

Abstract
Receptor-mediated activation of heterotrimeric G proteins leading to dissociation of the Gα subunit from Gβγ is a highly conserved signaling strategy used by numerous extracellular stimuli. Although Gβγ subunits regulate a variety of effectors, including kinases, cyclases, phospholipases, and ion channels (Clapham, D.E., and E.J. Neer. 1993. Nature (Lond.). 365:403–406), few tools exist for probing instantaneous Gβγ-effector interactions, and little is known about the kinetic contributions of effectors to the signaling process. In this study, we used the atrial muscarinic K+ channel, which is activated by direct interactions with Gβγ subunits (Logothetis, D.E., Y. Kurachi, J. Galper, E.J. Neer, and D.E. Clap. 1987. Nature (Lond.). 325:321–326; Wickman, K., J.A. Iniguez-Liuhi, P.A. Davenport, R. Taussig, G.B. Krapivinsky, M.E. Linder, A.G. Gilman, and D.E. Clapham. 1994. Nature (Lond.). 366: 654–663; Huang, C.-L., P.A. Slesinger, P.J. Casey, Y.N. Jan, and L.Y. Jan. 1995. Neuron. 15:1133–1143), as a sensitive reporter of the dynamics of Gβγ-effector interactions. Muscarinic K+ channels exhibit bursting behavior upon G protein activation, shifting between three distinct functional modes, characterized by the frequency of channel openings during individual bursts. Acetylcholine concentration (and by inference, the concentration of activated Gβγ) controls the fraction of time spent in each mode without changing either the burst duration or channel gating within individual modes. The picture which emerges is of a Gβγ effector with allosteric regulation and an intrinsic “off” switch which serves to limit its own activation. These two features combine to establish exquisite channel sensitivity to changes in Gβγ concentration, and may be indicative of the factors regulating other Gβγ-modulated effectors.
Keywords: signal transduction, GTP binding proteins, muscarinic receptor, inward rectifier K+ channel, atrial myocytes
introduction
Heterotrimeric GTP-binding proteins (G proteins) transduce signals from an extensive family of receptors to a variety of cellular effectors which include plasma membrane–localized enzymes and ion channels (Clapham and Neer, 1993). Molecular dissection of G protein–mediated signal transduction pathways has revealed roles for both of the products of receptor-mediated G protein activation, namely Gα-GTP and Gβγ subunits (Birnbaumer et al., 1990; Birnbaumer, 1992; Clapham and Neer, 1993). The distinctive functional properties of Gα and Gβγ subunits suggest the possibility for regulatory mechanisms unique to each subunit class.
Resolution of the kinetic features of Gβγ interactions with cellular effectors may provide insights into the distinctive features of Gβγ-mediated signaling. The atrial G protein–gated inwardly rectifying K+ channel (muscarinic K+ channel), comprised of a heterotetramer (Yang et al., 1995) of GIRK1 and CIR (GIRK4) (Krapivinsky et al., 1995; Hedin et al., 1996), has been extensively studied as a direct G protein effector (Kurachi et al., 1992; Yamada et al., 1994a ). Both Gα and Gβγ subunits have been implicated in its activation mechanism (Birnbaumer et al., 1990; Kurachi et al., 1992; Clapham and Neer, 1993), although recent mutational analysis and biochemical studies have clearly defined a primary role for Gβγ subunits (Huang et al., 1995; Slesinger et al., 1995; Pessia et al., 1995; Kunkel and Peralta, 1995). In this study, we used the atrial muscarinic K+ channel as a prototype Gβγ effector to probe alterations in effector activity in response to agonist-induced alterations in Gβγ concentrations. We find that atrial muscarinic K+ channels gate in bursts of activity, and although burst duration is not a function of Gβγ concentration, the heterogeneous gating kinetics within bursts are characterized by Gβγ-mediated shifts in modal preference. These results demonstrate that muscarinic K+ channel activity can be “titrated” by binding of variable numbers of Gβγ, and provide evidence for muscarinic K+ channel inactivation or desensitization.
materials and methods
Atrial myocytes were obtained from the bullfrog Rana catesbeiana as described previously (Scherer and Breitwieser, 1990). Single-channel currents were recorded with standard high-resolution patch-clamp techniques (Hamill et al., 1981) using 1 mm O.D. square bore glass (Glass Co. of America, Millville, NJ). The membrane potential was zeroed with the following bath solution (in mM): 150 KCl, 5 EGTA, 5 glucose, 1.6 MgCl2, 5 HEPES (pH adjusted to 7.4). Pipettes contained (in mM): 150 KCl, 1 CaCl2, 1.6 MgCl2, 5 HEPES (pH adjusted to 7.4). Acetylcholine (ACh)1 was added to the pipette solution. The excised patch configuration was used to examine channel activity in the presence of GTPγS; bath solution for these experiments contained pipette solution plus 100 μM GTPγS ± ATP or AppNHp (as described in the individual experiments). Whole cell currents were recorded as previously described (Scherer and Breitwieser, 1990). Currents were recorded with a LIST EPC-7 amplifier, filtered at 2 kHz (8-pole Bessel filter; 17 μs rise time), and stored on computer. Data acquisition and analysis were performed using PCLAMP software (versions 5.5.1 and 6.0; Axon Instruments, Foster City, CA). Channel opening and closing transitions were identified with the half-amplitude threshold-crossing algorithm. Bursts were defined as a series of openings (single events were eliminated from the analysis) separated by closed intervals shorter than some critical interval, tc . In most records the occurrence of bursts was infrequent so the choice of tc was not critical and the division of the record into bursts was unambiguous. In a few cases of high activity, when the separation was less clear, tc was calculated as described previously (Magleby and Pallotta, 1983) and had values between 120 and 140 ms. For each defined burst, the duration, the number of apparent open intervals and the probability of a channel being open within a burst, po, were calculated. The po value was calculated as the ratio of the total open time in the burst to the burst length. When indicated, the individual events from selected bursts were compiled to generate histograms of open- and closed-interval durations. All experiments were done at a holding potential of −90 mV (unless otherwise noted) and at 20–22°C.
results
Heterogeneity of Unitary Muscarinic K+ Currents
Acetylcholine (ACh)1 released upon stimulation of the vagus nerve causes slowing of heart rate by activation of muscarinic receptors and the subsequent opening of muscarinic K+ channels in the sinoatrial node and atrium. Unitary currents through muscarinic K+ channels were recorded from cell-attached patches on frog atrial myocytes in the presence of different concentrations of ACh in the pipette. Fig. 1 A shows an example of a continuous record of single-channel currents activated by 1 μM ACh; the membrane potential of the patch was held at −90 mV. Channel gating behavior is heterogeneous, characterized by both apparent clustering of openings into bursts and spontaneous changes in the pattern of channel activity.
Figure 1.

Bursting activity of muscarinic K+ channels in atrial myocytes. (A) Continuous single-channel record from a cell-attached patch. The patch pipette contained 1 μM ACh. Downward deflections correspond to inward currents. Calibration: 5 pA, 100 ms. (B) Distribution of burst lengths for 1 μM ACh (combined data from seven patches), recorded at a membrane potential of −90 mV. The continuous line represents the monoexponential fit used to estimate the time constant, τb, which was 193.4 ms.
Muscarinic K+ Channels Gate in Bursts
Records, such as that illustrated in Fig. 1 A, indicate that bursts of muscarinic K+ channel openings are separated by long silent periods; individual bursts were therefore considered to reflect the activity of a single ion channel and analyzed further. Fig. 1 B illustrates the burst length histogram obtained from activity elicited by 1 μM ACh, plotted on a logarithmic time scale. The histogram was best fitted by a single exponential term with τb = 193.4 ms. Burst length distributions were also obtained at 50 nM and 0.5 μM ACh (Table I). No significant differences between burst length time constants were evident, indicating that agonist concentration does not determine burst length. Furthermore, the holding potential (−60 or −90 mV) did not significantly affect the burst length distribution obtained at 0.5 μM ACh (Table I).
Table I.
Burst Duration Time Constants for Atrial Muscarininc K+ Channels Determined under a Variety of Experimental Conditions
| Recording configuration | Agonist concentration/ bath additions | Holding potential | Burst duration time constant | Number of bursts analyzed | ||||
|---|---|---|---|---|---|---|---|---|
| (mV) | (ms) | |||||||
| Cell-attached | 50 nM ACh | −90 | 196.5 | 571 | ||||
| Cell-attached | 0.5 μM ACh | −60 | 187.6 | 273 | ||||
| Cell-attached | 0.5 μM ACh | −90 | 258.1 | 350 | ||||
| Cell-attached | 1 μM ACh | −90 | 193.4 | 623 | ||||
| Inside-out | 100 μM GTPγS | −90 | 193.0 | 209 | ||||
| Inside-out | 100 μM GTPγs 5mM ATP | −90 | 186.8 | 234 | ||||
| Inside-out | 100 μM GTPgs 1mM AppNHp | −90 | 190.4 | 273 |
Agonist concentration/bath additions indicates contents of a pipette for cell-attached recording condition, or additions to the bath under inside-out recording conditions. Burst duration time constant was obtained by a single exponential fit to the burst duration histogram, comprised of 200–600 individual bursts, as indicated.
The transient nature of Gβγ interactions with effectors presents the possibility that the dynamics of G protein turnover may limit channel burst length under physiological conditions. All heterotrimeric G proteins exploit a highly conserved signaling strategy in which the metastable GTP-bound state of the Gα subunit is used as a molecular clock (Bourne et al., 1990, 1991). When GTP is hydrolyzed to GDP, Gα is inactivated, and its affinity for free Gβγ is increased. The potential contributions of G protein turnover to the kinetics of G protein–mediated signaling can be eliminated with the hydrolysis-resistant GTP analogue, GTPγS. Binding of GTPγS to Gα persistently activates G proteins, constraining both Gα and Gβγ to their active states, reducing the signaling pathway to diffusion-limited interactions between Gβγ and the channel. Fig. 2 A illustrates representative unitary K+ currents recorded from an excised, inside-out membrane patch in the presence of 100 μM GTPγS. In the presence of GTPγS, channel activity was similar to that observed in cell-attached patches in the presence of 1 μM ACh. Indeed, comparison of the burst length distributions displayed in Fig. 1 B and 2 B reveals no significant differences in τb for the two conditions (Table I). Thus, the combined data in Figs. 1 and 2 suggest that muscarinic K+ channel burst duration is not determined by agonist concentration, nor the dynamics of G protein turnover, i.e., GTP hydrolysis, presenting the possibility that burst length is determined by a property intrinsic to the muscarinic K+ channel itself, i.e., inactivation or desensitization in the continued presence of Gβγ, or may be a feature of the Gβγ-channel interaction.
Figure 2.

Gating of muscarinic K+ channels in the presence of GTPγS. (A) Continuous record of single channel activity observed in inside-out patch following the application of 100 μM GTPγS. AMP-PNP (1 mM) was also present in the bath solution to inhibit ATP-sensitive K+ channels. Membrane potential was held at −90 mV. Calibration: 5 pA, 100 ms. (B) Burst length histogram for the bursts recorded in the presence of 100 μM GTPγS. The continuous line indicates the monoexponential fit to the data; τb = 190.4 ms.
Heterogeneity of Muscarinic K+ Channel Gating within Bursts
Although channel burst duration is independent of the agonist concentration, inspection of current records reveals two features of muscarinic K+ channel gating worthy of further exploration. First, gating is heterogeneous over a wide range of conditions including different agonist concentrations (Fig. 4 A), in excised patches following GTPγS-mediated activation of the channel (Fig. 2 A), and at different membrane potentials over the range from −110 to −40 mV (Fig. 3). Second, despite the apparent gating heterogeneity, there is a systematic increase in the open probability during individual bursts as the ACh concentration is increased, as illustrated in Fig. 4 A for individual bursts recorded in the presence of two different ACh concentrations. To determine whether these features of muscarinic K+ channel gating could be related to the level of activated Gβγ, we employed several complementary approaches, at low (50 nM) and high (1 μM) ACh concentrations, to separate bursts into two or more discrete and kinetically simplified gating modes. First, we examined the distributions of the number of apparent openings for bursts composed of two or more openings. These distributions should consist of a sum of geometric components, the number of components being equal to the number of open states, although the contributions of some components may be too small to resolve (Colquhoun and Hawkes, 1983). Examples of histograms of the apparent openings per burst are presented in Fig. 4 B for 50 nM and 1 μM ACh. Both distributions were best fitted by a sum of two geometrics, μ1 and μ2: the first component had a mean value of approximately three openings per burst, while the mean of the second component was ~15. Comparison of the fits to these distributions reveals that the agonist concentration determines the proportion of bursts in the two groups: the fraction of bursts containing a larger number of apparent openings increased from 36 ± 6% to 62 ± 5% when comparing bursts obtained in 50 nM and 1 μM ACh, respectively, while the mean values for μ1 and μ2 were similar for the two concentrations.
Figure 4.
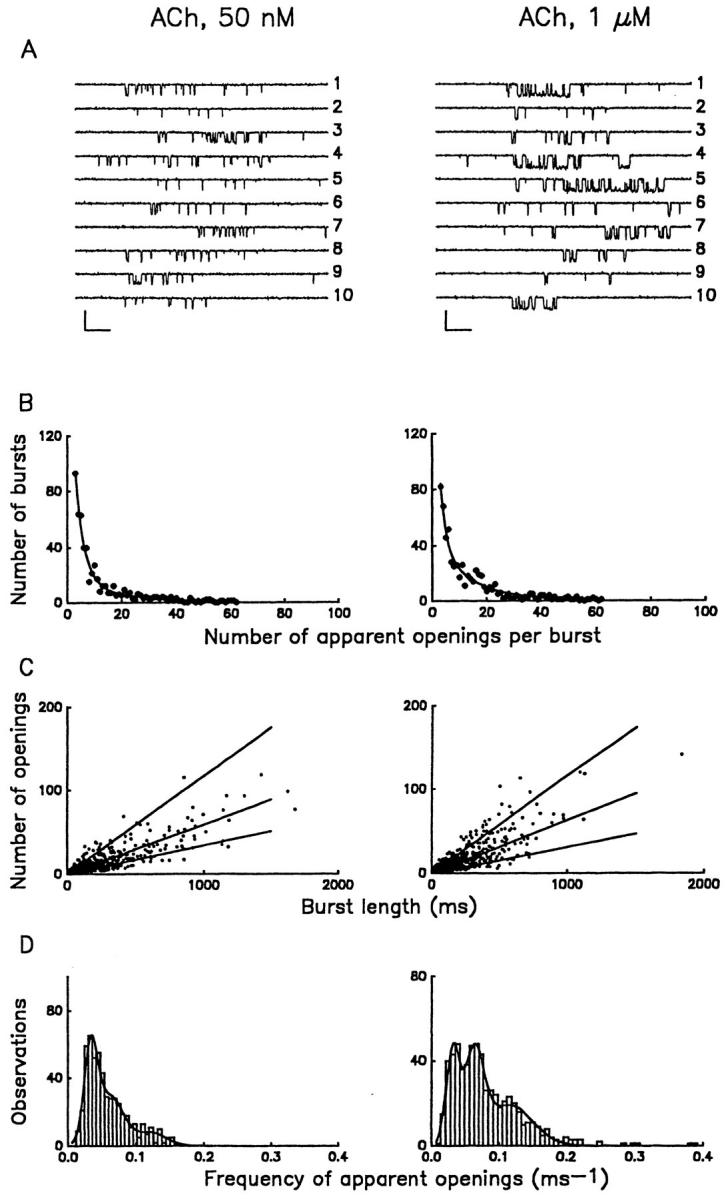
Kinetic heterogeneity of individual muscarinic K+ channel bursts. (A) Multiple gating patterns of the channel at low (50 nM) and saturating (1 μM) concentrations of ACh. Two sets of bursts illustrate the scope of kinetic heterogeneity of gating. The probability that a channel resides in an open conformation during the burst, po, is as follows for each trace: 50 nM ACh (1) 0.078; (2) 0.02; (3) 0.164; (4) 0.099; (5) 0.026; (6) 0.071; (7) 0.166; (8) 0.079; (9) 0.231; and (10) 0.060; 1 μM ACh (1) 0.31; (2) 0.057; (3) 0.152; (4) 0.361; (5) 0.559; (6) 0.034; (7) 0.173; (8) 0.202; (9) 0.065; and (10) 0.652. Calibration: 5 pA, 50 ms. (B) Distributions of the number of apparent openings per burst. Distributions are shown for ACh at concentrations of 50 nM (left panel) and 1 μM (right panel) and were fitted by the sum of two geometrics (Colquhoun and Hawkes, 1981): P(r) = a 1μ1 −1(1-μ1 −1)r -1 + a 2μ2 −1 (1-μ2 −1)r -1 (continuous line). The means, μI (and percentage areas (a i)) are 3.7 ± 0.4 (64 ± 6%) and 19.2 ± 5.6 (36 ± 6%) at 50 nM ACh and 2.8 ± 0.5 (38 ± 5%) and 14.4 ± 2.0 (62 ± 5%) at 1 μM ACh. In the presence of 1 μM ACh, a small population of very long bursts was observed. These long bursts contained too many openings (200–1,000) to be fitted by the distributions described above. Although such long bursts were too infrequent to be analyzed, they contributed substantially to the total single channel activity because of their duration. (C) Frequency of channel activations within bursts. Plots of apparent number of openings, N, vs. burst duration, B are shown for the experiments illustrated in B. Each symbol represents N and B values determined for an individual burst. Lines in the figures were derived from the peaks of the frequency distributions plotted in D. (D) Distributions of f values in 50 nM (left panel) and 1 μM (right panel) ACh for the data shown in C. Both histograms are fitted by the sum of three Gaussian distributions. The frequency of openings within bursts, f, was estimated as N/B. Peak frequencies of channel activation were independent of the agonist concentration and have values of 0.03 ms−1 (low-f), 0.06 ms−1 (medium-f) and 0.12 ms−1 (high-f). At 50 nM ACh, 40% of bursts were low-f, 47% medium-f and 13% the high-f type. The proportion of high-frequency bursts increased with ACh concentration; at 1 μM ACh the relative areas of low-, medium-, and high-frequency bursts were 25%, 35% and 40%, respectively.
Figure 3.

Bursts of muscarinic K+ channel activity at various holding potentials. Representative records from an inside-out patch following application of 100 μM GTPγS plus 1 mM adenylylimidodiphosphate. Holding potential was varied as indicated. Calibration: 5 pA, 100 ms.
Classification of bursts into groups according to the two geometric components in Fig. 4 B hinted at the existence of distinct gating modes, but even among bursts with an equivalent number of openings, the gating behavior remained heterogeneous with respect to both mean open time and open probability. Therefore, as an alternative approach to grouping bursts and identifying distinct patterns of gating, we plotted the number of apparent openings in the burst, N, vs. the length of the burst, B, for each burst. Illustrated in Fig. 4 C are plots for bursts recorded in the presence of either 50 nM ACh or 1 μM ACh. Each symbol in the figure represents the kinetic behavior of an individual burst. Histograms of frequencies of openings within individual bursts (f = N/B) were generated from the same bursts illustrated in Fig. 4 C to empirically determine the most populated regions of the N-B plots. The histograms, illustrated in Fig. 4 D, define three distinct peaks centered around f values of 0.03 ms−1, 0.06 ms−1, and 0.12 ms−1. These frequencies were therefore used as the criteria for separation of channel gating behavior into low-, medium-, and high-f modes (and were used to generate the lines in Fig. 4 C). Although the predominant frequencies of channel openings were independent of ACh concentration, comparison of the two plots in Fig. 4 D indicates that modal preference is a function of the ACh concentration. At 50 nM ACh, low- and medium-f behavior dominate the f -histogram, while saturating ACh concentrations favor the high-f mode. To determine the maximal effect on modal preference, we also analyzed the frequency of openings within individual bursts, f, for a total of 273 bursts, recorded in excised patches exposed to 100 μM GTPγS. The frequency distribution after GTPγS activation revealed three distinct peaks, corresponding to the low-f (0.032 ms−1; relative area 13.5%), medium-f (0.064 ms−1; relative area 36.5%), and high-f (0.116 ms−1; relative area 50%) modes characterized previously in the cell-attached configuration in the presence of ACh (Fig. 4 D). Thus, the heterogeneous kinetic behavior observed in the presence of GTPγS reflects sojourns of the channel in the same dominant gating modes found in the presence of ACh.
Kinetics of Muscarinic K+ Channel Gating within Distinct Gating Modes
Although ACh concentrations establish modal preference for muscarinic K+ channels, all modes are represented at any given ACh concentration. To characterize the kinetic behavior of the channel in each gating mode, we analyzed histograms of open and closed interval durations generated from low-, medium-, and high-f burst populations recorded in 1 μM ACh (Fig. 5). For this analysis, only bursts with f values within 2 SD of the mean of a particular f population were included (eliminating bursts in which there was an obvious switch in gating mode during the burst). Each open and closed state adds an additional exponential component to these distributions (Colquhoun and Hawkes, 1981). Within each mode, a sum of three exponentials provided a satisfactory fit to the open time distributions (Fig. 5, left). The fast, intermediate, and slow components showed little variation from one mode to the next, and had time constants (τo1 − τo3) of ~0.18, 1.5, and 5.0 ms, respectively. The relative areas under the individual exponentials, however, were quite different for the three modes and verified the general observation that the low-f bursts were predominantly made of short openings while high-f bursts were dominated by longer ones.
Figure 5.

Distributions of open and closed times for individual gating modes. Bursts were compiled from those obtained in 1 μM ACh. Three populations of bursts (labeled low-f, medium-f, and high-f), selected from bursts within 2 SD of the peak frequencies of 0.03 ms−1, 0.06 ms−1 and 0.12 ms−1, were used to generate open and closed time histograms. The histograms constructed from bursts within each mode are fitted by the sum of three exponentials (continuous lines) and are plotted for each mode to facilitate comparisons. Also plotted are the individual fits to each exponential (dashed lines). The relative proportion for each component is given in parentheses after the time constant. In the low-f mode the fast, intermediate, and slow open time constants (τo1–τo3) are 0.19 ms (52%), 1.7 ms (32%), and 5.6 ms (16%) and the closed time constants are 0.34 ms (34%), 1.6 ms (9%), and 67.8 ms (57%). In the medium-f mode the open time constants are 0.16 ms (45%), 1.2 ms (33%), and 4.8 ms (22 %) and the closed time constants are 0.17 ms (40%), 1.3 ms (25%), and 23.9 ms (35%). In the high-f mode the open time constants are 0.2 ms (26%), 1.7 ms (29%), and 4.6 ms (45%) and the closed time constants are 0.17 ms (56%), 1.2 ms (28%), and 12.3 ms (16%).
Closed intervals were distributed over a wider time range than open times, and fitting of closed-time histograms obtained from randomly compiled bursts required more than three exponentials. After modal classification of the bursts at 1 μM ACh, however, three exponential terms were sufficient to describe the closed times in each mode (Fig. 5, right). The fast and intermediate components had similar time constants for the three kinetic modes (τc1 = 0.23 ms and τc2 = 1.4 ms), whereas the third time constant, τc3 , decreased as the frequency increased, being 67.8 ms for the low-f bursts, 23.9 ms for medium-f, and 12.3 ms for the high-f bursts.
Similar analysis of mode-segregated bursts recorded from GTPγS-activated patches was performed (data not shown). The fits to the open-time distributions yielded fast, intermediate, and slow time constants which were similar in all modes and comparable to those observed in the presence of ACh (τo1 = 0.14 ms; τo2 = 1.4 ms; τo3 = 4.0 ms). The fast and intermediate closed time constants had mean values of τc1 = 0.15 ms and τc2 = 1.3 ms in all modes, while the slow closed time constant, τc3, was once again the primary discriminator between modes, being 80.0 ms for the low-f mode, 23.1 ms for the medium-f mode, and 12.4 ms for the high-f mode. These results suggest that the channel enters the same gating modes whether the G proteins are activated by GTP or GTPγS, with a minimum of three open (O1-O2-O3) and three closed (C1-C2-C3) states within each mode. The values for all three open time constants (τo1– τo3) as well as the fast and intermediate closed time constants (τc1 and τc2) are similar for the three modes. As long as the conducting and nonconducting states of the channel can be regarded as reporters of distinct conformational states of the protein, our results imply that gating in any mode arises from a common set of five conformational states O1 . . . C2 and one additional nonconducting state C3 which is the main kinetic discriminator between the modes. The concentration of agonist (and presumably the concentration of Gβγ) has the unique role of establishing modal preference, i.e., determining the ease with which the channel enters the higher frequency gating modes.
Physiological Relevance of Modal Shifts in Gating
Activation of whole cell muscarinic K+ current (IK[ACh]) by application of ACh incorporates both the kinetic changes within bursts which have been described in previous sections, as well as potential alterations in interburst intervals and the number of functional channels. To determine the physiological relevance of the modal shifts in gating within bursts, three measures of muscarinic K+ channel activity were compared at a range of ACh concentrations, Fig. 6. As a standard for comparison, the steady state whole cell dose response for ACh was plotted (data normalized to the current at 1 μM ACh). To assess the dose response relation for ACh-mediated shifts in p o within bursts, the average p o within 150 random (i.e., non-mode-segregated) bursts was tabulated, at three ACh concentrations (50 nM, 0.5 μM, and 1 μM). The average p os were normalized to the value obtained at 1 μM ACh (p o = 0.3). Finally, to determine the overall dose response relation for ACh at the single channel level (bursts plus interburst intervals), the p o of representative recordings in the cell-attached configuration was determined at 50 nM, 0.5, and 1 μM ACh (normalized to the p o obtained at 1 μM ACh ≈ 0.047). All three measures of the dose response relation are similar, suggesting that ACh does not differentially affect the interburst intervals, and therefore that the ACh-induced shifts in modal gating within bursts are the primary determinant of the overall, whole cell current response.
Figure 6.

Acetylcholine (ACh) titration of muscarinic K+ channel activity. Three measures of muscarinic K+ channel activity are compared at varying ACh concentrations, from 5 × 10−8 to 10−4 M. Open circles indicate the whole cell steady state current, IK[ACh], at 60 s after application of various ACh concentrations, normalized to the steady state (60 s) current obtained in 10−6 M ACh. Each point represents the mean ± SEM of at least four cells. The line indicates the fit obtained with the Hill equation, with V max = 111.7%, K = 1.7 × 10−7 M, and n = 0.99. Filled circles represent the p o of representative recordings obtained in the cell-attached configuration at various ACh concentrations (normalized to 10−6 M ACh, p o = 0.0468). Estimates of p o were obtained from at least three patches at each concentration (recording periods greater than 2 min in each condition). Filled triangles represent the average p o for representative bursts (non-mode-segregated) obtained at various ACh concentrations, normalized to the p o of bursts obtained at 10−6 M ACh (p o = 0.301). For each concentration, 150 random bursts from multiple patches were averaged.
discussion
This study reveals two novel aspects of muscarinic K+ channel gating. First, muscarinic K+ channels gate in discrete bursts of activity. Burst duration is monoexponentially distributed and insensitive to a variety of factors which modulate muscarinic K+ channel activity, including agonist concentration, membrane potential, and G protein turnover (i.e., GTP hydrolysis). Second, the gating of muscarinic K+ channels within bursts is heterogeneous, but discrete modes of gating can be defined based on the frequency of channel opening within a burst. Agonist concentration determines modal preference, although all modes are represented at all agonist concentrations. Agonist-induced shifts in modal preference are the main determinant of whole cell IK[ACh]. We will consider these results in light of extensive studies which suggest Gβγ to be an integral activator of muscarinic K+ channels (Logothetis et al., 1987; Kurachi et al., 1992; Wickman et al., 1994; Yamada et al., 1994a ; Krapivinsky et al., 1995), with direct binding to the channel recently demonstrated in vitro (Huang et al., 1995; Slesinger et al., 1995; Pessia et al., 1995; Kunkel and Peralta, 1995). It is also possible that additional, muscarinic receptor- or G protein–activated signaling pathways modulate muscarinic K+ channel activation (e.g., Scherer and Breitwieser, 1990; Yamada et al., 1994b ). Since these additional pathways are presumably also activated in a dose-dependent manner by ACh, our results at present cannot distinguish between the primary Gβγ interaction with the muscarinic K+ channel and these modulatory influences. We thus limit our discussion to the minimal model which requires only direct Gβγ interaction with the channel heteromultimer.
Burst Duration Is Independent of Gβγ Concentrations
The simplest model for determination of muscarinic K+ channel burst duration consistent with the data is one in which the channel enters a discrete inactivated or desensitized state (independent of Gβγ), as indicated in the following scheme.
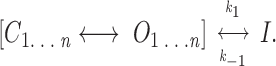 |
The channel undergoes repeated transitions between closed and open states (as determined by Gβγ interactions) and, with a rate constant k 1 (reflected in the time constant for burst duration), enters the inactivated state. k −1 represents the time constant for exit from the inactivated state, and can in principal be obtained from interburst intervals. In practice, however, this is not possible since it is difficult to unequivocally establish the number of channels in the patch (maximal patch open probability is 0.08–0.1). Nevertheless, the existence of an inactivated, nonresponsive state limits the activity of the channel, and, if Gβγ subunits interact directly with muscarinic K+ channels, has implications for Gβγ-mediated signaling.
Gβγ subunits lack an intrinsic enzymatic activity which serves to periodically alter the conformational state of the complex (Sondek et al., 1996; Neer and Smith, 1996) and hence the affinity for either effectors or Gα subunits. Dissociation of Gβγ from effector binding sites may therefore be the rate-limiting step in termination of Gβγ-mediated signaling. Inactivation of muscarinic K+ channels presents the possibility that effectors themselves may contribute to termination of Gβγ-mediated responses. Channel inactivation may be accompanied by either a transient decrease in channel affinity for Gβγ (i.e., a conformational change may occur in the Gβγ binding site), or it may occur despite the continued presence of bound Gβγ, with subsequent dissociation of Gβγ while the channel is inactivated.
Modal Preference Determination by Gβγ Concentrations
The native muscarinic K+ channel in atrial myocytes contains multiple structurally heterogeneous, Gβγ binding sites within what is most likely a tetrameric channel structure (Krapivinsky et al., 1995; Yang et al., 1995). In this context, the simplest model of channel gating would require the occupancy of all Gβγ binding sites before channel activation. The ability of ACh (i.e., Gβγ) to titrate modal preference, however, favors more complex models, in which distinct functional consequences, i.e., modal shifts, accompany binding of successive Gβγ. While our data at present cannot be constrained to a model which imposes a one-for-one correlation between the number of bound Gβγ and a particular gating mode, a reaction scheme similar to the Monod-Wyman-Changeux allosteric model (Monod et al., 1965) in which all modes are interconnected, and binding of increasing numbers of Gβγ subunits facilitates transitions to the high-f mode is entirely consistent with our observations. A similar mechanism has been proposed for G protein–mediated inhibition of neuronal N-type Ca2+ channels (Boland and Bean, 1993; Delcour et al., 1993; Delcour and Tsien, 1993), which has recently been shown to be mediated by Gβγ (Ikeda, 1996; Herlitze et al., 1996).
Modal Shifts as a Means of Increasing Gβγ Signaling Specificity
Some effectors are activated in a highly specific manner by particular subtypes of Gβγ (Inguez-Liuhi et al., 1992; Schmidt et al., 1992; Kleuss et al., 1993; Wu et al., 1993) while other effectors are activated equally well by all Gβγ subtypes (Clapham and Neer, 1993; Wickman et al., 1994; Ueda et al., 1994). Muscarinic K+ channels fall into the second class, i.e., all Gβγ subtype combinations are equally effective at the activation of the channel (Wickman et al., 1994), with the exception of transducin βγ (Yamada et al., 1994a ). This presents a conceptual difficulty, since it implies that activation of any G protein–coupled receptor within the cell should contribute to the population of activated Gβγ and thus muscarinic K+ channel activation. Signaling specificity must therefore arise on the basis of other mechanisms. Our results suggest a possible means for increasing signaling specificity and/or sensitivity in spite of a lack of Gβγ subtype specificity, namely, the existence of multiple functional states of an effector, which are dependent upon the number of bound Gβγ. The efficiency of signal transduction via a population of Gβγ would thus depend not only on the amount of free Gβγ and the relative abundance of effectors, but also on the number of functional modes accessible to the various effectors and on the ability of Gβγ to control the equilibrium between these modes. In the case of the muscarinic K+ channel, the combination of an intrinsic inactivation mechanism which may serve to limit the duration of the Gβγ interaction plus allosteric regulation via multiple Gβγ subunits (which is poised to provide significant whole cell K+ current only at high Gβγ concentrations) produces a system which is rapidly regulated and highly sensitive to dynamic changes in Gβγ concentration.
Acknowledgments
We thank E. Nikolova, F. Sigworth, M. Li, and W. Agnew for helpful comments at different stages of the work. We thank C. Frederick Lo for providing the whole cell acetylcholine dose response data.
This work was supported by the National Institutes of Health (HL41972), a National Science Foundation Career Advancement Award (9407251) and an E.I. from the American Heart Association (AHA) National Center (900126) to G.E. Breitwieser. T.T. Ivanova-Nikolova was partially supported by a Fellowship from the AHA Maryland Affiliate.
Abbreviation used in this paper
- ACh
acetylcholine
references
- Birnbaumer L. Receptor-to-effector signaling through G proteins: roles for βγ dimers as well as α subunits. Cell. 1992;71:1069–1072. doi: 10.1016/s0092-8674(05)80056-x. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Abramowitz J, Brown AM. Receptor- effector coupling by G proteins. Biochim Biophys Acta. 1990;1031:163–224. doi: 10.1016/0304-4157(90)90007-y. [DOI] [PubMed] [Google Scholar]
- Boland LM, Bean BP. Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-releasing hormone: kinetics and voltage dependence. J Neurosci. 1993;13:516–533. doi: 10.1523/JNEUROSCI.13-02-00516.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature (Lond) 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature (Lond) 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Clapham DE ,, and E.J. Neer. New roles for G-protein βγ-dimers in transmembrane signalling. Nature (Lond) 1993;365:403–406. doi: 10.1038/365403a0. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. On the stochastic properties of single ion channels. Proc R Soc B. 1981;211:205–235. doi: 10.1098/rspb.1981.0003. [DOI] [PubMed] [Google Scholar]
- Colquhoun, D., and A.G. Hawkes. 1983. The principles of the stochastic interpretation of ion-channel mechanisms. In Single Channel Recording. B. Sakmann and E. Neher, editors. (Plenum Press New York, NY), pp. 135–175.
- Delcour AH, Lipscombe D, Tsien RW. Multiple modes of N-type calcium channel activity distinguished by differences in gating kinetics. J Neurosci. 1993;13:181–194. doi: 10.1523/JNEUROSCI.13-01-00181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour AH, Tsien RW. Altered prevelance of gating modes in neurotransmitter inhibition of N-type calcium channels. Science (Wash DC) 1993;259:980–984. doi: 10.1126/science.8094902. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hedin KE, Lim NF, Clapham DE. Cloning of a Xenopus laevis inwardly rectifying K+ channel subunit that permits GIRK1 expression of IK[ACh]currents in oocytes. Neuron. 1996;16:423–429. doi: 10.1016/s0896-6273(00)80060-4. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+channels by G-protein βγ subunits. Nature (Lond) 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Huang C-L, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature (Lond) 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Inguez-Liuhi JA, Simon MI, Robishaw JD, Gilman AG. G-protein βγ subunits synthesized in Sf9 cells. J Biol Chem. 1992;267:23409–23417. [PubMed] [Google Scholar]
- Kleuss C, Scherubl H, Hescheler J, Schultz G, Wittig B. Selectivity in signal transduction determined by γ subunits of heterotrimeric G proteins. Science (Wash DC) 1993;259:832–834. doi: 10.1126/science.8094261. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IK[ACh] is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature (Lond) 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- Kunkel MT, Peralta EG. Identification of domains conferring G protein regulation on inward rectifier potassium channels. Cell. 1995;83:443–449. doi: 10.1016/0092-8674(95)90122-1. [DOI] [PubMed] [Google Scholar]
- Kurachi Y, Tung RT, Ito H, Nakajima T. G protein activation of cardiac muscarinic K+channels. Prog Neurobiol. 1992;39:229–246. doi: 10.1016/0301-0082(92)90017-9. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+channel in heart. Nature (Lond) 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- Magleby KL, Pallotta BS. Burst kinetics of single calcium-activated potassium channels in cultured rat muscle. J Physiol. 1983;344:605–623. doi: 10.1113/jphysiol.1983.sp014958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Neer EJ, Smith TF. G protein heterodimers: new structures propel new questions. Cell. 1996;84:175–178. doi: 10.1016/s0092-8674(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Pessia M, Bond CT, Kavanaugh MP, Adelman JP. Contributions of the C-terminal domain to gating properties of inward rectifier potassium channels. Neuron. 1995;14:1039–1045. doi: 10.1016/0896-6273(95)90342-9. [DOI] [PubMed] [Google Scholar]
- Scherer RW, Breitwieser GE. Arachidonic acid metabolites alter G protein-mediated signal transduction in heart: effects on muscarinic K+channels. J Gen Physiol. 1990;96:735–755. doi: 10.1085/jgp.96.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Thomas TC, Levine MA, Neer EJ. Specificity of G protein β and γ subunit interactions. J Biol Chem. 1992;267:13807–13810. [PubMed] [Google Scholar]
- Slesinger PA, Reuveny E, Jan YN, Jan LY. Identification of structural elements involved in G protein gating of the GIRK1 potassium channel. Neuron. 1995;15:1145–1156. doi: 10.1016/0896-6273(95)90102-7. [DOI] [PubMed] [Google Scholar]
- Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Crystal structure of a G protein βγ dimer at 2.1 A resolution. Nature (Lond) 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- Ueda N, Iniguez-Liuhi JA, Lee E, Smrcka AV, Robishaw JD, Gilman AG. G protein βγ subunits. Simplified purification and properties of novel isoforms. J Biol Chem. 1994;269:4388–4395. [PubMed] [Google Scholar]
- Wickman K, Iniguez-Liuhi JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein βγ-subunits activate the muscarinic-gated atrial potassium channel. Nature (Lond) 1994;368:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- Wu D, Katz A, Simon MI. Activation of phospholipase C-β 2 by the α and βγ subunits of trimeric GTP-binding protein. PNAS. 1993;90:5297–5301. doi: 10.1073/pnas.90.11.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Ho Y-K, Lee RH, Kontani K, Takahashi K, Katada T, Kurachi Y. Muscarinic K+channels are activated by βγ subunits and inhibited by the GDP-bound form of subunit of transducin. Biochem Biophys Res Commun. 1994a;200:1484–1490. doi: 10.1006/bbrc.1994.1618. [DOI] [PubMed] [Google Scholar]
- Yamada M, Terzic A, Kurachi Y. Regulation of potassium channels by G-protein subunits and arachidonic acid metabolites. Methods Enzymol. 1994b;238:394–422. doi: 10.1016/0076-6879(94)38036-8. [DOI] [PubMed] [Google Scholar]
- Yang J, Jan NY, Jan LY. Determination of the subunit stoichiometry of an inwardly rectifying potassium channel. Neuron. 1995;15:1441–1447. doi: 10.1016/0896-6273(95)90021-7. [DOI] [PubMed] [Google Scholar]