR component (see below).
Figure 7.

Effects of BAPTA-AM on the kinetics of caffeine- induced release. (A) Recordings of Ca2+ transients elicited by three applications of high K+ (1 s each, vertical arrows) and two applications of 10 mM caffeine (25 s each, horizontal bars) in a fura-2– loaded sympathetic neuron. (B) The same experiment was repeated after incubating the cell for 10 min in the presence of 10 μM of BAPTA-AM. Incubation with BAPTA-AM slowed the decay of the K+-Ca2+ transients and eliminated the TR component. (C) Example of an experiment where the TR component was spared. Note changes in the kinetics of decay of both K+- and C- Ca2+ transients. Horizontal arrows indicate resting [Ca2+].
All experiments depicted so far support the hypothesis that CICR underlies the fast release component initiated by caffeine in nerve cells: This component results entirely from intracellular Ca2+ release and is inhibited by tetracaine and intracellular BAPTA, which interfere with Ca2+-mediated positive feedback loops by two completely different mechanisms. The suppression by intracellular BAPTA or tetracaine of a fast kinetic component of calcium release in skeletal muscle is one of the strongest evidences supporting the assertion that it constitutes a release component controlled by calcium (Jacquemond et al., 1991; Pizarro et al., 1992). An important property of the TR component is that its appearance requires the simultaneous activation of a significant fraction of Ca2+-release channels together with an adequate loading of intracellular Ca2+ stores. This property suggests a dependence on the rate of rise of trigger calcium.
Mechanisms Involved in the Termination of the TR Component: Sr2+ Substitution
The kinetics of Ca2+ release depicted in this study are consistent with a Ca2+-dependent dual feedback control mechanism proposed for cardiac cells (Fabiato, 1985). According to this model, the abrupt termination of the TR component could be explained by Ca2+-dependent inactivation of release. However, other factors, such as adaptation of RyR/Ca2+ release channels (Györke and Fill, 1993; Stern, 1996) or rapid depletion of releasable Ca2+ from intracellular stores cannot be ruled out. Ca2+ substitution with permeant divalent cations like Sr2+ or Ba2+ has provided valuable insights on Ca2+ dependent phenomena, such as Ca2+-mediated inactivation of voltage-gated Ca2+ channels (Tillotson, 1979). For the most part, these ionic replacement experiments have been useful because Ba2+ and Sr2+ do not interact as efficiently as Ca2+ with regulatory binding sites (Oberhauser et al., 1988). Since Sr2+ binds to fura-2 with relatively high affinity, and the spectral properties of the complex are similar to those of fura-2 bound with Ca2+ (Kwan and Putney, 1990), we decided to examine the effects of substituting Sr2+ forCa2+ on the kinetics of TR and PR components. We were hoping that this maneuver would provide clues to better understand the mechanisms that underlie onset and termination of the release process.
Fig. 8 shows fura-2 fluorescence changes obtained from a rat sympathetic neuron after undergoing the Sr2+ replacement procedure described in materials and methods. As shown in the first half of Fig. 8, both depolarization and caffeine-induced fluorescence changes continue in Sr2+-containing saline. This demonstrates that Sr2+ substitutes well for Ca2+ both in voltage-gated influx and agonist-mediated intracellular release. Control experiments showed that responses to both stimuli were eliminated after manipulating the cells in a similar fashion but with solutions lacking both Ca2+ and Sr2+(data not shown). Sr2+ transients reach much higher concentrations than Ca2+ transients do. This could be due in part to the fact that Ca2+ binding proteins and Ca2+ extrusion mechanisms bind Sr2+ less efficiently than Ca2+. Nonlinearity of the dye signal should not be a problem here, since the maximum concentrations reached (about 10 μM) are similar to the k d of fura-2 for Sr2+ (7.6 μM; Kwan and Putney, 1990). The kinetics of Sr2+ transients shown in Fig. 8 is quite different from that recorded in the presence of Ca2+. Most noticeably, Sr2+ excursions decay more slowly, reflecting a less efficient Sr2+ uptake and extrusion by metabolic pumps (Rasgado-Flores et al., 1987; Berman and King, 1990). Further diminution of buffering capacity is observed when the cell is stimulated repeatedly. The time constants of monoexponential functions fitted to the decay phases (τd) of depolarization-induced (small arrows) and caffeine-induced (large arrows) Sr2+ signals, increased progressively from their initial values 7.3 and 6.8 s, respectively, (first pair of responses in Fig. 8) to 10.2 and 11.84 s (sixth pair of responses). Near the middle of the recording, 20 μM ryanodine was added to the bath solution and alternate stimulation with high-K+ and caffeine was restated 2 min later. Shortly after its application, ryanodine produced a step increase in resting [Sr2+]and reduced the rate of decay of the next caffeine response (τd = 19.7 s). Once the use-dependent suppression of caffeine responses was established (asterisks), the rate of decay of depolarization-induced Sr2+ transients increased significantly, from a τd= 10.0 s (first deflection after stimulation reinitiated) to 17.0, 17.1, 18.7, and 19.9 s, in the following four responses. The effects of continuous ryano-dine exposure are similar to those observed in the presence of Ca2+ (data not shown) and are consistent with a locking of the release channels in an open state (Pessah and Zimanyi, 1991).
Fig. 9 compares fluorescence changes recorded from another cell, first, when it was bathed in Ca2+-containing saline (Fig. 9 A) and then in Sr2+-containing saline (Fig. 9 B), immediately after undergoing the Sr2+ replacement procedure (see materials and methods). The stimulation protocol consisted of four conditioning 0.5-s pulses of high K+ with 10 mM of either Ca2+ or Sr2+ to preserve the filling status of intracellular stores, followed by two pulses of 10 mM caffeine in saline containing either Ca2+ or Sr2+, to examine the kinetics of release. Besides the differences mentioned before, the most notorious dissimilarities were in the dynamics of release from intracellular stores. In the presence of Ca2+, the first caffeine application elicited TR and PR components, with distinct rates of decay (τdTR = 4.2 s, τdPR = 43.9 s). In contrast, only a single component of release with a monoexponential decay (τdPR= 11.5 s) was apparent after the Sr2+ replacement procedure. Fundamental differences also existed in the response to the second caffeine application: While in Ca2+the TR component was abolished with only the PR component remaining (τdPR = 59 s), a significant initial fast release persisted in Sr2+. Moreover, the rate of rise of the second caffeine response in Sr2+ was almost as large as the first one (compare Fig. 9, C and D). The decay of the second caffeine-induced Sr2+ release was also fitted with a single exponential function (τd= 11.1 s).
Figure 9.

Effects of replacing Ca2+ with Sr2+ (A) Ca2+ concentration changes recorded from a sympathetic neuron bathed with normal, Ca2+-containing saline. The stimulation protocol consisted of four conditioning applications of high K+ solution (0.5 s each, vertical arrows), followed by two applications of 10 mM caffeine in saline containing 2 mM Ca2+ (60 s each, horizontal bars). (C) First derivative of records shown in A. (B) Fluorescence changes obtained from the same cell immediately after undergoing the Sr2+ replacement procedure (see materials and methods). The stimulation protocol was repeated under conditions where EGTA (500 μM) was added to all bathing solutions, and Ca2+ was equimolarly replaced with Sr2+. (D) First derivative of records shown in B. Asterisks indicate corresponding responses to transmembrane flux and intracellular release before and after Sr2+ substitution (see text). Horizontal arrows indicate resting [Ca2+] and [Sr2+].
Fig. 9 C shows the first derivatives of the signals resulting from transmembrane Ca2+ influx and intracellular Ca2+ release. If we focus our attention in the last K-Ca2+ transient and in the ensuing C-Ca2+ transient, when the Ca2+ content of the store is similar (Fig, 9 C, asterisks), it is clear that the latter rises faster, possibly because of the positive feedback amplification that underlies the generation of the TR response, as opposed to the modest amplification exerted on Ca2+ signals originated from Ca2+ influx. The same comparison in Fig. 9 D (asterisks) reveals that the differences are smaller in Sr2+. These results are consistent with the notion that Sr2+-induced Sr2+ release may operate with lower amplification gain than CICR in rat sympathetic neurons.
The kinetics of caffeine-induced Sr2+ mobilization is consistent with Sr2+ release operating with lower feedback loop gain (both positive and negative) than the release of Ca2+. Consequently, a distinct TR response was not produced in Sr2+, and the diminution of the initial fast release during the second caffeine challenge was less pronounced. When Ca2+ was the divalent cation involved, substantial amplification by CICR determined a conspicuous TR response, whose abrupt termination may result of both rapid depletion of releasable Ca2+ and Ca2+-dependent inactivation/adaptation of RyR channels (see discussion).
The Main Kinetic Aspects of Caffeine-induced Release Can Be Reproduced with a Simple Mathematical Model
Fig. 10 illustrates the result of a simulation obtained with our mathematical model. Traces shown from top to bottom are: mean cytosolic [Ca2+]; luminal [Ca2+] in the ER compartment that takes up Ca2+ (thick line) and in the ER compartment that releases it (thin line); normalized Ca2+ occupancy of the activating (thin line) and inactivating (thick line) regulatory sites of the RyRs. Caffeine application was simulated by increasing the association rate constant for Ca2+ binding to the activation sites, which greatly increases the open probability of release channels. When intracellular reservoirs are filled with sufficient Ca2+ (first caffeine response), a steep initial release of Ca2+ is produced (Fig. 10 B, thin line). Because of their faster association rate constant, the activation sites are occupied more quickly by the step-like increase in [Ca2+] than the inactivation sites (Fig. 10 C, left inset), promoting further release that in turn may act as trigger Ca2+ to activate additional RyRs. This positive feedback loop is responsible for the regenerative response here termed TR component. The early phase of fast release is terminated by way of two independent processes: (a) slower but more prolonged occupation of the inactivation Ca2+ binding sites of the RyRs, leading to a decrease in open probability (Fig. 10 C, thick line), and (b) depletion of [Ca2+] in the ER compartment (Fig. 10 B, thin line), which reduces the driving force for calcium efflux. A more sustained release at a lower rate is maintained throughout the caffeine application (PR component). When caffeine is reapplied after a relatively short delay, a smaller Ca2+ efflux is produced because the stores have not refilled completely (Fig. 10 B, thick line). Consequently, Ca2+ accumulates more slowly at the cytoplasmic side of the RyRs, with the change in [Ca2+] resembling more a ramp than a step. In this case, the inactivating sites of the RyR are occupied with Ca2+ almost as quickly as the activation sites (Fig. 10 C, right inset), thereby suppressing the regenerative TR response. The situation is comparable to that of voltage-gated sodium channels induced to open with a slowly rising voltage ramp. Inactivation overcomes activation, thus preventing the generation of an action potential. As shown in Fig. 11, our model also predicts the recuperation of the TR response if additional Ca2+ is supplied by way of voltage-gated Ca2+ influx before the second caffeine application. These episodes of additional Ca2+ influx increase significantly the Ca2+ content of intracellular stores (Fig. 11 B, thick line), but since they produce relatively slow changes in [Ca2+] at the RyR regulatory sites, inactivation dominates activation and no CICR is produced (Fig. 11 C; compare thick and thin lines). The production of a TR component during the second caffeine application results from the increased initial Ca2+ efflux, which, by allowing a faster occupation of the activation sites with Ca2+ than the inactivation sites (Fig. 11 C, inset), provide the means for triggering a regenerative release response.
Figure 10.

Simulation of Ca2+ mobilization obtained by integrating the diffusion mathematical model: (A) mean cytosolic [Ca2+], (B) lumenal [Ca2+] in the ER compartment that takes up Ca2+ (Ca ERs; thick line) and in the ER compartment that releases it (Ca ERt; thin line), (C) normalized Ca2+ occupancy of activating (thin line) and inactivating (thick line) regulatory sites on the RyR channels. The effects of two separate 60 s caffeine applications are shown. Initial parameters used in the integration were as follows: [Ca2+] ext = 2 mM, [Ca2+] ERt = 75 μM, [Ca2+] ERs = 75 μM, B Tot = 50 μM, aRyR Tot = 0.1 μM, iRyR Tot = 0.1 μM, lRyR Tot = 0.1 μM, Rate ppm = 100 s−1μm−2, Rate perm = 100 s−1μm−2 , PUMP ermTot = 0.1 μM, Pump pmTot = 0.1 μM, D Ca = 200 μm2s−1, dx = 0.25 μm, ds = 0.8 μm2, ds erm = 0.4 μm2, vol ER = 100 μm3, k offB = 1 s−1, k onB = 0.5 μM−1 s−1, RyR max = 70,000 s−1μm−2, k offiRyR = 0.176 s−1, k oniRyR = 0.28 μM−1 s−1, k offiRyR = 1,300 s−1 , k onlRyR = 10 μM−1 s−1 , k on-Caff = 0.01 μM−1 s−1, k on + Caff = 2.5 μM−1 s−1, k offaRyR = 2 s−1, N = 7, Caf f Dur = 60 s. The inserts shown on an expanded time scale allow a comparison of the kinetics of Ca2+ binding to the activation and inactivation sites during the first and second caffeine applications.
Figure 11.

Simulation similar to that of Fig. 10, but with eleven depolarizing pulses given during the interval between the two caffeine applications to rapidly refill intracellular Ca2+ stores. The initial parameters used were the same as those in the simulation of Fig. 10, with the addition of Ca2+ flux pulses of Ca ChDur = 0.5 s in duration, interstimulus interval Ca Chint = 5 s and a rate Ca Chmax = 40 s−1μm−2.
According to this very simplified model, the filling status of intracellular stores is one of the most relevant aspects governing the kinetics of Ca2+ release, by determining the rate of change of cytosolic [Ca2+] and the occupation rate of activation Ca2+ binding sites of the RyRs. Undoubtedly, a multiplicity of other parameters, such as spatial organization of Ca2+ stores and buffering mechanisms, as well as interaction between neighboring release channels and Ca2+ stores, must come into play to determine the spatial distribution and release dynamics of Ca2+ signals in real cells (Stern, 1992). Nevertheless, the conclusions from this work provide certain constraints as to which functional roles neuronal Ca2+ stores can and cannot perform. In this regard, ryanodine-sensitive stores appear to be organized to emphasize subcellular Ca2+ gradients initiated by agonist-induced intracellular release, rather than to produce widespread amplification of Ca2+ signals originated from transmembrane Ca2+ influx. Naturally, this does not exclude the possibility that ryanodine-sensitive stores located near the plasma membrane may produce substantial local amplification of Ca2+ signals originated from transmembrane Ca2+ influx.
discussion
Voltage-gated Ca2+ influx in nerve cells is not very effective to initiate widespread CICR under physiological conditions (Verkhratsky and Shmigol, 1996). A likely explanation is that Ca2+ must diffuse from the plasmalemma to the endoplasmic reticulum over relatively long distances, and [Ca2+] cannot rise sufficiently fast near the RyRs to effectively activate them (Hernández-Cruz, et al., 1995; Shmigol et al., 1995). Conversely, Ca2+ released directly from intracellular reservoirs is more likely to efficiently change [Ca2+] in the microenvironment surrounding the RyRs. This implies that both positive and negative feedback regulation of Ca2+ release by Ca2+ (Fabiato, 1985) should be more apparent after intracellular release than after transmembrane calcium entry. These assumptions are supported by the experiments described here. One possible exception to this scheme would be the RyRs located on specialized endoplasmic reticulum organelles, called subsurface cisternae (Watanabe and Burnstock, 1976). In fact, the most convincing examples of physiologically relevant CICR phenomena after transmembrane Ca2+ influx in nerve cells involve the regulation of plasmalemmal channels, presumably by Ca2+ released from submembrane domains (Kawai and Watanabe, 1989; Sah and McLachlan, 1991; Yoshizaki et al, 1995).
This study was aimed to identify the physiological parameters that determine the dynamics of Ca2+ release from ryanodine-sensitive Ca2+ stores in rat sympathetic neurons. For simplicity, we focused on Ca2+ release phenomena triggered by caffeine, but the same conclusions should be applicable to Ca2+ release initiated by natural agonists (Hua et al., 1994). Caffeine-induced release comprises two ryanodine-sensitive kinetic components, a fast initial release (TR component), and a delayed, persistent release (PR component). The early event shows refractoriness resulting from the depletion of releasable Ca2+, and requires both the adequate activation of Ca2+ release channels and sufficient loading of intracellular stores. This behavior suggests a strong dependence on the rate of rise of trigger calcium. Tetracaine and BAPTA-AM, which interfere with Ca2+-mediated positive feedback loops by two different mechanisms, suppress the TR component, supporting the notion that it is a CICR phenomenon. Our data also suggest that the abrupt termination of the TR component results both from rapid depletion of releasable Ca2+ and Ca2+-dependent inactivation or adaptation of RyRs.
Caffeine-induced Intracellular Ca2+ Mobilization in Nerve Cells Comprises Two Kinetic Components, One of which Is a CICR-dependent Phenomenon
This work provides evidence for two separate components of caffeine-induced Ca2+ release. The TR component is expected from the regenerative nature of CICR if released Ca2+ can promote its own release by a positive feedback mechanism (see below). The PR component may result from a sustained nonregenerative activation of release balanced with Ca2+ uptake (Kuba, 1994).
The TR component can only be observed when Ca2+ stores are replete. A similar loading-dependent property has been described for InsP3-induced Ca2+ release in smooth muscle cells (Iino and Endo, 1992) and Ca2+ release from the sarcoplasmic reticulum of intact cardiac myocytes (Bassani et al., 1995). Several circumstances may combine to explain the effects of increasing the Ca2+ content of the store: (a) an increased transmembrane Ca2+ gradient, (b) an increased rate of Ca2+ release (Donoso et al., 1995), (c) an increased open probability of release channels (Vélez and Suárez-Isla, 1992; Sitsapesan and Williams, 1994; Tripathy and Meissner, 1996), and (d) an increased caffeine sensitivity of overfilled neuronal Ca2+ stores (Verkhratsky and Shmigol, 1996). Also, CICR is strongly dependent on the rate of rise of trigger calcium, a phenomenon that probably involves fast competition between Ca2+- dependent activation and inactivation of release channels (Fabiato, 1985, 1992; see below). Thus, the filling status of the Ca2+ store may affect the kinetics of release to the extent that it affects the initial rate of Ca2+ release. Accordingly, a second TR response cannot be generated (even with a high caffeine concentration) when Ca2+ stores are partially depleted, probably because [Ca2+] does not rise sufficiently fast to initiate CICR. Experiments in cardiac myocytes have demonstrated that the fractional Ca2+ release (the percentage of SR Ca2+ content released by a twitch) increases steeply as the SR Ca2+ load increases. Conversely, Ca2+ release is almost abolished when SR content falls below 40% of its maximum (Bassani et al., 1995).
Our results are inconsistent with the “quantal Ca2+ release” hypothesis according to which individual stores release their content in an all-or-none manner once their threshold for caffeine is reached (Cheek et al., 1994). If this scheme were true, application of caffeine concentrations <10 mM should elicit CICR phenomena involving low-threshold caffeine-sensitive Ca2+ stores. Likewise, the recruitment of Ca2+ stores with higher caffeine threshold should give rise to TR responses of their own, regardless of prior activation of low-threshold Ca2+ stores. This type of behavior was not observed (see Fig. 3 C). Recent experiments in a skeletal muscle cell line (Györke and Györke, 1996), suggest that the incremental Ca2+ release phenomenon cannot be accounted for by “quantal” Ca2+ release but by adaptive control of release units (Györke and Fill, 1993).
The TR component has properties expected from a CICR phenomena: (a) it exhibits refractoriness, (b) it requires the activation of minimum fraction of Ca2+-r elease channels (threshold), (c) it requires sufficient loading of intracellular Ca2+ reservoirs (i.e., it depends on the rate of rise of trigger Ca2+), (d) it behaves as an all-or-none regenerative process once a critical amount of loading is achieved, and (e) it is inhibited by drugs that restrict or abolish Ca2+-mediated positive feedback loops. One important conclusion of these experiments is that CICR in nerve cells is possible whenever the conditions are met which allow the generation of adequate trigger Ca2+ signals. Interestingly, freshly dissociated sympathetic neurons, or cells that have been in culture for 1–3 d, maintain their caffeine-sensitive Ca2+ stores occupied nearly to the limit of their capacity, despite the absence of spontaneous electrical activity. This physiological status supports an effective Ca2+ efflux whenever RyR open, either in response to Ca2+ or any other endogenous agonist (Bassani et al., 1995). This differs from the situation in older cultures of sympathetic neurons or in most studies with central neurons, where in order to obtain full-sized caffeine responses, ER stores have to be charged with releasable Ca2+ beforehand (Shmigol et al., 1994).
Tetracaine and BAPTA-AM Suppress the TR Component of the Release by way of Different Mechanisms
The local anesthetic tetracaine reduces Ca2+-induced Ca2+ release in several preparations (Endo, 1985). Presumably, these effects result from direct inhibition of channel activity by binding to high-affinity, regulatory sites on the RyRs (Xu et al., 1993). In our experiments, tetracaine reached the cell only during the interval between the last conditioning K+-Ca2+ transient and the following caffeine application. This was necessary to prevent tetracaine effects on voltage-gated Ca2+ influx and Ca2+ loading of intracellular stores that would have resulted from inhibition of voltage-gated Ca2+ channels with the tetracaine concentrations used here (Sugiyama and Muteki, 1994). In preliminary experiments, when tetracaine was applied continuously, rather than for a short period of time, we observed that the TR responses were always blocked. The brief exposure to tetracaine in the experiments reported here underestimates its effects on caffeine-induced Ca2+ release. In fact, they clearly show that CICR is more sensitive to tetracaine than voltage-gated Ca2+ influx, since it was able to delay, and in some cases suppress the TR component, sparing the PR component. These results are consistent with the reported inhibition by tetracaine of Ca2+-gated channel activity by an allosteric mechanism rather than by blockade of the RyR channel (Xu et al., 1993).
In contrast to tetracaine, BAPTA does not interact directly with the RyRs. We used this high-affinity fast diffusible Ca2+ buffer to try to prevent the interaction between released Ca2+ and RyRs regulatory Ca2+ binding sites. A measure of the ability of exogenous Ca2+ buffers to compete with other Ca2+ binding sites is their association rate constant (k on). Since BAPTA's k on is almost diffusion limited (∼6.2 × 108 M−1 s−1; see Kao and Tsien, 1988) and free BAPTA is likely to equilibrate with cytosolic Ca2+ in <3 μs (Adler et al., 1991), it is expected to compete effectively for Ca2+ with the RyR activating sites, thus limiting the feedback loop gain that underlies CICR phenomena. In some experiments, BAPTA was able to suppress the TR component without affecting Ca2+ transients induced by Ca2+ influx (see Fig. 7 B). In other cases, the TR component was spared, but the separation between TR and PR components became less discernible (see Fig. 7 C, dotted trace). These effects of BAPTA on the kinetics of decay of the caffeine-induced release are reminiscent of the effects of Sr2+ substitution (see Fig. 9 B). In both cases, the decay of the TR component becomes slower, conceivably as a result of a decreased interaction of the divalent cation with inhibitory regulatory binding sites on the RyRs (see below). Cells massively loaded with fura-2, (1-h incubation or more) often display alterations of Ca2+ dynamics similar to those produced by incubation with BAPTA AM.
Substitution of the Permeant Divalent Cation Provides Kinetic Information on the Onset and Termination of the TR Component of the Caffeine Response
A model of RyR dynamics has been postulated for cardiac cells, whereby the cytoplasmic side of the channel possess two regulatory Ca2+ binding sites, one that is fast and positively regulatory and another that is slow and negatively regulatory (Fabiato, 1985). A feedback control by Ca2+ on IP3-mediated Ca2+ release has been demonstrated in flash-photolysis experiments of caged IP3 and caged Ca2+ in permeabilized smooth muscle fibers (Iino and Endo, 1992). Caffeine-induced release in mammalian sympathetic neurons exhibits a bell-shaped dependence on cytosolic Ca2+ concentration, which could give rise to positive feedback in the earlier phase of release followed by negative feedback due to Ca2+-dependent inactivation (Hernández-Cruz et al., 1995). Although the kinetics of Ca2+ release depicted in the present study was seemingly consistent with this dual feedback control mechanism, it was uncertain to what extent the termination of the TR component could depend on Ca2+-dependent inactivation since for the most part, the reported biphasic Ca2+ dependence is more closely related to steady-state conditions. Furthermore, other factors related to the release process, such as adaptation of RyRs (Györke and Fill, 1993) and Ca2+ depletion of intracellular stores, could also influence this process.
The Ca2+ content of the caffeine-sensitive store diminishes after the generation of a TR response (see Figs. 1 and 3 A). We can speculate that if local depletion of releasable Ca2+ occurs at a sufficiently fast rate, the feedback gain that sustains the activity of release channels would be significantly reduced, thus playing a role in the termination of the TR. Ca2+-dependent inactivation or adaptation, described for RyRs isolated from cardiac sarcoplasmic reticulum (Györke and Fill, 1993), could also participate in terminating the release. Direct evidence for the relative contribution of these mechanisms to the termination of release is difficult to obtain in intact cells, because manipulations that affect Ca2+ binding to the inactivation site(s) are likely to affect binding to the activation site(s) as well. Also, cytoplasmic Ca2+ removal mechanisms (buffering, sequestration, and extrusion of Ca2+) can become affected in unpredictable ways.
Sr2+ permeates through both plasmalemmal and RyR Ca2+ channels (Hagiwara and Ohmori, 1982; Williams, 1992) and is taken up by neuronal intracellular stores (Rasgado-Flores et al., 1987). Moreover, Sr2+ influx can induce Ca2+ release from caffeine-sensitive stores in smooth muscle cells (Grégoire et al., 1993) and binds to fura-2 with high affinity, producing the same type of changes as Ca2+ in spectral properties of the dye (Kwan and Putney, 1990). Nevertheless, in part because of its larger ionic radius (Hille, 1992), Sr2+ is not expected to interact with Ca2+ binding sites on the RyR channel as efficiently as Ca2+. An impaired interaction of Sr2+ with regulatory binding sites has been well documented for Ca2+-activated K+ channels, where Sr2+ is ∼160 times less effective than Ca2+ as a channel agonist (Oberhauser et al., 1988). Given these properties, we reasoned that by examining the effects of substituting Ca2+for Sr2+ we could gain clues to better understand the mechanisms that underlie the dynamics of caffeine-induced Ca2+ release.
As shown in Fig. 8, Sr2+ can substitute for Ca2+ both in voltage-gated influx and caffeine-induced intracellular release in rat sympathetic neurons. Caffeine-induced signals were abolished by ryanodine, demonstrating that they indeed represent Sr2+ release from intracellular stores. A gross contamination by Ca2+ in these experiments was ruled out because (a) external Ca2+ concentration was maintained below 100 nM throughout the experiment by excluding Ca2+ and adding 0.5 mM EGTA to all external solutions, and (b) fura-2 fluorescence signals disappeared altogether when Sr2+ was omitted from the external medium. A potentially significant problem could result if a small amount of intracellular Ca2+ persists after the Sr2+ replacement procedure. The presence of a residual resting Ca2+ would seriously affect [Sr2+]determined with fura-2, because of its higher affinity for Ca2+ (k d = 0.3 μM). Nevertheless, this problem should not affect significantly the kinetics of intracellular Sr2+ transients.
Fluorescence changes recorded in the presence of Sr2+ were different from those obtained in Ca2+. For instance, they decayed more slowly, reflecting a less efficient handling of Sr2+ by metabolic pumps (Rasgado-Flores et al., 1987). Most importantly, the kinetics of release from intracellular stores was markedly dissimilar. Rather than the TR and PR components elicited by the first caffeine application when the stores are replete with Ca2+, only a single component of release was apparent in Sr2+, with a decay time constant (τd= 11.5 s) intermediate between those of TR and PR components in Ca2+-containing saline (τdTR = 4.2 s, τdPR = 43.9 s). Two possible explanations exist for this result: (a) the decay of the TR component slows as a result of deficient buffering and (b) TR and PR components fused into one.
The simplest interpretation for a monoexponential decay of caffeine-induced Sr2+ release is that Sr2+ inactivates its own release much less efficiently than Ca2+ and that the kinetics of decay are now dominated by the rate of depletion of intracellular stores. If Sr2+ were as effective as Ca2+ at activating RyRs, then caffeine should deplete more because of uninterrupted activation in the absence of counteracting inactivation. Nevertheless, as shown in Fig. 9 B, a substantial amount of fast Sr2+ release persists during the second caffeine challenge. This could indicate that stores become less depleted of Sr2+ at the end of the first caffeine application. This would be possible if feedback activation of RyR is also less effective when Sr2+ is the flux carrier. Alternatively, intracellular stores could become equally or even more depleted in the presence of Sr2+, but as inactivation is less effective, Sr2+ activation of the RyR channels could still evoke fast release in spite of the reduced intraluminal Sr2+ concentration. Comparison of activation and inactivation kinetics of single neuronal RyRs incorporated into lipid bilayers, upon step applications of Ca2+ and Sr2+, should help us decide among these possibilities.
When the first derivatives of signals associated to transmembrane Ca2+ influx and intracellular Ca2+ release are compared under similar conditions of Ca2+ content of the intracellular pool (Fig. 9 C, asterisks), it is apparent that the latter rises faster. This difference could result from the positive feedback amplification that underlies the generation of TR responses. However, when Sr2+ substitutes for Ca2+, the differences in rate of rise between signals associated with influx and release are smaller (Fig. 9 D, asterisks). The tentative conclusion of these results is that Sr2+-induced Sr2+ release operates with lower amplification gain than CICR in sympathetic neurons.
In summary, the kinetics of caffeine-induced Sr2+ mobilization is consistent with the idea that the release of Sr2+ operates with a lower feedback loop gain (both positive and negative) than the release of Ca2+. When Ca2+ is the flux carrier, a regenerative TR response can be generated as a result of amplification by CICR. The abrupt termination of this fast release, in spite of the continuous presence of the agonist, appears to be the outcome of both rapid depletion of releasable Ca2+ and Ca2+-dependent inactivation or adaptation of release. Single channel recordings of neuronal RyRs would be required to determine more precisely the contribution of Ca2+-dependent inactivation and adaptation to this process.
Mathematical Modeling of Caffeine-induced Ca2+ Mobilization
Our mathematical model attempts to describe qualitatively how the macroscopic phenomena observed in real cells relate to the basic properties of CICR. The model belongs to the class of common pool models (see Stern, 1992), meaning that trigger calcium reaches the RyRs via the same cytosolic calcium pool into which the calcium is released. These models are intrinsically unstable. If they are conferred with low loop gain (i.e., calcium release relatively insensitive to cytosolic [Ca2+]), they produce only a moderate amplification of transients produced by the trigger calcium. When the gain is set to a higher value, more amplification is produced, but all-or-none Ca2+ transients develop that evolve autonomously when cytoplasmic calcium reaches a certain threshold. In our variant of common pool model, Ca2+ entering through plasmalemmal Ca2+ channels is insufficient to induce CICR because of the effects of diffusion over several cytosolic compartments. Nevertheless, Ca2+ entering through this pathway is readily taken up by intracellular stores, greatly affecting their filling status.
Judging by the modest amplification of Ca2+ signals resulting from transmembrane Ca2+ influx, CICR in nerve cells constitute an example of low loop gain systems. However, as shown in this study, trigger Ca2+ originating from intracellular stores can produce regenerative responses characteristic of high loop gain models. The solution to this apparent discrepancy resides in the rate of rise of trigger Ca2+, which determines the effective gain of CICR: If Ca2+ rises sufficiently fast, activation can overcome inactivation and regenerative responses are generated. Conversely, for slowly developing changes in Ca2+ concentration, CICR operates with low gain. The notion that the rate of rise of trigger Ca2+ is the most important parameter determining the extent of release is supported by experimental and theoretical studies (Fabiato, 1985, 1992; Tang and Othmer, 1994; Stern 1996). The main conclusion of our results with the model is that the loading status of intracellular stores determines the effective gain of CICR, regardless of the source of trigger Ca2+. Since it is conceivable that this physiological parameter undergoes cellular control, both globally and locally, it may represent a potentially important mechanism for regulating CICR.
Interaction between neighboring RyRs and calcium stores is intrinsic to common pool models provided that they are sufficiently close to one another so that they respond to each other's released Ca2+. Our model does not specifically consider interactions between release channels. In heart cells, RyRs are organized in discrete release units coupled to single sarcolemmal dihydropyridine channels, but largely uncoupled from neighboring release units, an arrangement that provides high gain but prevents uncontrolled regenerative behavior (Stern, 1992).
In avian heart cells, where excitation-contraction coupling depends on Ca2+ diffusion from one release unit to the next one (because of the lack of t-tubules; Sommer et al., 1991), the distance between individual release units and its nearest neighbors is about 135 nm. Presumably this distance allows effective propagation of a wave of Ca2+ release between neighboring release units (Protasi and Franzini-Armstrong, 1996). The spatial organization of neuronal intracellular calcium pools and release units is largely unknown (Pozzan et al., 1994). Sympathetic neurons possess abundant ryanodine binding sites (Hernández-Cruz et al., 1995) but lack privileged communication pathways (i.e., t-tubules) between the plasmalemmal voltage-gated Ca2+ channels and the majority of their Ca2+ release channels. The necessity of using caffeine concentrations close to the half-maximal dose in order to elicit regenerative responses in nerve cells (this study), suggest that overall, the interaction between release channels is limited. It is possible that the distance between neighboring release units does not favor a significant interaction. Alternatively, Ca2+-dependent inactivation and local depletion of releasable Ca2+ could extinguish channel activity shortly after it has been initiated by the stimulus. A detailed study on the spatial organization of release units in nerve cells is required to understand the reasons for their apparently limited interaction.
This study suggests that neuronal CICR is designed to favor amplification of Ca2+ signals originated from intracellular sources like IP3 or ryanodine-sensitive Ca2+ stores themselves, rather than amplification of voltage-gated Ca2+ influx. The gain of this amplification mechanism may be regulated by the density of RyRs, the filling status of the Ca2+ stores and the local concentration of physiological activators of release (Mészáros et al., 1993; Hua et al., 1994). We propose that the primary role of ryanodine-sensitive intracellular stores in nerve cells (and possibly of other nonmuscular cells) is to improve signal to noise ratio by intensifying subcellular Ca2+ gradients generated by agonist-induced intracellular Ca2+ release. This local Ca2+ signaling function of CICR may be particularly relevant for confined cellular compartments, like synaptic terminals, dendritic spines, or submembrane domains (Verkhratsky and Shmigol, 1996).
Acknowledgments
The authors wish to thank Drs. Agustín Guerrero and Luis Vaca for reviewing an earlier version of this manuscript, Dr. Carlos Sevcik for helpful discussion of numerical methods for integrating parabolic partial differential equations, and Drs. Gonzalo Pizarro and Francisco Sala for many helpful suggestions. We also thank Teresa Santos for preparing and maintaining neuronal cultures.
This work was supported by grants from DGAPA-UNAM (IN212194, IN206395) and CONACyT 400346-5-2366PN (México; A. Hernández-Cruz) and CONICIT (S1-95000493; Venezuela, A.L. Escobar).
appendix
Mathematical Model
To compute Ca2+ concentration changes in the different compartments of the nerve cell, the cytosolic space was divided into three regions: an outmost shell (compartment 0 ; closer to the plasma membrane), a region composed of n − 1 intermediate, nonborder compartments, and an inmost shell (compartment n; closer to the intracellular store membrane). The endoplasmic reticulum was represented by two diffusionally connected compartments, one that takes up Ca2+ from the cytosol and another one that releases it into the cytosol.
The net change in Ca2+ concentration in any compartment c at all times is given by the difference between Ca2+ influx and efflux:
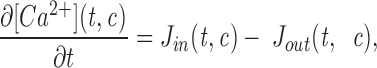 |
A1 |
where [Ca2+](t,c) is the Ca2+ concentration of compartment c, and J in (t,c), J out (t,c) are the net Ca2+ influx and efflux into/from compartment c. In general, for an arbitrary compartment c, Ca2+ fluxes resulting from diffusion exchange with adjacent compartments, DF(c) and exchange with fixed buffers, BF(c) are given by:
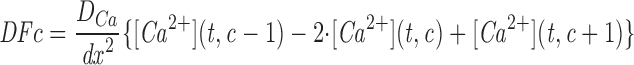 |
A2 |
and
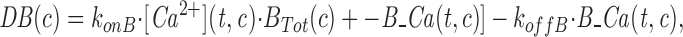 |
A3 |
where D Ca is the Ca2+ diffusion coefficient and dx the thickness of compartment c, B Tot (c) and B_Ca(t,c) are total and Ca2+-bound fixed buffers in compartment c, k onB and k offB are association and dissociation rate constants of fixed buffers with Ca2+. The Ca2+ pump activity responsible for Ca2+ extrusion from compartment 0 through the plasma membrane (Pump pm) and Ca2+ removal from compartment n through the ER membrane (Pump erm) is represented as follows:
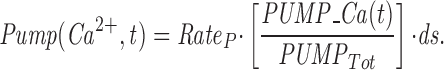 |
A4 |
The magnitude of this Ca2+ transfer function is given by the pump maximal transfer rate per unit of area, Rate p, multiplied by the fraction of the total number of Ca2+ binding regulatory sites which are occupied with Ca2+: PUMP_Ca(t)/PUMP Tot. Here, PUMP Tot is the total number of Ca2+ binding sites and PUMP_Ca(t) the number of sites bound with Ca2+. ds is the area section of plasma or endoplasmic reticulum membrane involved. Occupation with Ca2+ of regulatory sites is governed by a first order mass action law:
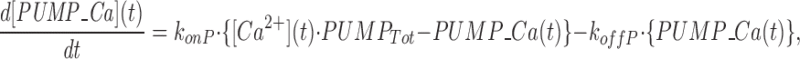 |
A5 |
where k onP and k offP represent association and dissociation rate constants for Ca2+ binding, respectively.
[Ca2+] changes in the outmost shell (compartment 0).
These changes are given by:
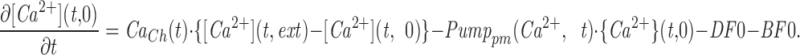 |
A6 |
In this expression, Ca Ch (t) describes the depolarization-induced opening of a voltage-gated Ca2+ conductance, given by:
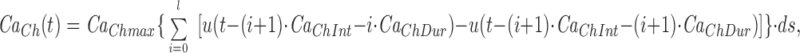 |
A7 |
where Ca Chmax is the maximum Ca2+ permeability per membrane area unit, Ca ChInt is the time interval between l successive depolarizations, Ca ChDur is the duration of each stimulus, and ds is the plasma membrane section area of compartment 0. u(t − a) is a square pulse Heavyside function defined as follows:
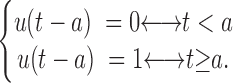 |
Ca2+ efflux from the outmost shell (compartment 0) by the plasmalemmal Ca2+ pump is given by:
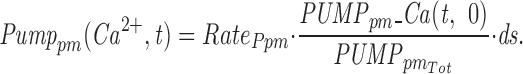 |
A8 |
Since Ca2+ diffusion through the plasma membrane is negligible, the diffusional fluxes of the outmost shell (compartment 0), can be written as follows:
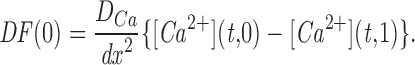 |
A9 |
[Ca2+] changes in nonborder shells (compartments 1 to n − 1)
In each of these compartments, Ca2+ fluxes due to the opening of Ca2+ channels and Ca2+ pump activity are absent. Therefore, the changes in Ca2+, are simply given by:
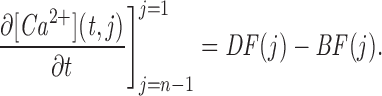 |
A10 |
[Ca2+] changes in the inmost shell (compartment n).
The Ca2+ concentration changes in compartment n can be computed as follows:
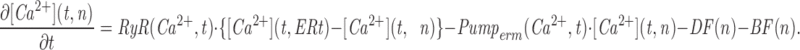 |
A11 |
The first term represents Ca2+ influx due to Ca2+ mobilization through RyR release channels. Here, the function RyR(Ca2+,t) represents the macroscopic Ca2+ permeability per unit membrane area of the endoplasmic reticulum compartment capable of releasing Ca2+to the cytosol (ERt). Release channels possess three regulatory Ca2+ binding sites: two cytosolic sites, which regulate activation and inactivation of the channel, and a third luminal site, which regulates channel conductance. The macroscopic Ca2+ permeability of these channels is given by:
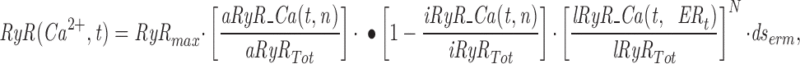 |
A12 |
where RyR max is the peak macroscopic permeability per unit of area, aRyR_Ca(t, n)/ARyR Tot is the fraction of activating sites occupied with Ca2+. Likewise, iRyR_Ca(t, n)/iRyR Tot is the fraction of inactivating sites occupied with Ca2+, and lRyR_Ca(t, ER t )/lRyR Tot is the fraction of luminal sites occupied with Ca2+. N is the cooperativity of the lumenal regulatory site and ds erm is the area of the section of the ER membrane. These regulatory Ca2+ binding sites are occupied with Ca2+ according to the law of mass action:
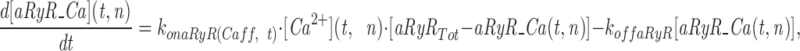 |
A13 |
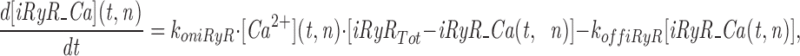 |
A14 |
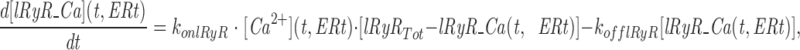 |
A15 |
where k onaRyR(caff, t), k offaRyR, k oniRyR, k offiRyR, k onlRyR, and k offRyR are the association and dissociation rate constants of cytosolic and lumenal regulatory Ca2+ binding sites of the release channel. A caffeine dependence has been introduced to the association rate constant of the activating site. This parameter is used to simulate agonist-induced stimulation of the release channel as follows:
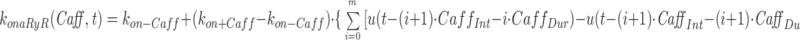 |
A16 |
where k on − Caff and k on + Caff are the association rate constants before and after and during caffeine application, respectively. Caff Int is the interval between m caffeine stimuli and Caff Dur is the duration of each caffeine stimulus.
The Ca2+ uptake from the inmost shell (compartment n) by the ER Ca2+ pump is given by a transfer function similar to that of the plasmalemmal Ca2+ pump.
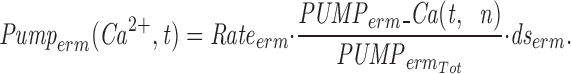 |
A17 |
Since Ca2+ diffusion through the ER membrane is negligible, the diffusional fluxes of the inmost cell (compartment n), can be written as follows:
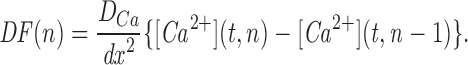 |
A18 |
Ca2+ distribution within the ER compartment.
In this model the ER comprises two diffusionally connected compartments. The first one (ERs), removes Ca2+ from the cytosol by way of the ER Ca2+ pump. The second one (ERt) releases Ca2+ into the cytosol via Ca2+ release channels. The change in Ca2+ concentration in compartment ERs is given by:
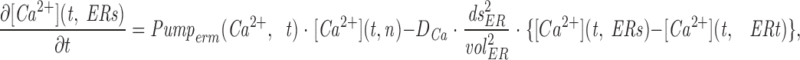 |
A19 |
where ds ER is the thickness of the ER compartment and vol ER is the volume of each ER compartment, and the change in Ca2+ concentration in the ER compartment that releases Ca2+ into the cytosol is given by:
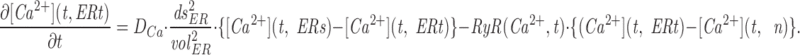 |
A20 |
All equations were numerically integrated using a finite difference approximation (Euler method) with the aid of the Scope 3.5 simulation package.
Footnotes
Abbreviations used in this paper: BAPTA, 1,2-bis(2-aminophenoxy) ethane-NNN′N′-tetraacetic acid; C-Ca2+ transient, Ca2+ transient elicited by caffeine application; CICR, Ca2+-induced Ca2+ release; ER, endoplasmic reticulum; K+-Ca2+ transient, Ca2+ transient initiated by depolarization with a high K+ solution; PR, persistent component of caffeine-induced Ca2+ release; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; TR, transient component of caffeine-induced Ca2+ release; τd, time constant of decay; τdPR, time constant of decay of PR component; τdTR, time constant of decay of TR component.
In recent experiments, we found that the fluorescent ryanodine derivative BODIPY© FL-X ryanodine (1 μM; Molecular Probes), produces a use-dependent, partial inhibition of caffeine-induced Ca2+ transients and the abolition of the TR component (González, C., and A. Hernández-Cruz. 1996).
The experiment shown in Fig. 5 summarizes results from over 12 similar experiments that showed the described phenomenon, but that could not be consummated because of a gradual and irreversible increase in resting Ca2+levels followed by depression of Ca2+ transients.
references
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike N, Sadoshima J-I. Caffeine affects four different ionic currents in the bull-frog sympathetic neurone. J Physiol (Lond) 1989;412:221–244. doi: 10.1113/jphysiol.1989.sp017612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani JWM, Weilong Y, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–C1329. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- Berman MC, King SB. Stoichiometries of calcium and strontium transport coupled to ATP and acetyl phosphate hydrolysis by skeletal sarcoplasmic reticulum. Biochim Biophys Acta. 1990;1029:235–240. doi: 10.1016/0005-2736(90)90159-l. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Erlich BE. Bell-shaped calcium response curves of Ins(1,4,5)P3and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature (Lond) 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science (Wash DC) 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Cheek TR, Berridge MJ, Moreton RB, Stauderman KA, Murawsky MM, Bootman MD. Quantal Ca2+mobilization by ryanodine receptors is due to all-or-none release from functionally discrete intracellular stores. Biochem J. 1994;301:879–883. doi: 10.1042/bj3010879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso P, Prieto H, Hidalgo C. Luminal calcium regulates calcium release in triads isolated from frog and rabbit skeletal muscle. Biophys J. 1995;68:507–515. doi: 10.1016/S0006-3495(95)80212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryer, S.E., M. Drake, T. D'Souza, and S. Raucher. 1995. Caffeine causes direct blockade of delayed rectifier potassium currents in vertebrate neurons and secretory cells. Soc. Neurosci. Abst. 21:505. (Abstr.).
- Endo M. Calcium release from the sarcoplasmic reticulum. Curr Top Membr Transp. 1985;25:181–230. [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced calcium release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato, A. 1992. Two kinds of calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cardiac cells. In Excitation-contraction Coupling in Skeletal, Cardiac and Smooth Muscle. G.B.G. Frank, P. Bianchi, and H. Keurs, editors. Plenum Press, New York. 245–263. [DOI] [PubMed]
- Favre CJ, Schrenzel J, Jaquet J, Lew DP, Krause K-H. Highly supralinear feedback inhibition of Ca2+ uptake by the Ca2+load of intracellular stores. J Biol Chem. 1996;271:14925–14930. doi: 10.1074/jbc.271.25.14925. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. A caffeine and ryanodine-sensitive Ca2+-store in bullfrog sympathetic neurons modulates effects of Ca2+ entry on [Ca2+]i . J Physiol (Lond) 1992;430:217–246. doi: 10.1113/jphysiol.1992.sp019125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, C.E., L. Granados, A. Cárabez, and A. Hernández-Cruz. 1996. Bodipy FL-X ryanodine labeling of ryanodine receptor channels in rat sympathetic neurons. Soc. Neurosci. Abstr. 22:340. (Abstr.).
- Grégoire G, Loirand G, Pacaud P. Ca2+ and Sr2+ entry induced Ca2+ release from the intracellular Ca2+store in smooth muscle cells of rat portal vein. J Physiol (Lond) 1993;474:483–500. doi: 10.1113/jphysiol.1993.sp019957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, Fay FS, Singer JJ. Caffeine activates a Ca2+permeable nonselective cation channel in smooth muscle cells. J Gen Physiol. 1994;104:375–394. doi: 10.1085/jgp.104.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke I, Györke S. Adaptive control of intracellular Ca2+release in C2C12 mouse myotubes. Pflüg Arch. 1996;431:838–843. doi: 10.1007/s004240050075. [DOI] [PubMed] [Google Scholar]
- Györke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca2+-induced Ca2+release in heart. Science (Wash DC) 1993;260:807–809. doi: 10.1126/science.8387229. [DOI] [PubMed] [Google Scholar]
- Györke S, Palade P. Ca2+-dependent negative control mechanism for Ca2+-induced Ca2+release in crayfish muscle. J Physiol (Lond) 1994;476:315–322. doi: 10.1113/jphysiol.1994.sp020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara S, Ohmori H. Studies of calcium channels in rat clonal pituitary cells with patch electrode voltage clamp. J Physiol (Lond) 1982;331:231–252. doi: 10.1113/jphysiol.1982.sp014371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Cruz A, Díaz-Muñoz M, Gómez-Chavarín M, Cañedo-Merino R, Protti DA, Escobar AL, Sierralta J, Suárez-Isla BA. Properties of the ryanodine-sensitive release channels that underlie caffeine-induced Ca2+mobilization from intracellular stores in mammalian sympathetic neurons. Eur J Neurosci. 1995;7:1684–1699. doi: 10.1111/j.1460-9568.1995.tb00690.x. [DOI] [PubMed] [Google Scholar]
- Hernández-Cruz A, Sala F, Adams PR. Subcellular calcium transients visualized by confocal microscopy in a voltage-clamped vertebrate neuron. Science (Wash DC) 1990;247:858–862. doi: 10.1126/science.2154851. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic Channels in Excitable Membranes, 2nd Ed. Sinauer Associates, Inc. Sunderland MA. 261–290.
- Hua SY, Tokimasa T, Takasawa S, Furuya Y, Nohmi M, Okamoto H, Kuba K. Cyclic-ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+entry in bullfrog sympathetic neurons. Neuron. 1994;12:1073–1079. doi: 10.1016/0896-6273(94)90315-8. [DOI] [PubMed] [Google Scholar]
- Iino M, Endo M. Calcium-dependent immediate feedback control of inositol 1,4,5-trisphosphate-induced Ca2+-release. Nature (Lond) 1992;360:76–78. doi: 10.1038/360076a0. [DOI] [PubMed] [Google Scholar]
- Ivanenko A, Baring MD, Airey JA, Sutko JL, Kenyon JL. A caffeine and ryanodine-sensitive Ca2+store in avian sensory neurons. J Neurophysiol. 1993;70:710–722. doi: 10.1152/jn.1993.70.2.710. [DOI] [PubMed] [Google Scholar]
- Jacquemond V, Csernoch L, Klein MG, Scheider MF. Voltage-gated and calcium-gated calcium release during depolarization of skeletal muscle fibers. Biophys J. 1991;60:867–874. doi: 10.1016/S0006-3495(91)82120-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JPY, Tsien RY. Ca2+binding kinetics of fura-2 and azo-1 from temperature-jump relaxation measurements. Biophys J. 1988;53:635–639. doi: 10.1016/S0006-3495(88)83142-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Watanabe M. Effects of ryanodine on the spike after-hyperpolarization in sympathetic neurones of the rat superior cervical ganglion. Pflüg Arch. 1989;413:470–475. doi: 10.1007/BF00594175. [DOI] [PubMed] [Google Scholar]
- Konishi M, Olson A, Hollingworth S, Baylor SM. Myoplasmic binding of fura-2 investigated by steady-state fluorescence and absorbance measurements. Biophys J. 1988;54:1089–1104. doi: 10.1016/S0006-3495(88)83045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuk P, Verkhratsky A. Calcium stores in neurons and glia. Neurosci. 1994;63:381–404. doi: 10.1016/0306-4522(94)90537-1. [DOI] [PubMed] [Google Scholar]
- Kuba K. Ca2+ induced Ca2+release in neurons. Jpn J Physiol. 1994;44:613–650. doi: 10.2170/jjphysiol.44.613. [DOI] [PubMed] [Google Scholar]
- Kuba K, Hua S-Y, Hayashi T. A UV laser-scanning confocal microscope for the measurement of intracellular Ca2+ . Cell Calcium. 1994;16:205–218. doi: 10.1016/0143-4160(94)90023-x. [DOI] [PubMed] [Google Scholar]
- Kwan C, Putney JW. Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated lachrymal acinar cells. J Biol Chem. 1990;265:678–684. [PubMed] [Google Scholar]
- Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje neurons. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Meissner G. Ryanodine receptor/Ca2+release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- Mészáros L, Bak J, Chu A. Cyclic-ADP-ribose as an endogenous regulator of the non-skeletal type ryanodine receptor Ca2+channel. Nature (Lond) 1993;364:76–79. doi: 10.1038/364076a0. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Campbell KP. Characterization of the major brain form of the ryanodine receptor/Ca2+release channel. J Biol Chem. 1993;268:19785–19790. [PubMed] [Google Scholar]
- Nohmi M, Hua S-Y, Kuba K. Intracellular calcium dynamics in response to action potentials in bullfrog sympathetic ganglion cells. J Physiol (Lond) 1992;458:171–190. doi: 10.1113/jphysiol.1992.sp019412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhauser A, Alvarez O, Latorre R. Activation by divalent cations of a Ca2+-activated K+channel from skeletal muscle membrane. J Gen Physiol. 1988;92:67–86. doi: 10.1085/jgp.92.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill SC, Donoso P, Eisner DA. The role of [Ca2+] and [Ca2+] sensitization in the caffeine contracture of rat myocytes: Measurements of [Ca2+] and [caffeine] J Physiol (Lond) 1990;425:55–70. doi: 10.1113/jphysiol.1990.sp018092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah IN, Zimanyi I. Characterization of multiple [3H] ryanodine binding sites on the Ca2+release channel of sarcoplasmic reticulum from skeletal and cardiac muscle: evidence for a sequential mechanism in ryanodine action. Mol Pharmacol. 1991;39:679–689. [PubMed] [Google Scholar]
- Pizarro G, Csernoch L, Uribe I, Ríos E. Different effects of tetracaine on two kinetic components of calcium release in frog skeletal muscle fibers. J Physiol (Lond) 1992;457:525–538. doi: 10.1113/jphysiol.1992.sp019392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Medolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Protasi, F., and C. Franzini-Armstrong 1996. Significance of the distance between calcium release units in cardiac and skeletal muscle. Biophys. J. 70:245a.
- Rasgado-Flores H, Sánchez-Armass S, Blaunstein MP, Nachshen DA. Strontium, barium and manganese metabolism in isolated presynaptic nerve terminals. Am J Physiol. 1987;252:C604–C610. doi: 10.1152/ajpcell.1987.252.6.C604. [DOI] [PubMed] [Google Scholar]
- Russeau E, Smith JS, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+release channel. Am J Physiol. 1987;253:C364–C368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Ca2+-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca2+-activated Ca2+release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-z. [DOI] [PubMed] [Google Scholar]
- Schneider M, Simon BJ. Inactivation of calcium release from the sarcoplasmic reticulum in frog skeletal muscle. J Physiol (Lond) 1988;405:727–745. doi: 10.1113/jphysiol.1988.sp017358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham JSK, Cleemann L, Morad M. Functional coupling of Ca2+channels and ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci USA. 1995;92:121–125. doi: 10.1073/pnas.92.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Kirischuk S, Kostyuk P, Verkhratsky A. Different properties of caffeine-sensitive Ca2+stores in peripheral and central mammalian neurones. Pflüg Arch. 1994;426:174–176. doi: 10.1007/BF00374686. [DOI] [PubMed] [Google Scholar]
- Shmigol A, Svichar N, Kostyuk P, Verkhratsky A. Gradual caffeine-induced Ca2+ release in mouse dorsal root ganglion neurons is controlled by cytoplasmic and lumenal Ca2+ . Neuroscience. 1996;73:1061–1067. doi: 10.1016/0306-4522(96)00108-x. [DOI] [PubMed] [Google Scholar]
- Shmigol A, Verkhratsky A, Isengerb G. Calcium-induced calcium release in rat sensory neurons. J Physiol (Lond) 1995;489:627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Mechanisms of caffeine activation of single calcium release channels of sheep cardiac sarcoplasmic reticulum. J Physiol (Lond) 1990;423:425–439. doi: 10.1113/jphysiol.1990.sp018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca2+ release channel by luminal Ca2+ . J Membr Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Sommer JR, Bossen E, Nassar R. Avian extended JSR (EJSR): a challenge to direct contact signal transduction (DCT) for coupling excitation to calcium-release (ECR) in cardiac muscle. Physiologist. 1991;34:109. [Google Scholar]
- Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD. “Adaptive” behavior of ligand-gated ion channels: constraints by thermodynamics. Biophys J. 1996;70:2100–2109. doi: 10.1016/S0006-3495(96)79776-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama K, Muteki T. Local anesthetics depress the calcium current of rat sensory neurons in culture. Anesthesiology. 1994;80:1369–1378. doi: 10.1097/00000542-199406000-00025. [DOI] [PubMed] [Google Scholar]
- Tang Y, Othmer HS. A model of calcium dynamics in cardiac myocytes based on the kinetics of ryanodine-sensitive calcium channels. Biophys J. 1994;67:2223–2235. doi: 10.1016/S0006-3495(94)80707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SA, Hirning LD, Miller RJ. The role of caffeine-sensitive calcium stores in the regulation of intracellular free calcium concentration in rat sympathetic neurons in vitro. Mol Pharmacol. 1988;34:664–673. [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol (Lond) 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillotson D. Inactivation of Ca conductance dependent on entry of Ca ions in molluscan neurons. Proc Natl Acad Sci USA. 1979;76:1497–1500. doi: 10.1073/pnas.76.3.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy A, Meissner G. Sarcoplasmic reticulum lumenal Ca2+ has access to cytosolic activation and inactivation sites of skeletal muscle Ca2+release channel. Biophys J. 1996;70:2600–2615. doi: 10.1016/S0006-3495(96)79831-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uneyama H, Munakata M, Akaike N. Caffeine response in pyramidal neurons freshly dissociated from rat hippo-campus. Brain Res. 1993;604:24–31. doi: 10.1016/0006-8993(93)90348-q. [DOI] [PubMed] [Google Scholar]
- Usachev Y, Shmigol N, Pronchock N, Kostyuk P, Verkhratsky A. Caffeine-induced calcium release from internal stores in cultured rat sensory neurons. Neuroscience. 1993;57:845–859. doi: 10.1016/0306-4522(93)90029-f. [DOI] [PubMed] [Google Scholar]
- Vélez, P., and B.A. Suárez-Isla. 1992. Intraluminal Ca2+ sites regulate the sarcoplasmic reticulum (SR) channel. Biophys. J. 61:430a.
- Verkhratsky A, Shmigol A. Calcium-induced calcium release in neurones. Cell Calcium. 1996;19:1–14. doi: 10.1016/s0143-4160(96)90009-3. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Burnstock G. Junctional subsurface organs in frog sympathetic ganglion cells. J Neurocytol. 1976;5:125–136. doi: 10.1007/BF01176186. [DOI] [PubMed] [Google Scholar]
- Wier WG. Cytoplasmic [Ca2+]i in mammalian ventricle: dynamic control by cellular processes. Annu Rev Physiol. 1990;52:467–488. doi: 10.1146/annurev.ph.52.030190.002343. [DOI] [PubMed] [Google Scholar]
- Williams AJ. Ion conduction and discrimination in the sarcoplasmic reticulum ryanodine receptor/calcium release channel. J Musc Res Cell Motil. 1992;13:7–26. doi: 10.1007/BF01738423. [DOI] [PubMed] [Google Scholar]
- Xu L, Jones R, Meissner G. Effects of local anesthetics on single channel behavior of skeletal muscle calcium release channel. J Gen Physiol. 1993;101:207–233. doi: 10.1085/jgp.101.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki K, Hoshino T, Sato M, Koyano H, Nohmi M, Hua S-Y, Kuba K. Ca2+ induced Ca2+release and its activation in response to a single action potential in rabbit otic ganglion cells. J Physiol (Lond) 1995;486:177–187. doi: 10.1113/jphysiol.1995.sp020801. [DOI] [PMC free article] [PubMed] [Google Scholar]