

Abstract
Dihydropyridines (DHPs) are well known for their effects on L-type voltage-dependent Ca2+ channels. However, these drugs also affect other voltage-dependent ion channels, including Shaker K+ channels. We examined the effects of DHPs on the Shaker K+ channels expressed in Xenopus oocytes. Intracellular applications of DHPs quickly and reversibly induced apparent inactivation in the Shaker K+ mutant channels with disrupted N- and C-type inactivation. We found that DHPs interact with the open state of the channel as evidenced by the decreased mean open time. The DHPs effects are voltage-dependent, becoming more effective with hyperpolarization. A model which involves binding of two DHP molecules to the channel is consistent with the results obtained in our experiments.
Keywords: potassium channels, dihydropyridines
introduction
Dihydropyridines (DHPs)1 with a 1,4 dihydropyridine ring and a phenyl ring are clinically used to treat a variety of cardiovascular disorders, primarily acting as vasodilators. Effects of the DHPs on L-type Ca2+ channels are especially interesting in that the DHPs can be either agonists or antagonists (Reuter et al., 1988; Hess, 1990). These drugs appear to act allosterically to modify the channel function by promoting different modes of gating (Hess, 1990). They have greater affinities for the inactivated state of the channel, which is favored by depolarization (Hess, 1990).
Although DHPs have profound effects on L-type Ca2+ channels, it was shown that these compounds also affected voltage-dependent Na+ channels (Yatani et al., 1988) and voltage-dependent K+ channels (Jacobs and DeCoursey, 1990; Gotoh et al., 1991). Using the whole cell voltage-clamp method, Jacobs and DeCoursey (1990) tested a series of clinically relevant agents, including nifedipine, on the K+ currents in alveolar epithelial cells and found that nifedipine produced apparent inactivation. They suggested that nifedipine, at least in part, works by acting as an open channel blocker. Gotoh et al. (1991) studied the effect of nicardipine on the K+ channel current in rabbit heart cells. They found that nicardipine accelerated the time course of the outward K+ current decline with the 50% inhibitory concentration of 630 nM. This is fourfold higher than that for the Ca2+ current in the same cells (160 nM).
The structural elements involved in the DHP binding to the Ca2+ channels are not thoroughly understood. Biochemical experiments (Nakayama et al., 1991; Regulla et al., 1991) using antibody mapping indicated that DHPs may bind to the amino acid residues in the S6 segment of the α1 subunit of the Ca2+ channel. Some investigators (Glossmann et al., 1984; Triggle et al., 1989) have suggested that Ca2+ channels may have two DHP binding sites, a high-affinity site (nM range) and a low-affinity site (μM range). A mutagenesis study using chimeric channels based on two distinct Ca2+ channels with different DHP affinities shows that the S5, Pore, and S6 segments may be important in determining the DHP sensitivity (Grabner et al., 1996). A molecular simulation study showing how a DHP may bind to the Ca2+ channel pore with Ca2+ ions bound suggests that DHPs occlude the channel pore (Zhorov and Ananthanarayanan, 1996).
We investigated the effects of DHPs on the Shaker K+ channels expressed in Xenopus oocytes. Native cells may express more than one class of K+ channels and the interpretations of the results of the drug action are usually not straightforward. Heterologous expression of a single class of channel can largely overcome this problem. Since the biophysical gating properties of the Shaker channel have been studied extensively (Bezanilla et al., 1994; Hoshi et al., 1994; Stefani et al., 1994; Zagotta et al., 1994 a,b), any alterations in the channel gating can be more easily detected and studied. Furthermore, molecular manipulations of the Shaker channel affecting some of the gating transitions, such as inactivation rates, have been described (Hoshi et al., 1990, 1991; Zagotta et al., 1990; Lopez-Barneo et al., 1993), and they can be used to probe the interactions of the channel with DHPs. The results obtained in this study indicate that DHPs interact with the open state of the Shaker channel inducing apparent inactivation although the drugs do not act as simple open-channel blockers. The results also show that the overall DHP efficacy can be modulated by a single amino acid mutation in the S6 segment.
materials and methods
Channel Expression
Wild-type and mutant Shaker K+ channels were expressed in Xenopus oocytes essentially as described previously (Hoshi et al., 1990). The following channels were used in this study. ShD (Timpe et al., 1988) is characterized by intact N-type inactivation and C-type inactivation. ShBΔ6-46 contains a 41-residue deletion in the amino terminus and lacks N-type inactivation (Hoshi et al., 1990). In addition to the amino terminus deletion, ShBΔ6-46 T449V contains a single amino acid substitution at position 449 (using the ShB numbering) from T to V. The ShBΔ6-46 T449V channel has disrupted N-type inactivation and drastically slowed or disrupted C-type inactivation (Lopez-Barneo et al., 1993). ShBΔ6-46 T449K has a point mutation at position 449 from T to K to accelerate C-type inactivation to a more experimentally manageable range (Lopez-Barneo et al., 1993). ShBΔ6-46 T449V: A463I has an additional point mutation at position 463 that has been shown to be involved in C-type inactivation (Hoshi et al., 1991). The RNAs were transcribed using T7 RNA polymerase (Ambion, Austin, TX) and injected into the oocytes (40 nl/cell). Recordings were typically made 1 to 14 d after injection.
Electrophysiological Recording
In most experiments, the single channel and macroscopic patch currents were recorded using excised configurations of the patch-clamp technique (Hamill et al., 1981; Methfessel et al., 1986) with an AxoPatch 200A amplifier (Axon instruments, Foster City, CA). The macroscopic patch currents were low-pass filtered through an eight-pole Bessel filter unit with 2 kHz corner frequency (Frequency Devices, Haverhill, MA) and digitized at 10 kHz using an ITC16 computer interface (Instrutech, Great Neck, NY). The whole oocyte currents were recorded using a Warner OC-725B (Warner, Hamden, CT) two-electrode voltage clamp amplifier. The electrodes filled with 3 M KCl had a typical resistance of less than 0.5 MΩ. The data were collected and analyzed using Pulse/PulseFit (HEKA, Lambrecht, Germany), IGOR PRO (Wavemetrics, Lake Oswego, OR), TAC (SKALAR, Seattle, WA), and DataDesk (DataDescriptions, Ithaca, NY) programs running on Apple Macintosh computers. Unless otherwise indicated, the linear capacitative and leak currents have been subtracted from the macroscopic currents presented using a modified P/n protocol. The single channel data were idealized using the half amplitude threshold method. For the analysis of the single channel dwell times, additional exponential components were included only if the probability of requiring one additional component was >0.95 using the likelihood ratio test (Hoshi et al., 1994). Numerical simulations of the ionic currents predicted by the kinetic schemes were performed using the Q-matrix approach (Colquhoun and Hawkes, 1995) implemented in IGOR. The parameters of the schemes were optimized by minimizing the chi-squared value using the downhill simplex algorithm (Press et al., 1994) implemented in IGOR. When appropriate, the data values are presented as mean ± standard deviation. The error bars are not shown when smaller than the symbol size. All experiments were performed at room temperature (20–24°C).
Solutions
The intracellular solution typically contained (in mM): 140 KCl, 11 EGTA, 10 HEPES, pH 7.2 (titrated with n-methyl-d-glucamine [NMG]). The extracellular solution typically contained (in mM): 140 NaCl, 2 MgCl2, 2 KCl, 10 HEPES (NMG), pH 7.2. The high K+ extracellular solution contained (in mM): 140 KCl, 2 MgCl2, 10 HEPES (NMG), pH 7.2. Other solutions used are indicated in the legends.
Nifedipine (Sigma Chemical Co., St. Louis, MO), Bay K 8644 (RBI, Natick, MA), and nimodipine (Sigma) were dissolved in 100% ethanol (10 mM). Nicardipine (Sigma) was dissolved in 100% methanol (5 mM). The stock solutions were kept in the dark at <4°C. The drug solutions were prepared fresh from these stock solutions and vortexed immediately before each use.
results
The ShBΔ6-46 T449V channel contains a deletion in the amino terminus to disrupt N-type inactivation (Hoshi et al., 1990). In addition, threonine at position 449 (in ShB numbering) in the P-segment is mutated to valine to disrupt or drastically slow C-type inactivation (Lopez-Barneo et al., 1993). Effects of DHPs were primarily assayed using this ShBΔ6-46 T449V channel since the gating transitions are simplified with the N- and C-type inactivation mechanisms disrupted. Because internal Mg2+ ions are known to induce voltage-dependent block of currents through Shaker -like potassium channels (Ludewig et al., 1993), all data presented were recorded in the absence of internal Mg2+ ions to exclude interference with divalent block in interpreting the DHP effects.
Nifedipine Induces Apparent Inactivation
Application of nifedipine to the intracellular side induced a time-dependent decline in the ionic currents through the ShBΔ6-46 T449V channels. Fig. 1 A shows the effects of intracellular nifedipine (50 μM) on the ShBΔ6-46 T449V K+ currents elicited in response to depolarizing voltage pulses. The amplitudes of the control ShBΔ6-46 T449V currents did not decline appreciably with maintained depolarization. Most of the reduction in the control current amplitude observed with maintained depolarization is attributable to accumulation of K+ ions in the extracellular space immediately adjacent to the channels as judged by the changes in the reversal potential of the tail currents with time (data not shown). In the presence of nifedipine, the currents underwent marked apparent inactivation at the positive voltages where the activation process is fast, suggesting that nifedipine exerts its action after the channel opens. The current-voltage (I-V) curves indicate that the reduction of the current amplitude by nifedipine is apparently voltage dependent (Fig. 1 B).
Figure 1.

Nifedipine block of K+ currents through ShBΔ6-46 T449V channels. (A) Current traces obtained from the inside-out patch in response to voltage steps from −100 mV to −20 mV, 0 mV, and +50 mV with (bottom) and without (top) nifedipine (50 μM) in the internal solution are shown. (B) Current amplitudes measured at the end of 200-ms pulses with (closed circles) and without (open circles) nifedipine (50 μM) in the internal solution.
The effect of nifedipine application to the intracellular side to induce apparent inactivation had a very fast onset, much faster than the limit of the manual bath perfusion system employed. The effect was readily and fully reversible. Applications of nifedipine to the extracellular side were less effective than those to the intracellular side (Fig. 2 A). Extracellular applications of nifedipine (100 μM) often induced a time-independent decrease in the current amplitude (inset traces in Fig. 2 A). These observations indicate that nifedipine works preferentially from the intracellular side. Considering the nonpolar nature of nifedipine, the effects of extracellular application were probably mediated by its movement through the cell membrane.
Figure 2.

(A) Nifedipine applied externally is less effective in blocking the ionic currents through the ShBΔ6-46 T449V channel. Peak amplitudes of the currents measured using the two-electrode voltage clamp during voltage pulses from −100 mV to +50 mV are plotted (filled circles: data measured during nifedipine (100 μM) application, open circles: wash out of drug). Representative current sweeps recorded at the end of the corresponding segment are shown in inset. (B) Nifedipine, exposed to ultraviolet light, is less effective in blocking the ShBΔ6-46 T449V current. Representative inside-out macro-patch currents in response to pulses from −100 mV to +50 mV. A half of the nifedipine solution (50 μM) was treated with UV light (254 nm 18W for 90 min at 1 cm) and the efficacy of the UV-treated nifedipine was compared with the other half of the nifedipine solution, which did not receive UV treatment.
DHPs are light-sensitive and the effects on voltage- dependent Ca2+ channels can be markedly diminished by UV-light (Meyer et al., 1984). We examined whether the nifedipine effects on the K+ channels could be diminished by UV-light. We found that UV-treated nifedipine (254 nm, 18 W, for 90 min at 1 cm) was much less effective in producing the apparent inactivation of the ShBΔ6-46 T449V currents (Fig. 2 B). The vehicles (ethanol, methanol) did not induce the inactivation at the concentrations used.
Other Dihydropyridines Are Also Effective
In addition to nifedipine, other DHPs are also effective in inducing apparent inactivation in the Shaker channels. Fig. 3 compares the intracellular applications of nimodipine, nicardipine, and BAY K 8644 (+,−) on the ShBΔ6-46 T449V currents elicited in response to the voltage pulses to +50 mV. As with nifedipine, these DHPs induced apparent inactivation in a reversible manner when applied to the intracellular side. Extracellular applications of these drugs were also much less effective (data not shown).
Figure 3.

Block of the ShBΔ6-46 T449V macroscopic currents by different DHPs. Currents were recorded in the inside-out configuration in response to depolarizing voltage steps to 0 mV. Indicated DHPs were applied to the internal side. The concentration was 100 μM for all DHPs.
Concentration Dependence of DHP Block
The currents recorded at +50 mV in the presence of various concentrations of nifedipine are shown in Fig. 4 A. Fig. 4 C–F show the concentration dependence of the block of the macroscopic currents at +50 mV by nifedipine and nimodipine. The effect of nifedipine at +50 mV became noticeable at 3 μM with the half-maximal reduction in the steady-state current occurring at ∼30 μM (Fig. 4 C). For nimodipine, the half-maximal reduction was observed at a slightly lower concentration (Fig. 4 E). With greater concentrations, the apparent inactivation time course became faster and the steady-state current became progressively smaller. These results are again consistent with the drugs exerting the action after the channel opens. The fractions of the currents blocked at the end of the sweep are shown in Fig. 4 C (nifedipine) and E (nimodipine). The time constants of the current decline are shown in Fig. 4 D (nifedipine) and F (nimodipine). These plots illustrate a slight difference in the efficacies of nifedipine and nimodipine. The Hill plot transformations of the block data (Fig. 4 B) indicate that the concentration dependence of both nifedipine and nimodipine block has the Hill coefficient of 1.5, consistent with more than one DHP molecule binding to the channel.
Figure 4.

Concentration dependence of DHP block of the macroscopic currents through the ShBΔ6-46 T449V channels. (A) Current traces obtained in response to voltage steps to +50 mV with the indicated concentrations of nifedipine in the internal solution. (B) Hill plot of the concentration dependence of block of ShBΔ6-46 T449V channel by nimodipine (open circles) and nifedipine (closed circles). Block is given by Idrug/Icontrol. Dashed lines are the least square fits of the data with Hill equation. Both nifedipine and nimodipine fits give a Hill coefficient value of 1.5. (C) Relative reduction of the steady state current measured at the end of a 200-ms voltage pulse to +50 mV by different concentrations of nifedipine. Values on the vertical axis were calculated as 1-Inifedipine/ Icontrol. Data points represent mean ± standard deviation of six experiments. (D) Time constants of the current decline induced by nifedipine block. The currents elicited by 200-ms voltage pulses to +50 mV in the presence of nifedipine at the concentrations indicated were fitted with a sum of two exponentials. (E) Relative reduction of the steady-state current by nimodipine. The data were collected and analyzed as in C. (F) Time constants of the current decline in the presence of nimodipine (averages of four to seven experiments). The data were collected and analyzed as in D. In B–F, smooth curves show least square fits of the data obtained from the simulated currents as described in discussion (thin line, scheme SII, thick line, scheme SV).
Nifedipine Decreases the Mean Open Time
If nifedipine interacts with the open state of the channel, the mean open time should decrease in a concentration-dependent manner. To examine the effect of nifedipine on the single channel level, a mutant Shaker channel, ShBΔ6-46 T449V:A463I, which contains a single amino acid substitution at position 463 (Hoshi et al., 1991) in the ShBΔ6-46 T449V background was used. The A463I mutation increases the single channel amplitude by 50%, and it increases the mean open time by 20–30-fold. Thus, this channel is well suited to assay the DHP effects. Representative openings of a single ShBΔ6-46 T449V:A463I channel recorded at 0 mV are shown in Fig. 5 A. Application of nifedipine decreased the mean open time in a concentration-dependent manner (Fig. 5, B and C). The reduction of mean open time is consistent with nifedipine interacting with the open state of the channel. The amplitude of the main conductance state of the channel was not markedly affected by nifedipine application. Although we observed the substates more frequently in the presence of nifedipine, we did not systematically investigate this issue.
Figure 5.

Nifedipine decreases the mean open time in ShBΔ6-46 T449V:A463I. (A) Representative openings in the absence (upper traces) and in presence of 100 μM nifedipine (lower traces) in the internal solution. Inside-out configuration at 0 mV. Closed state is indicated by dashed line. The data were filtered at 1.8 kHz. The patch contained one functional channel. (B) Open time histograms with and without 100 μM nifedipine. Data are shown in square root scaling on vertical axis (Sigworth and Sine, 1987). Single exponential fits are shown superimposed. (C) Reciprocals of the mean open times at several concentrations of nifedipine. Each data point represents the mean ± standard deviation of four to six experiments. Solid line shows linear least square fit of first three points.
The closed durations recorded from the data shown in Fig. 5 are compared in Fig. 6. In the absence of the drug, the closed durations were described by a sum of at least three exponentials. The shortest exponential component with a time constant value of 200–300 μs reflects the short burst closures as observed for the ShBΔ6-46 channel, Cf (Hoshi et al., 1994). The second component with a time constant value of 2–4 ms reflects the intermediate closed state, Ci (Hoshi et al., 1994). In addition to these closed duration components, similar to those already documented for the ShBΔ6-46 (Hoshi et al., 1994), the closed durations from the ShBΔ6-46 T449V:A463I channel also showed an additional component with a time constant value of 50–200 ms with a small fractional amplitude (3% in the data shown). Nifedipine (100 μM) did not introduce an additional closed duration component in a statistically significant way but noticeably increased the relative amplitude of the third component (from 3% to 11% in Fig. 6). It is possible that nifedipine did induce an extra component, but the new time constant was too similar to one of the existing time constants to be detected. Similar results were obtained from two other single-channel patches.
Figure 6.
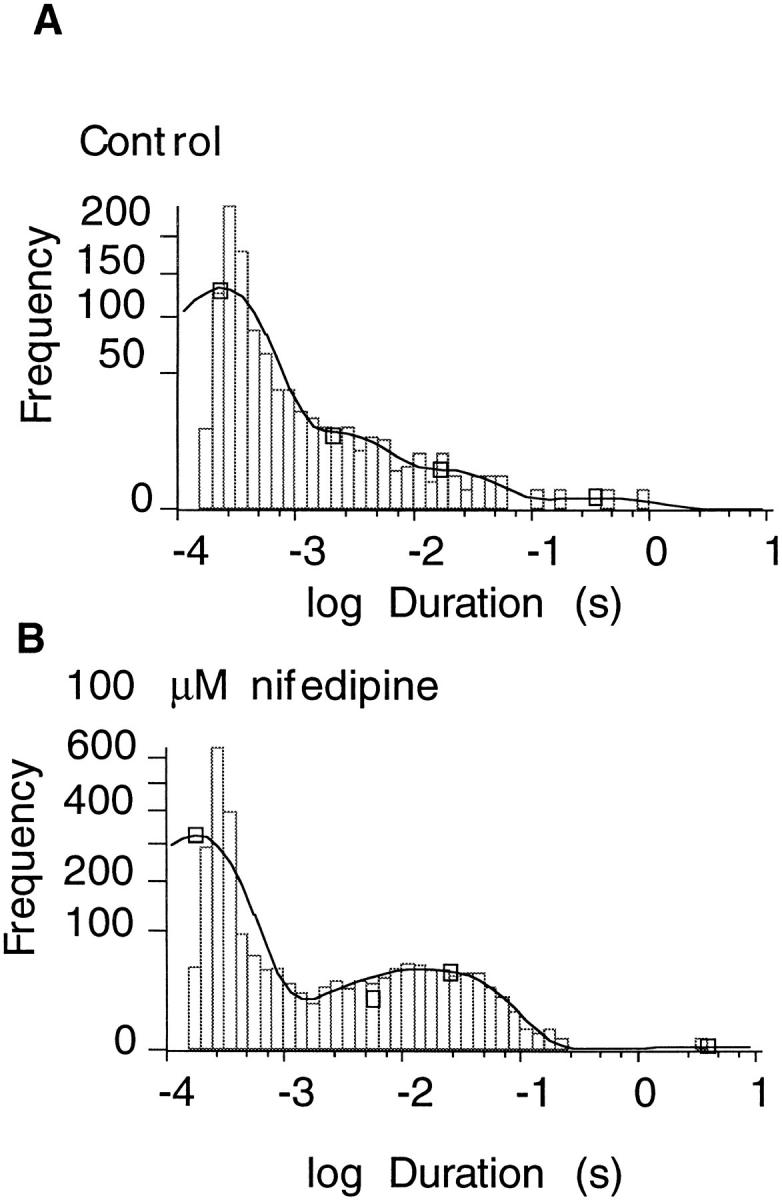
Nifedipine effects on the closed time distribution of ShBΔ6-46 T449V:A463I. Closed time distributions in the absence (A) and in the presence (B) of 100 μM nifedipine. Data are shown in square root scaling on vertical axis. Lines shown represent four exponential fits of the distributions. Under control conditions the time constants (fractional amplitudes) were 0.2 ms (0.871), 2.1 ms (0.097), 16.8 ms (0.029), and 352 ms (0.003). For nifedipine, the three time constants (fractional amplitudes) were 0.2 ms (0.84), 5.7 ms (0.048), and 26 ms (0.109).
Interaction between the Nifedipine Block and N- and C-type Inactivation
Since nifedipine induces apparent inactivation, we studied whether nifedipine interacts with N-type or C-type inactivation. We first examined the possible interaction between nifedipine and the N-type inactivation mechanism by using the ShD channel (Timpe et al., 1988), which exhibits fast N-type inactivation. As described earlier (Choi et al., 1991), in the presence of internal tetraethylammonium (TEA) (2 mM), the current decline is slower since the TEA-bound ShD channels cannot undergo N-type inactivation, and a cross-over of the scaled current time courses was observed (Fig. 7 A). In the absence of TEA, the time course of the current decline was well fitted by a single exponential with a time constant of 9 ms whereas with TEA, a sum of two exponentials with time constant values of 2 and 26 ms was required to fit the current decline. In contrast, decline of the ShD current in the presence of nifedipine was not slower than the control current and the scaled currents superimposed well (Fig. 7 B). Because the apparent on and off rates of nifedipine are slower than those of TEA (see Fig. 1), the results do not necessarily show that N-type inactivation and nifedipine do not compete.
Figure 7.

Nifedipine block in presence of either N- or C-type inactivation. (A) Internal TEA and N-type inactivation compete. The ShD currents obtained with and without internal TEA (2 mM) are shown at the top and the scaled currents are shown below. The scaled currents show a cross-over. (B) Nifedipine block of the macroscopic currents through ShD channels. (C) Macroscopic currents through the ShBΔ6-46 T449K channels with C-type inactivation (control and 100 μM nifedipine). Scaled currents (lower traces) show that the current in the presence of nifedipine inactivates faster. (D) Time constants of ShBΔ6-46 T449K current decline in control and in the presence of 100 μM nifedipine in internal solution. The current traces were fitted with a sum of two exponentials. Each point represents mean ± standard deviation of 12 experiments (Control) or 8 experiments (Nifedipine). (E) Nifedipine block (50 μM) of the ShBΔ6-46 T449V currents without N- or C-type inactivation recorded with different external K+ concentrations. The currents were elicited by depolarizing pulses to +50 mV.
We further examined whether nifedipine and the C-type inactivation mechanism compete by using the ShBΔ6-46 T449K channel. This channel mutant has a 41–amino acid deletion in the amino terminus to disrupt N-type inactivation (Hoshi et al., 1990). In addition, it also contains a single amino acid mutation at position 449 from threonine to lysine to accelerate C-type inactivation (Lopez-Barneo et al., 1993). If C-type inactivation and nifedipine compete, it is expected that the time course of the current decline in the presence of nifedipine is slower because the nifedipine-bound channels are prevented from undergoing C-type inactivation. Representative currents through the ShBΔ6-46 T449K channels in control and in presence of 100 μM nifedipine are shown in Fig. 7 C and rate constants obtained from two exponential current fits are shown in Fig. 7 D. In the presence of nifedipine (100 μM), the current decline was faster than that in the absence of the drug. The results suggest that nifedipine and C-type inactivation do not compete.
If nifedipine induces the apparent inactivation in the ShBΔ6-46 T449V channel by enhancing the residual C-type inactivation mechanism, the nifedipine-induced inactivation should display properties similar to those of C-type inactivation. For example, the nifedipine-induced inactivation should slow down in the presence of high external K+ as C-type inactivation is slowed by high external K+ (Lopez-Barneo et al., 1993). We tested this hypothesis by comparing the efficacies of the nifedipine block in the presence of different external K+ concentrations. As compared in Fig. 7 E, the time course of the nifedipine block with 140 mM external K+ was not slower than that observed with 2 or 50 mM external K+. Thus, the nifedipine-induced inactivation and C-type inactivation differ in their sensitivities to external K+, suggesting that they are mediated by distinct mechanisms. This does not, however, rule out the possibility that nifedipine and C-type inactivation interact as suggested by the results obtained with the ShBΔ6-46 T449K channel (Fig. 7, C and D).
Voltage Dependence of the DHP Block
Because nifedipine is not a charged molecule, it was not expected that its action would be voltage dependent even if it might position itself in the membrane electric field. We examined the possible voltage dependence of the DHP action as shown in Fig. 8. The channels were opened first in response to a voltage pulse to +50 mV to allow the nifedipine molecules to block the channels. Then, the membrane voltage was changed to a variety of voltages where the steady-state probability of the channel being open is still saturated (Zagotta et al., 1994 a). If the DHP action is voltage independent, the only instantaneous and time-independent changes in the current, reflecting the changes in the driving force, should be observed, as recorded under control conditions (Fig. 8 A). The presence of the time-dependent relaxation of the current would suggest that the drug action is voltage dependent. The results in Fig. 8, B–D do show such time-dependent relaxation. The time courses of the current relaxations were fitted with single exponentials and the ratios of the extrapolated instantaneous current amplitudes to the steady state currents are shown in Fig. 8 E. DHPs became less effective in blocking the current with greater depolarization. As a data description parameter for the voltage dependence, we chose the number of apparent equivalent charges (n). The value n was obtained by fitting the relative block data (Fig. 8 E) with the following function
 |
Figure 8.

Voltage dependence of DHP block. The currents were first elicited by voltage pulses to +50 mV followed by voltage steps to −20 to +90 mV in 10 mV increments. (A) The ShBΔ6-46 T449V channels do not show significant relaxation after the voltage change. (B) Slow relaxations of the macroscopic currents through the ShBΔ6-46 T449V channels in presence of 25 μM nimodipine. The currents relaxed to lower values at voltages more negative than +50 mV showing more effective block of nimodipine at these voltages. (C) Same as B in presence of 100 μM nifedipine. (D) Voltage-dependent relaxation of the ShBΔ6-46 T449V:A463I current with 100 μM nifedipine. (E) Voltage dependence of the block for ShBΔ6-46 T449V in the presence of 25 μM (diamonds) and 50 μM (circles) of nifedipine. Relative block was calculated as a ratio of the current value immediately after the voltage change over the current value after relaxation based on the single exponential fit of the current decline. The data sets were fitted with function exp [n e (V − 50) / k T] to obtain number of equivalent charges n. (F ) Concentration dependence of the equivalent charges associated with the voltage dependence of the block obtained as described in E for nifedipine block of the ShBΔ6-46 T449V current (circles), ShBΔ6-46 T449V:A463I current (squares), and nimodipine block of ShBΔ6-46 T449V current (triangles). Solid lines represent the scheme SV predictions of nifedipine (upper line) and nimodipine (lower line) block equivalent charges. The model predictions were calculated using the blocking rate constants obtained from the concentration dependence data shown in Fig. 4. Data points represent mean ± standard deviation of five to eight experiments.
where V is membrane voltage, other constants have their usual meanings. Examples of this fit for 25 and 50 μM of nifedipine block of ShBΔ6-46 T449V current are shown in Fig. 8 E as the solid lines. The apparent charge number is plotted against the DHP concentration in Fig. 8 F. The voltage dependence increased with the drug concentration. The data also show a difference in the voltage dependence of nifedipine and nimodipine blocks of the ShBΔ6-46 T449V currents as well as a difference in the voltage dependence of nifedipine block of the ShBΔ6-46 T449V current (Fig. 8 F, circles) and the ShBΔ6-46 T449V:A463I current (squares).
Nifedipine Interaction with Ion Flow through Channel
The results presented so far suggest that nifedipine interacts with the open state of the channel to impede the ion flow. One possible mechanism is that nifedipine physically occludes the ion conducting pore, acting as an open channel blocker. Several molecules have been shown to act as open channel blockers for voltage-dependent K+ channels, including TEA, and the Shaker inactivation peptide (Demo and Yellen, 1991). If nifedipine acts as an open channel blocker, K+ flux through the channel should regulate the block efficacy.
The effects of the direction of the net K+ flow on the nifedipine efficacy are shown in Fig. 9. With 140 mM K+ outside, the effects of nifedipine on the net inward and outward currents were measured at +30 mV with 0 mM and 140 mM K+ inside, respectively. The reduction in the steady state current caused by nifedipine was not markedly altered by the changes in the net current flow, suggesting that DHP efficacy is independent of the direction of the net current flow.
Figure 9.

Nifedipine block does not depend on the current flow direction. (A) Macroscopic currents through the ShBΔ6-46 T449V channels (control and 100 μM nifedipine) at +30 mV. Outward currents were obtained with 140 mM K+ out / 140 mM K+ in, and the inward currents with 140 mM K+ out/ no internal K+ (substituted with NMG). (B) Comparison of nifedipine block (100 μM) at various voltages with high K+ (140 mM) inside (circles) and no K+ ions inside (squares). External K+ concentration was 140 mM in both cases. Data points show the fraction of unblocked current at the end of 200-ms voltage pulse to the voltages indicated (mean ± standard deviation of five experiments).
Regulation of Nifedipine Efficacy by Mutation A463I in the S6 Segment
Residue 463 in the S6 segment of the ShB channel has been shown to be involved in regulation of C-type inactivation (Hoshi et al., 1991; Lopez-Barneo et al., 1993). In ShBΔ6-46 T449V, where both the N- and C-type inactivation mechanisms are disrupted, mutating the residue 463 from A to I (ShBΔ6-46 T449V:A463I) causes the mean open time and the single channel amplitude to increase (see Fig. 5). Consistent with the slow channel closing rate, the macroscopic G(V) curve of this channel is shifted to a more negative direction by ∼40 mV compared with that of ShBΔ6-46 or ShBΔ6-46 T449V (data not shown). We found that the effects of nifedipine on ShBΔ6-46 T449V:A463I are distinct from those on ShBΔ6-46 T449V. The effects of nifedipine on the ShBΔ6-46 T449V:A463I channel differ in three major areas: steady-state fraction of the current blocked, recovery from the nifedipine block, and voltage dependence of the block. Nifedipine (100 μM) was less effective in blocking the ShBΔ6-46 T449V:A463I channel than the ShBΔ6-46 T449V channel. Fig. 10 B compares the concentration dependence of nifedipine block of the ShBΔ6-46 T449V (circles) and ShBΔ6-46 T449V: A463I (squares) currents, also showing that nifedipine is less effective on the ShBΔ6-46 T449V:A463I channel. A difference in the voltage dependence of nifedipine block of these two channel types is shown in Fig. 8 F using the same symbols.
Figure 10.

Differential effects of nifedipine on the ShBΔ6-46 T449V and ShBΔ6-46 T449V:A463I channels. (A) Effects of nifedipine (100 μM) on the macroscopic currents recorded at +50 mV from ShBΔ6-46 T449V (top) and ShBΔ6-46 T449V:A463I (bottom). (B) Comparison of the concentration dependence of nifedipine block of the ShBΔ6-46 T449V (circles) and ShBΔ6-46 T449V: A463I (squares) currents. Solid line represents the model prediction of scheme SV for ShBΔ6-46 T449V:A463I. Values of the block constants of nifedipine for ShBΔ6-46 T449V were used in simulation. ShBΔ6-46 T449V:A463I was simulated by stabilizing the open state in scheme SI by 1.3 kcal/mol (see discussion). Each data point represents mean ± standard deviation of six to nine experiments. (C) Scaled tail currents obtained at −100 mV for ShBΔ6-46 T449V and at −120 mV for ShBΔ6-46 T449V:A463I after pulses to +50 mV with 140 mM K+ out. Nifedipine (100 μM) slowed the tail current in ShBΔ6-46 T449V and accelerated in ShBΔ6-46 T449V: A463I.
Nifedipine differentially altered the tail currents through the ShBΔ6-46 T449V and ShBΔ6-46 T449V: A463I channels. The tail current was slowed by nifedipine in the ShBΔ6-46 T449V channels (Fig. 10 C). In contrast, nifedipine accelerated the time course of deactivation in the ShBΔ6-46 T449V:A463I channel at all the voltages examined (−100 to −140 mV).
discussion
The results presented show that DHPs, such as nifedipine and nimodipine, reduce the ionic currents through the Shaker K+ channels. Although higher concentrations of DHPs are required for inhibition of the Shaker channels than for some voltage-dependent Ca2+ channels (Hess, 1990), the DHP effects on the Shaker channels are still specific and require the intact dihydropyridine structure since the UV-treated DHPs largely lose their ability to reduce the current. The DHP efficacy is dependent on voltage, but independent of the direction of the K+ flow.
Our consideration of the possible biophysical and molecular mechanisms underlying the DHP action on the Shaker channels is based on the description of their gating process in Zagotta et al. (1994 b). This scheme can be presented in a simplified form as the following:
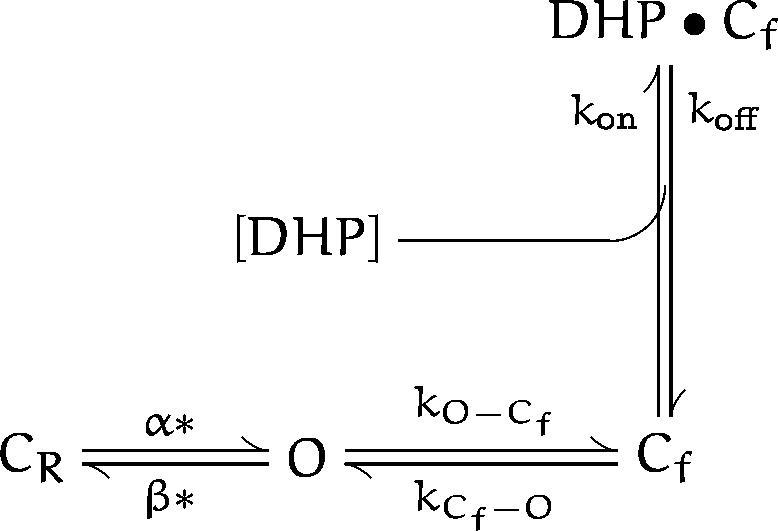
where C R represents the numerous closed states that the channel visits before opening, and C f represents the closed state(s) that the channel enters after opening. At positive voltages, the value of the reverse rate constant β* is small, and the fast transitions between Cf and O can be assumed to be at equilibrium. This scheme SIs well suited for the description of the gating behavior of ShBΔ6-46 T449V, which shows neither C- or N-type inactivation in Xenopus oocytes (Hoshi et al., 1990; Lopez-Barneo et al., 1993). Although in this scheme, activation of the channel is represented with a single transition for simplicity, in numerical simulations we used the complete model as described in Zagotta et al. (1994 b). Values of all the rate constants were taken from Zagotta et al. (1994 b). All the blocking schemes considered below are based on scheme SI as they are reduced to this scheme SIn the absence of drug.
Scheme I.

The simplest hypothesis that the DHP molecule binds to the open conducting state of the channel (O) only can be represented by the following scheme:
where DHP·O represents the drug-bound non-conducting open state. According to this scheme, the mean open time should decrease in a concentration-dependent manner in the presence of nifedipine. The mean open time was indeed decreased by nifedipine as shown in Fig. 5. Furthermore, according to scheme SII, the reciprocal of the mean open time should be linearly related to the DHP concentration by the following relationship:
 |
Scheme II.

assuming that β* = 0 at positive voltages. This linear prediction did not hold for all the concentrations of nifedipine examined (Fig. 5 C). At the high concentrations tested (>40 μM), the block was more than predicted, raising some implications about the nature of the block (see below). Furthermore, this model does not provide any mechanism for the voltage dependence of the block. To improve this model, we could assume that k on and/or k off in scheme SII are voltage dependent. Alternatively, we can assume that the voltage dependence of the block comes from a voltage-dependent gating property of the channel rather than voltage-dependent binding of the drug. The rate constant for the Cf to O transition is voltage dependent (Zagotta et al., 1994 b), decreasing with hyperpolarization with an equivalent charge of 0.17, similar to the voltage dependence of the DHP action (Fig. 8 F). Thus, the following scheme where the DHP molecule binds to the Cf state from which the exit rate constant γ is voltage dependent could be considered:
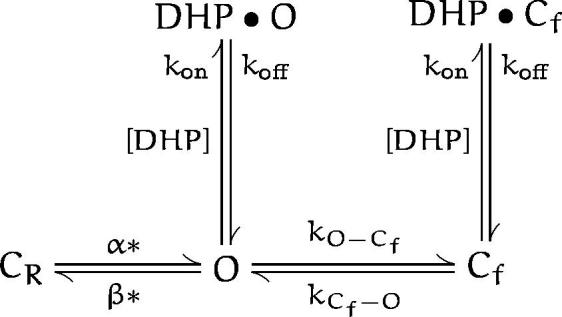
An obvious drawback of this scheme SIs that it does not predict the concentration-dependent mean open time. This can be resolved by combining schemes ii and iii into a single scheme:
This model has four more free parameters than scheme SI, and it can account for the effect of DHP on the mean open time and the voltage dependence of the block. This model, however, does not describe the concentration dependence curve well (Fig. 4) and it predicts a Hill coefficient of 1 as opposed to the observed higher value (Fig. 4 B), which requires binding of more than one DHP molecules to the channel. Thus, we introduce the following model:
(scheme v)
Time constants of the current decline, voltage dependence of the block and the concentration dependence predicted by schemes ii and v were simulated and shown superimposed on the experimental data (Figs. 4, 8, and 10). For these computations, all parameters in the absence of drug were fixed at the values given in Zagotta et al. (1994 b). Only the block/unblock rates included in each scheme were adjusted by minimizing the chi square value to simultaneously fit the steady-state fractional block (Fig. 4, C and E) and the time constant of the current decline in the presence of nifedipine and nimodipine (Fig. 4, D and F). Table I shows the parameter values used to simulate the data. The predictions of scheme SV with two DHP binding steps are markedly better than those of scheme SII with one binding step (Fig. 4). With the rate constant values obtained from fitting the concentration dependence, scheme SV also reproduces the voltage dependence of the block even though the parameters were not specifically optimized to fit the voltage dependence (Fig. 8 F).
Table I.
Numerical Values of the Rate Constants Used to Simulate DHP Block
| k on (s−1 μM−1) | 2.42 | 3.8 | — | — | ||||
| k off (s−1) | 31.9 | 24.1 | — | — | ||||
| k on A (s−1 μM−1) | — | — | 0.56 | 0.72 | ||||
| k off A (s−1) | — | — | 31.9 | 48.3 | ||||
| k on B (s−1 μM−1) | — | — | 49.4 | 72.2 | ||||
| k off B (s−1) | — | — | 229 | 109 |
nimodipine
Scheme V.

The prediction of scheme SV about the concentration dependence of the mean open time, which is determined by constant k on A, is consistent with initial slope of the curve shown in Fig. 5 C. The DHP binding to the fast closed state Cf in scheme SV could also account for the non-linear concentration dependence of the mean open time (Fig. 5 C). Because the mean dwell time in Cf is short, many of the transitions to this state are unresolved. The DHP binding to the Cf state will increase its mean dwell time, thus reducing the number of unresolved events, which in turn could result in an apparent non-linear dependence of the mean open time on DHP concentration at high concentrations.
The model in scheme SV can be used to explain the altered drug sensitivity of the ShBΔ6-46 T449V:A463I channel. In the ShBΔ6-46 T449V:A463I channel, the occupancy probability in the Cf state is lower than that in the ShBΔ6-46 T449V channel as this channel has a greater mean open time. Thus, according to scheme SV, where the second DHP molecule binds only to the Cf state, the ShBΔ6-46T449V:A463I channel should be less sensitive to DHP than the ShBΔ6-46 T449V channel, as opposed to scheme ii, in which the increased occupancy of the open state should favor increased blocking. This prediction is consistent with the data obtained (see Fig. 10), where ShBΔ6-46T449V:A463I is less sensitive to DHP. The shift in G(V) curve of the ShBΔ6-46T449V:A463I channel could be well represented by decreasing the rate constants of the transitions away from the open state (δ and O-Cf as given in the model of Zagotta et al., 1994 b) by a factor of 10, thus stabilizing the open state by ∼1.3 kcal/mol. The concentration dependence given by scheme SV with the block rates obtained from the ShBΔ6-46T449V data with the open state stabilized by 1.3 kcal/mol is shown in Fig. 10 B. With those values, scheme SV produces less block, which is consistent with the observed effect. We consider the results of the simulations based on scheme SVto be in qualitative agreement with the observed effects. scheme SV reasonably describes the concentration and voltage dependence of DHP block as well as the effect of A463I mutation. There are ways to further increase the agreement between the model prediction and the experimental data. For example, it is possible to improve the general fit by assuming that allosteric interactions increase the rate of DHP binding to the Cf (DHP•B) state compared with that to the O state. Since the available experimental data did not specifically discriminate the various possible allosteric models, we did not consider them further.
The observation that DHP is not likely to compete with C-type inactivation (Fig. 6) suggests that C-type inactivation itself can occur from the fast closed state, which is stabilized by the bound DHP molecule. Because C-type inactivation is slowed by K+ ions (Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1996), it is reasonable to speculate that C-type inactivation occurs from the fast closed state or other nonconducting states (Baukrowitz and Yellen, 1996). Baukrowitz and Yellen (1996) showed that internal TEA derivatives enhance C-type inactivation by emptying K+ ions from the pore. Similarly, DHP may have two effects on the Shaker channel. First, binding of the DHP itself to the channel decreases the K+ flux. The reduced K+ ion occupancy in the channel pore in turn increases the residual C-type inactivation rate.
Although a mutation in the S6 segment affected the DHP efficacy (Fig. 10), it is likely that other amino acid residues are also involved in regulating the DHP efficacy. For example, considering that DHP preferentially works from the intracellular side, the residues in the internal mouth of the channel may also be involved. Those amino acid residues in the S5, P, and S6 segments shown to be important for the 4-aminopyridine block (Kirsch et al., 1993) are also good candidates for involvement in the DHP sensitivity. However, since the DHP efficacy does not appear to be markedly influenced by the net K+ flux (Fig. 9), the DHP molecules probably do not lie directly in the K+ flux pathway.
A large number of dihydropyridines have been synthesized and these drugs appear to have different potencies in blocking the Shaker potassium channel (Fig. 3; unpublished observation). At the same concentration, nimodipine appears be more effective than nifedipine (Fig. 4). However, minor quantitative changes in the rate constants of the model in scheme SV can account for the observed differential potency, suggesting that the underlying biophysical mechanisms are the same. Furthermore, by comparing the effects of different dihydropyridines with different side groups, it should be possible to gain information on which structural components of the drug are critical in determining the different rate constants of the model.
Several μM of nifedipine was required to observe noticeable effects on the ShB channel currents at +50 mV whereas the drug's effects on L-type Ca2+ channels may be observed in the nM range. Despite the higher concentration required to affect the Shaker potassium channels, the effects are specific in that the UV-treated nifedipine is ineffective. Because the DHP action on Shaker channels is enhanced by hyperpolarization, it is possible that the efficacy may be greater under physiological conditions where the transmembrane potential may not reach +50 mV. It is also possible that other homologues of Shaker channels show much higher DHP sensitivities than the channels examined in this study. Given the structural similarities found in different voltage-dependent ion channels, it will be interesting to rigorously compare the biophysical and molecular mechanisms of the DHP action on voltage-dependent Ca2+ and K+ channels.
Scheme III.
Scheme IV.
Acknowledgments
We thank J. Kabat and J. Thommandru for technical assistance. We also thank K. Larson and A. Kamath for performing some of the early experiments and Ray Dietrich for the instrument design.
Footnotes
T. Hoshi was supported in part by Klingenstein Foundation, McKnight Foundation and American Heart Association (96014400). E.F. Shibata is an Established Investigator of the American Heart Association.
Abbreviations used in this paper: DHP, dihydropyridine; NMG, n-methyl- d-glucamine; TEA, tetraethylammonium.
references
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-pore of a K+channel. Science (Wash DC) 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Perozo E, Stefani E. Gating of Shaker K+channels: II. The components of gating currents and a model of channel activation. Biophys J. 1994;66:1011–1021. doi: 10.1016/S0006-3495(94)80882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+channels. Proc Natl Acad Sci USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D., and A.G. Hawkes. 1995. A Q-matrix cookbook: how to write only one program to calculate the single-channel and macroscopic predictions for any kinetic mechanism. In Single-channel recording. Plenum, New York. 589–633.
- Demo SD, Yellen G. The inactivation gate of the Shaker K+channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Glossmann H, Ferry DR, Goll A, Rombusch M. Molecular pharmacology of the calcium channel: evidence for subtypes, multiple drug-receptor sites, channel subunits, and the development of a radioiodinated 1,4-dihydropyridine calcium channel label, 125Iiodipine. J Cardiovasc Pharmacol. 1984;6:S608–621. [PubMed] [Google Scholar]
- Gotoh Y, Imaizumi Y, Watanabe M, Shibata EF, Clark RB, Giles WR. Inhibition of transient outward K+ current by DHP Ca2+antagonists and agonists in rabbit cardiac myocytes. Am J Physiol. 1991;260:H1737–1742. doi: 10.1152/ajpheart.1991.260.5.H1737. [DOI] [PubMed] [Google Scholar]
- Grabner M, Wang ZY, Hering S, Striessnig J, Glossmann H. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 1996;16:207–218. doi: 10.1016/s0896-6273(00)80037-9. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflug Arch Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hess P. Calcium channels in vertebrate cells. Ann Rev Neurosci. 1990;13:337–356. doi: 10.1146/annurev.ne.13.030190.002005. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shakerpotassium channel inactivation. Science (Wash DC) 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Shakerpotassium channel gating. I: Transitions near the open state. J Gen Physiol. 1994;103:249–278. doi: 10.1085/jgp.103.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs ER, DeCoursey TE. Mechanisms of potassium channel block in rat alveolar epithelial cells. J Pharmacol Exp Ther. 1990;255:459–472. [PubMed] [Google Scholar]
- Kirsch GE, Shieh CC, Drewe JA, Vener DF, Brown AM. Segmental exchanges define 4-aminopyridine binding and the inner mouth of K+pores. Neuron. 1993;11:503–512. doi: 10.1016/0896-6273(93)90154-j. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shakerpotassium channels. Receptors & Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Ludewig U, Lorra C, Pongs O, Heinemann SH. A site accessible to extracellular TEA+ and K+ influences intracellular Mg2+block of cloned potassium channels. Eur Biophys J. 1993;22:237–247. doi: 10.1007/BF00180258. [DOI] [PubMed] [Google Scholar]
- Methfessel C, Witzemann V, Takahashi T, Mishina M, Numa S, Sakmann B. Patch clamp measurements on Xenopus laevisoocytes: currents through endogenous channels and implanted acetylcholine receptor and sodium channels. Pflügers Archiv. 1986;407:577–588. doi: 10.1007/BF00582635. [DOI] [PubMed] [Google Scholar]
- Meyer, H., F. Bossert, E. Wehinger, and D. Scherling. 1984. Chemistry of nitrendipine and its metabolites. In Nitrendipine. Urban & Schwarzenberg, Baltimore. 1–9.
- Nakayama H, Taki M, Striessnig J, Glossmann H, Catterall WA, Kanaoka Y. Identification of 1,4-dihydropyridine binding regions within the alpha 1 subunit of skeletal muscle Ca2+ channels by photoaffinity labeling with diazipine. Proc Natl Acad Sci USA. 1991;88:9203–9207. doi: 10.1073/pnas.88.20.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W.H., S.A. Teukolsky, W.T. Vetterling, and B.P. Flannery. 1994. Numerical recipes in C: the art of scientific computing. Cambridge University Press, New York.
- Regulla S, Schneider T, Nastainczyk W, Meyer HE, Hofmann F. Identification of the site of interaction of the dihydropyridine channel blockers nitrendipine and azidopine with the calcium-channel alpha 1 subunit. EMBO (Eur Mol Biol Organ) J. 1991;10:45–49. doi: 10.1002/j.1460-2075.1991.tb07919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter H, Porzig H, Kokubun S, Prod'hom B. Calcium channels in the heart. Properties and modulation by dihydropyridine enantiomers. Ann NY Acad Sci. 1988;522:16–24. doi: 10.1111/j.1749-6632.1988.tb33338.x. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani E, Toro L, Perozo E, Bezanilla F. Gating of Shaker K+channels: I. Ionic and gating currents. Biophys J. 1994;66:996–1010. doi: 10.1016/S0006-3495(94)80881-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpe LC, Jan YN, Jan LY. Four cDNA clones from the Shakerlocus of Drosophila induce kinetically distinct A-type potassium currents in Xenopus oocytes. Neuron. 1988;1:659–667. doi: 10.1016/0896-6273(88)90165-1. [DOI] [PubMed] [Google Scholar]
- Triggle DJ, Langs DA, Janis RA. Ca2+channel ligands: structure-function relationships of the 1,4-dihydropyridines. Med Res Rev. 1989;9:123–180. doi: 10.1002/med.2610090203. [DOI] [PubMed] [Google Scholar]
- Yatani A, Kunze DL, Brown AM. Effects of dihydro-pyridine calcium channel modulators on cardiac sodium channels. Am J Physiol. 1988;254:H140–147. doi: 10.1152/ajpheart.1988.254.1.H140. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science (Wash DC) 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Dittman J, Aldrich RW. a. Shakerpotassium channel gating. II: Transitions in the activation pathway. J Gen Physiol. 1994;103:279–319. doi: 10.1085/jgp.103.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. b. Shakerpotassium channel gating. III: Evaluation of kinetic models for activation. J Gen Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhorov BS, Ananthanarayanan VS. Structural model of a synthetic Ca2+ cahnnel with bound Ca2+ions and dihydro-pyridine ligand. Biophysical J. 1996;70:22–37. doi: 10.1016/S0006-3495(96)79561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]