

Abstract
Osteopontin is one of the major noncollagenous bone matrix proteins produced by osteoblasts and osteoclasts, bone cells that are uniquely responsible for the remodeling of mineralized tissues. Osteoclasts express the αvβ3 integrin, which is one of the receptors for osteopontin. Recent knockout studies revealed that noncollagenous bone matrix proteins are functionally important in regulation of bone metabolism. However, the significance of the presence of osteopontin in in vivo has not been known. We report here that osteopontin knockout mice are resistant to ovariectomy-induced bone resorption compared with wild-type mice. Microcomputed tomography analysis indicated about 60% reduction in bone volume by ovariectomy in wild-type mice, whereas the osteopontin-deficient mice exhibited only about 10% reduction in trabecular bone volume after ovariectomy. Reduction in uterine weight was observed similarly in both wild-type and osteopontin-deficient mice, indicating the specificity of the effect of osteopontin deficiency on bone metabolism. We propose that osteopontin is essential for postmenopausal osteoporosis in women. Strategies to counteract osteopontin’s action may prove effective in suppressing osteoporosis.
Postmenopausal osteoporosis (1–4) is one of the most common diseases affecting aged women. It is a major health problem with regard to not only the high fracture rates and loss of quality of life of women but also the economic loss to society. In Japan and the United States, the number of patients is estimated to be approximately 5% of the population. It is well established that withdrawal of estrogen causes loss of bone mineral because of an increase in osteoclastic bone resorption, and that supplementation with estrogen can reduce bone loss not only in humans but also in animal models.
One of the critical steps in osteoclastic bone resorption is the attachment of osteoclasts to bone and the subsequent formation of a sealing zone, which can be visualized as a clear zone by electron microscopy (5–8). This attachment is a prerequisite for bone resorption because it creates a sequestered microenvironment into which osteoclasts secrete protons, creating an acidic milieu suitable for the dissolution of bone mineral. Osteoclasts also secrete proteases into this sealed environment to digest collagen and other bone proteins.
Integrins are dimeric cell-surface receptors that function in development of osteoclasts in the migration of osteoclasts to sites of resorption and in the attachment of osteoclasts to bone and the formation of the sealing zone (9). One of the characteristics of osteoclasts is the high level of the αvβ3 integrin on the cell surface (10, 11). The functional importance of integrins has been indirectly suggested by the inhibitory effects of disintegrins, such as echistatin, which are RGD containing αvβ3 integrin peptide antagonists. These peptides have been shown to block osteoclast development, osteoclast attachment, and subsequent bone resorption in vitro. Importantly, these disintegrins also block bone resorption in vivo (12–16). These observations indicate that integrins play a critical role in bone resorption.
Osteopontin (OPN), which is produced by osteoblasts and osteoclasts, is one of the more abundant noncollagenous proteins in bone matrix (17, 18). It is also produced by the cells in nonskeletal tissues and has been implicated in tumorigenesis (17, 18). Substrate-bound OPN promotes attachment of osteoclasts (19, 20), whereas soluble OPN can alter calcium levels in osteoclasts (21, 22), suppress inducible nitric-oxide synthase induction in kidney cells, and macrophages and serve as a chemoattractant (17, 18, 23). These observations suggest that OPN may play a key role both in cell attachment and in controlling subsequent bone cell functions such as resorption. OPN is present at high levels in bone, with particularly high concentrations found in cement (renewal) lines and the lamina limitans (24, 25). OPN contains GRGD sequence motif and binds to integrins, especially to αvβ3 subtype. However, the role of OPN in vivo in bone metabolism has not yet been elucidated.
Recently, Rittling et al. (26) and Liaw et al. (27) used homologous recombination in mouse embryonic stem cells to create mouse lines that are null for OPN. These mice mature normally with apparently unaltered development of tissues and organs. The OPN-deficient (OPN−/−) mice exhibit normal skeletal sizes, and the skeletal patterning is largely indistinguishable from that of control mice. They are fertile and give birth to litters of normal size.
Bone resorption after ovariectomy is a model of postmenopausal osteoporosis. To examine the role of OPN in this process, we removed the ovaries of OPN−/− mice and compared the structure of the bones to those of comparable ovariectomized wild-type mice. Our results indicate that OPN deficiency makes the animal resistant to the bone loss induced by ovariectomy.
MATERIALS AND METHODS
Animals.
OPN−/− mice were created as described in Rittling et al. (26), in a 129/SV × C57BL/6 F2 background. Wild-type and OPN−/− mice derived from the original heterozygous crosses were maintained as separate colonies.
Ovariectomy.
OPN−/− mice or wild-type animals (4.5–6 months old) were ovariectomized or sham-operated, and all were sacrificed by overdosing of anesthetics 4 weeks after surgery. All the animals were injected with calcein at 4 mg/kg and xylenol orange at 100 mg/kg 4 and 2 days before sacrifice, respectively, to obtain dynamic parameters for bone formation. Uteri of all the animals were excised and were weighed to evaluate the effects of ovariectomy.
Histological Preparations.
Tibiae and femora were fixed in 4% paraformaldehyde and were subsequently decalcified at 4°C in 10% EDTA for 2 weeks. The bones were dehydrated in gradients of alcohol (70% to 100%) followed by xylene and then embedded in paraffin. Serial 6-μm sections were made and stained either according to the Masson-trichrome method or for tartrate-resistant acid phosphatase (TRAP). Histological specimens were examined under the light microscope. TRAP-positive cells were counted within an area of 0.25 mm2 in the epiphyseal regions. Histomorphometry using undecalcified sections stained according to the Masson-trichrome method was conducted in the proximal epiphyseal regions of tibiae in an area of 0.2 mm2, 0.4 mm away from the growth plate, by using an image analyzer equipped with software (System Supply, Nagano, Japan).
Microcomputed Tomography (μCT).
Fixed bones were subjected to x-ray microtomography by using a microcomputed–tomography apparatus(Nittetsu Elex, Osaka). The apparatus consists of a microfocus sealed x-ray tube. Slices were made with a thickness of 20 μm in the midsaggital planes of the bones. The image data were subsequently quantified by using an automated image analysis system (Nireco, Tokyo). Quantification of the bone volume was made in the area of 0.32 mm2, 0.1–0.4 mm distal to the growth plate of the proximal ends of tibiae.
Statistical Analyses.
The data were evaluated for statistical significance by using Fisher’s test.
RESULTS
OPN−/− and wild-type (OPN +/+) mice, 4.5–6 months old, were ovariectomized, whereas control animals were subjected to sham surgeries. To evaluate the systemic effects of ovariectomy on these animals, uteri were removed at 4 weeks after surgery and their weights determined. The results (Table 1) establish that the effect of ovariectomy was similar in both the OPN−/− and wild-type mice, with a reduction in uterine weight of 25–30% observed in both cases, as compared with the sham-operated controls. The total body weights of the animals in all groups were similar (data not shown). These results confirm that the uterine response to decreased estrogen levels was unaffected by a lack of OPN.
Table 1.
Uterine weight of control and ovariectomized mice
| Mice | Uterine weight, g
|
|
|---|---|---|
| Mean | SD | |
| Wild type; ovariectomized | 0.026* | 0.006 |
| Wild type; sham operated | 0.081 | 0.021 |
| Osteopontin deficient; ovariectomized | 0.024* | 0.008 |
| Osteopontin deficient; sham operated | 0.105 | 0.039 |
n = 4 in all cases.
Difference between the ovariectomized and sham-operated animals is statistically significant (P < 0.05).
Simple x-ray examination of the bones of these animals revealed, as expected, reduced radiopacity in the epiphyses of the tibiae in ovariectomized wild-type mice (Fig. 1A), as compared with the sham-operated controls (Fig. 1B). This reduced radiopacity in the wild-type animals was observed mainly in the epiphyseal and metaphyseal bone areas. Ovariectomy did not reduce the radiopacity in the corresponding regions of the tibiae in the OPN−/− mice (Fig. 1C) nearly as much as it did in the ovariectomized wild-type mice; the radiopacity remained at the level of the sham-operated controls (Fig. 1D).
Figure 1.

Simple x-ray pictures of the tibiae. Wild-type (WT) (A and B) or OPN−/− (KO) (C and D) mice were either ovariectomized (OVX, A and C) or sham-operated (SHAM, B and D). X-ray pictures were taken after dissection of the tibiae. Arrowheads indicate the ends of trabecular bones.
The morphology of the trabecular bone in the proximal epiphyses of the tibiae was examined in the midsaggital planes by using μCT as shown in Fig. 2. The exact planes of the tomographic slices used for μCT are indicated as straight lines in the corresponding panels in Fig. 1. In these midsaggital tomographic sections, the trabecular bones of the ovariectomized wild-type mice were sparse (Fig. 2A) compared with sham-operated wild type (Fig. 2B). The most striking feature in the OPN−/− mice was the similar morphology of the trabecular bones between the ovariectomized (Fig. 2C) and sham-operated animals (Fig. 2D). In addition, the trabecular bones were longer and more connected in the sham-operated OPN−/− mice (Fig. 2D), compared with the trabecular bones in sham-operated wild-type mice (Fig. 2B).
Figure 2.
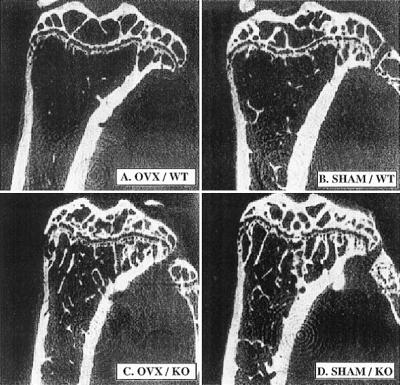
μCT analysis of the tibiae of mice. Wild-type (WT) (A and B) or OPN−/− (KO) (C and D) mice were either ovariectomized (OVX, A and C) or sham-operated (SHAM, B and D). Four weeks postoperatively, μCT pictures of the tibiae were taken in the midsaggital planes as indicated by solid white lines in Fig. 1 A–D, by using μCT Musashi (Nittetsu Elex ).
Quantification of the two-dimensional trabecular bone volume in the tibiae shown in Fig. 2 by using an automated image analyzer indicated that the bone volume of the wild-type mice, expressed as bone area per tissue area, was significantly reduced at 4 weeks after ovariectomy, as compared with the sham-operated wild-type animals (58% reduction, P < 0.05; Table 2). A slight reduction in bone volume was observed at 4 weeks in the ovariectomized OPN mice compared with the sham-operated mice (12%; Table 2), but this difference was not significant. Unexpectedly, the quantification revealed an approximate 2-fold greater trabecular bone volume in the sham-operated OPN mice compared with the sham-operated wild-type mice.
Table 2.
μCT-based quantification of the trabecular bone volume of sham-operated and ovariectomized mice
| Mice | Bone volume, %
|
|
|---|---|---|
| Mean | SD | |
| Wild type; ovariectomized | 2.59* | 0.62 |
| Wild type; sham operated | 6.16 | 0.54 |
| Osteopontin deficient; ovariectomized | 13.88 | 2.07 |
| Osteopontin deficient; sham operated | 15.80 | 2.34 |
n = 4 in all cases.
Difference between the ovariectomized and sham-operated animals is statistically significant (P < 0.05).
Histomorphometrical analysis of these trabecular bones confirmed the results of the μCT, demonstrating a large reduction in bone volume after ovariectomy in the wild-type mice, but only a slight nonsignificant reduction in the OPN mice. Thus, in wild-type animals, trabecular bone volume was decreased by 58% (P < 0.05; Table 3, Fig. 3) after ovariectomy, but only by 32% (not significant; Table 3, Fig. 3)in the OPN−/− mice. Mineral apposition rate was similar among the four experimental groups (Table 3). Bone formation rate was increased by 73% (P < 0.05; Table 3) after ovariectomy in wild-type animals, as expected; the 27% increase observed in the OPN−/− mice was again not significant. Also, the bone formation rate was not affected by OPN status in sham-operated animals (606 vs. 600 μm3/μm2/day for OPN−/− and wild type, respectively; Table 3). These results confirm the resistance of the OPN−/− mice to bone resorption after ovariectomy.
Table 3.
Histomorphometry-based parameters observed in sham-operated and ovariectomized mice
| Parameters | Mice | Mean | SD |
|---|---|---|---|
| BV/TV, % | Wild type: ovariectomized | 3.35* | 1.75 |
| Wild type: sham operated | 7.94 | 3.51 | |
| Osteopontin deficient: ovariectomized | 7.28 | 2.03 | |
| Osteopontin deficient: sham operated | 10.58 | 2.90 | |
| MAR, μm/day | Wild type: ovariectomized | 3.00 | 0.15 |
| Wild type: sham operated | 2.72 | 0.73 | |
| Osteopontin deficient: ovariectomized | 3.03 | 0.15 | |
| Osteopontin deficient: sham operated | 3.04 | 0.73 | |
| BFR/BV, μm3/μm2/day | Wild type: ovariectomized | 1042* | 388 |
| Wild type: sham operated | 600 | 284 | |
| Osteopontin deficient: ovariectomized | 774 | 165 | |
| Osteopontin deficient: sham operated | 606 | 260 |
BV/TV, bone volume/tissue volume; MAR, mineral apposition rate; BFR/BV, bone formation rate/bone volume; n = 4 in all cases.
Difference between the ovariectomized wild type and sham-operated wild type is statistically significant (P < 0.05).
Figure 3.

Histology of the tibiae of the mice. Wild-type (WT) (A and B) or OPN−/− (KO) (C and D) mice were either ovariectomized (OVX) (A and C) or sham-operated (SHAM) (B and D). Tibiae of the mice were subjected to undecalcified histological preparation. Sections were made in the saggital planes of the tibiae and stained according to the Masson-trichrome method.
The number of TRAP-positive cells in the bones of wild-type animals was increased about 3-fold after ovariectomy, compared with sham-operated animals (Fig. 4), consistent with the increased bone resorption that occurs because of ovariectomy in wild-type mice. In sharp contrast, ovariectomy did not significantly increase the number of osteoclasts in OPN−/− mice, which interestingly was about 3-fold higher than in wild-type mice (Fig. 4). These results are consistent with the increased osteoclast development in vitro that we reported previously (26).
Figure 4.

Quantification of TRAP-positive cells attached to the bone surface. Wild-type (wt) or OPN−/− (KO) mice were ovariectomized (ovx) or sham-operated (sham). Four weeks postoperatively, tibiae were excised, and histological sections were stained for TRAP activity. TRAP-positive multinucleated cells within the epiphyseal regions were counted.
DISCUSSION
Mice Are Resistant to Ovariectomy-Induced Bone Loss in the Absence of OPN.
We report here that in the absence of OPN, mice are resistant to the bone loss that accompanies estrogen depletion subsequent to ovariectomy. Quantification of two-dimensional μCT (Table 2) and conventional histomorphometry (Table 3), as well as radiographical density investigations (Fig. 1) and also a three-dimensional μCT morphology study (data not shown) of the trabecular bones of the tibia of ovariectomized OPN−/− mice revealed that the bones of the ovariectomized OPN−/− animals did not differ significantly in mineral content from those of sham-operated OPN−/− mice. In contrast, the OPN+/+ mice exhibited the expected bone loss, with bone volume decreasing by about 60% 4 weeks after ovariectomy. These observations clearly indicated that OPN is required for the rapid bone loss induced by estrogen depletion caused by ovariectomy.
In wild-type mice, ovariectomy-induced bone loss was associated with an increase in bone formation (Table 3), indicating the presence of a high turnover state, as previously described. In contrast, in OPN−/− mice, resistance to ovariectomy-induced bone loss was associated with no increase in bone formation, indicating the absence of the high bone turnover state even after estrogen depletion (Table 3).
In the presence of physiological estrogen levels, the skeletal structure of the OPN−/− mice was normal (26), although we show here that trabecular bone volume is higher in OPN−/− mice than wild type. We have also recently observed resistance to bone loss in the OPN mice under other conditions, specifically, (i) after bone marrow ablation, and (ii) after interleukin 1 injection into the knee joint (H.Y. and M.N., unpublished data; Y. Takazawa and M.N., unpublished data). Taken together, these data argue that in the absence of OPN, bone loss is impaired in these mice. As mentioned above, our experiments revealed that sham-operated OPN−/− mice had a higher bone volume than sham-operated OPN+/+ mice at the age of 4–6 months. However, such a difference was not noticeable at 2 months, as we previously reported (26). These observations suggest an age-dependent phenomenon in the amount of trabecular bones in OPN−/− mice. The reasons for this age dependence of the trabecular bone volume remain to be elucidated.
In OPN−/− mice, the resistance to bone loss induced by ovariectomy was observed while histomorphometric evidence indicated that bone formation was normal in the OPN−/− animals (Table 3). These findings suggest, though do not prove, that the bone resorptive activity of the osteoclasts in the OPN−/− animals could be impaired. In fact, experiments using forearm bones of embryonic mice (either wild-type or OPN−/− mice) organ cultured in the presence or the absence of parathyroid hormone (PTH) indicated that PTH increased bone resorption in wild-type bones but not in OPN−/− mice bones (H. Ihara and M.N., data not shown). This suggests an impairment of osteoclastic function in the bones of OPN−/− mice.
Enhanced Osteoclastogenesis in the Absence of OPN.
Our data reveal that bone loss is impaired in the combined absence of OPN and estrogen and that in the control sham-operated OPN−/− mice, trabecular bone volume is higher than in wild-type mice, despite the increased number of osteoclasts. One interpretation of this result is the possible existence of impairment in osteoclast function and the presence of a feedback loop that could sense reduced osteoclast function and could up-regulate osteoclast differentiation and/or recruitment. In fact, similar combination of the resistance to bone loss in association with an increase in osteoclast number has been observed in the bones of bisphosphonate-treated animals (28). The enhanced number of osteoclasts in OPN−/− mice may result from an increase in a population of osteoclast precursors. This is suggested by our observation in vitro that several-fold more osteoclasts are generated in coculture systems by using spleen or marrow of OPN−/− mice compared with OPN+/+ mice (26).
The recent discovery of tumor necrosis factor (TNF) family members and their ligands OPG/OCIF, RANKL/OPGL/ODF/TRANCE, and ODFR/RANK/TRANCER (29–32), acting in concert to regulate osteoclast formation and function, has revolutionized the field of osteoclast biology. Our results with the OPN mice may shed light on how this system functions. One possible mechanism for the increased osteoclast generation in OPN−/− mice could be increased production of receptor activator of NFkB ligand (RANKL)/osteoclast differentiation factor (ODF) or decreased production of osteoprotegerin (OPG) by the stromal cells in these cocultures. As we reported, increased osteoclastogenesis by OPN−/− spleen cells is supported by wild-type stromal cells. Also, we have observed that RANKL/TNF-related activation induced cytokine (TRANCE)/ODF mRNA levels, as well as those for the receptor for this factor [RANK/ODF receptor (ODFR/TRANCER], are elevated OPN−/− vertebral mRNA (K. Tsuji and M.N., data not shown). Together, these observations suggest that the enhanced osteoclastogenesis in the OPN−/− mice occurs at an early stage of osteoclast differentiation, implicating these molecules in the proposed feedback mechanism.
How this mechanism senses decreased bone loss and the details of how the mechanism functions to increase osteoclast numbers are clearly important questions to be answered by future research. In spite of our observation (unpublished data) that calcium levels are normal in the OPN−/− mice, parathyroid hormone is probably involved as well as RANKL/TRANCE/ODF and/or its receptor.
Despite the increased osteoclast numbers observed in the absence of OPN in both in vitro cocultures and the sham-operated animals, estrogen deficiency fails to induce a further increase in osteoclast numbers in the OPN−/− mice. Although this observation may account for some of the resistance of these animals to ovariectomy-induced bone loss, the reason for this failure is unclear. It may be that in the face of already elevated osteoclast numbers in the estrogen-sufficient animals, a further increase is held in check by additional regulatory mechanisms, perhaps involving osteoprotegerin.
Is this feedback mechanism generally used in situations in which osteoclast function is impaired? That such a mechanism may be widely used is suggested by the observation that in other gene knockout models in which osteoclast function is impaired, osteoclast numbers are also elevated. In src−/− mice, osteoclast numbers in trabecular bone are elevated about 4-fold (33, 34), and a 3-fold increase in osteoclast numbers has been reported in β3 integrin knockout mice (35). Also increased osteoclast numbers were reported in a cathepsin K knockout model (36). These data are suggestive of a generalized mechanism acting to limit osteopetrosis by increasing osteoclast numbers when osteoclast activity is reduced. Clearly, such a mechanism would not be operative in osteopetrosis observed in mice lacking fos (37) or macrophage colony-stimulating factor (38), syndromes in which osteopetrosis results from decreased numbers of osteoclasts rather than impaired function of these cells.
Role of OPN.
Various studies have implicated IL-1, TNF, and IL-6 in bone resorption resulting from estrogen deficiency. Inhibition of TNF activity, either alone or in combination with IL-1 antagonists results in resistance to bone loss induced by ovariectomy (39–42). Mice lacking IL-6 are similarly unable to resorb bone after ovariectomy (43), although the role of IL-6 in this process remains controversial (44, 45). Our results are consistent with a role for OPN either upstream or downstream of these cytokines in the pathway regulating osteoclast function in the absence of estrogen. These cytokines are thought to act to regulate osteoclast development, and they have been shown to regulate OPN expression, at least in vitro (17, 18). Thus the role of OPN in ovariectomy-induced bone resorption may be downstream of these cytokines.
The reduced bone loss we have observed may be related to a requirement for efficient signaling through the αvβ3 integrin for optimal osteoclast function. OPN is a high-affinity ligand for this integrin (18), and we speculate this high-affinity interaction between OPN and the αvβ3 integrin is required for maximal bone resorption activity by osteoclasts. In the absence of OPN, osteoclasts can interact with other αvβ3 ligands present in bone, such as BSP (bone sialoprotein), but the lower affinity of these interactions may result in less efficient osteoclast function.
In summary, we have demonstrated that OPN−/− 129/SV × C57BL/6 mice are resistant to bone loss induced by ovariectomy. A similar resistance to ovariectomy-induced osteopenia was also observed in an inbred 129/SV background (data not shown). Whether OPN is required for human postmenopausal osteoporosis will require further investigation. We are not aware of information regarding OPN deficiency in humans, but such individuals might be expected to show moderate resistance to postmenopausal osteoporosis. Genetic analysis of OPN gene polymorphisms may predict certain patients who could have high or low risk of postmenopausal bone loss. If OPN does play a role in human postmenopausal osteoporosis, new approaches would be available for the development of anti-bone-resorptive drugs, particularly those designed to suppress the action of OPN.
Acknowledgments
We thank Drs. Teruo Amagasa, Koji Kino, Akira Nifuji, Kunikazu Tsuji, and Teruhito Yamashita for their support and advice for this research. This research was supported by the grants-in-aid received from the Japanese Ministry of Education (11877357, 10044246, 0930734), grants from the Marine and Fire Insurance Association of Japan, Inamori Foundation, and Japanese Foundation for Multidisciplinary Treatment for Cancer, grants from Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation, and a grant from the Research for the Future Program of the Japan Society for the Promotion of Science (JSPS) (96I00205) to M.N. Research at Rutgers was supported by grants awarded by the National Institutes of Health (DC01295 and AR44434 to D.T.D., CA72740 to S.R.R.) and by the New Jersey Cancer Commission (no. 795-035 and no.796-031 to S.R.R.).
ABBREVIATIONS
- OPN
osteopontin
- TRAP
tartrate-resistant acid phosphatase
- μCT
microcomputed tomography
References
- 1.Greendale G A, Judd H L. J Am Geriatr Soc. 1993;41:426–436. doi: 10.1111/j.1532-5415.1993.tb06953.x. [DOI] [PubMed] [Google Scholar]
- 2.Johnston C C, Hui S L, Witt R M, Appledorn R, Baker R S, Longcope C. J Clin Endocrinol Metab. 1985;61:905–911. doi: 10.1210/jcem-61-5-905. [DOI] [PubMed] [Google Scholar]
- 3.Cauley J A, Gutai J P, Sandler R B, LaPonte R E, Kuller L H, Sashin D. Am J Epidemiol. 1986;124:752–761. doi: 10.1093/oxfordjournals.aje.a114451. [DOI] [PubMed] [Google Scholar]
- 4.Slemenda C, Hui S L, Longcope C, Johnston C C. J Clin Invest. 1987;80:261–269. doi: 10.1172/JCI113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukushima O, Bekker P J, Gay C V. Anat Rec. 1991;231:298–315. doi: 10.1002/ar.1092310303. [DOI] [PubMed] [Google Scholar]
- 6.Holtrop M E, Raisz L G. Calcif Tissue Int. 1979;29:201. doi: 10.1007/BF02408081. [DOI] [PubMed] [Google Scholar]
- 7.Holtrop M E, Cox K A, Clark M B, Holick M F, Anast C S. Endocrinology. 1981;108:2293–2301. doi: 10.1210/endo-108-6-2293. [DOI] [PubMed] [Google Scholar]
- 8.Zambonin-Zallone A, Teti A, Grano M, Marchisio P C. J Bone Miner Res. 1988;3:517–523. doi: 10.1002/jbmr.5650030507. [DOI] [PubMed] [Google Scholar]
- 9.Rodan S B, Rodan G A. J Endocrinol. 1997;154:47–56. [PubMed] [Google Scholar]
- 10.Nesbitt S, Nesbit A, Helfrich M, Horton M A. J Biol Chem. 1993;268:16737–16745. [PubMed] [Google Scholar]
- 11.Horton M A, Taylor M L, Arnett T R, Helfrich M H. Exp Cell Res. 1991;195:368–375. doi: 10.1016/0014-4827(91)90386-9. [DOI] [PubMed] [Google Scholar]
- 12.Fisher J E, Caulfield M P, Sato M, Quartuccio H A, Gould R J, Garsky V M, Rodan G A, Rosenblatt M. Endocrinology. 1993;132:1411–1413. doi: 10.1210/endo.132.3.8440195. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto M, Fisher J E, Gentile M, Seeder J G, Leu C T, Rodan S B, Rodan G A. Endocrinology. 1998;139:1411–1419. doi: 10.1210/endo.139.3.5831. [DOI] [PubMed] [Google Scholar]
- 14.Horton M A, Rodan G A. In: Adhesion Receptors as Therapeutic Targets. Horton M A, Raton B, editors. Boca Raton, FL: CRC; 1996. pp. 223–224. [Google Scholar]
- 15.Crippes B A, Engleman V W, Settle S L, Delarco J, Ornberg R L, Helfrich M H, Horton M A, Nickols G A. Endocrinology. 1996;137:918–924. doi: 10.1210/endo.137.3.8603604. [DOI] [PubMed] [Google Scholar]
- 16.Engleman V W, Nickols G A, Ross F P, Horton M A, Griggs D W, Settle S L, Ruminski P G, Teitelbaum S L. J Clin Invest. 1997;99:2284–2292. doi: 10.1172/JCI119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler W T, Ridall A L, McKee M D. In: Principles of Bone Biology. Bilezikian J P, Raisz L G, Rodan G A, editors. 1997. pp. 167–181. [Google Scholar]
- 18.Denhardt D T, Noda M. J Cell Biochem. 1998;31:92–102. doi: 10.1002/(SICI)1097-4644(1998)72:30/31+<92::AID-JCB13>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 19.Ross F P, Chappel J, Alvarez J I, Sander D, Butler W T, Farach-Carson M C, Mintz K A, Robey P G, Teitelbaum S L, Cheresh D A. J Biol Chem. 1993;268:9901–9907. [PubMed] [Google Scholar]
- 20.van Dijk S, D’Errico J, Somerman M J, Farach-Carson M C, Butler W T. J Bone Miner Res. 1993;8:1499–1506. doi: 10.1002/jbmr.5650081213. [DOI] [PubMed] [Google Scholar]
- 21.Miyauchi A, Alvarez J, Greengield E M, Teti A, Grano M, Colucci S, Zambonin-Zallone A, Ross F P, Teitelbaum S M, Cheresh D, et al. J Biol Chem. 1991;266:20369–20374. [PubMed] [Google Scholar]
- 22.Zimolo Z, Wesolowski G, Tanaka H, Hyman J, Hoyer J R, Rodan G A. Am J Physiol. 1994;266:C376–381. doi: 10.1152/ajpcell.1994.266.2.C376. [DOI] [PubMed] [Google Scholar]
- 23.Hwang S-M, Lopez C A, Heck D E, Gardner C R, Laskin D L, Laskin J D, Denhardt D T. J Biol Chem. 1994;269:711–715. [PubMed] [Google Scholar]
- 24.McKee M D, Glimcher M J, Nanci A. Anat Rec. 1992;234:479–492. doi: 10.1002/ar.1092340404. [DOI] [PubMed] [Google Scholar]
- 25.McKee M D, Farach-Carson M C, Butler W T, Hauschka P V, Nanci A. J Bone Miner Res. 1993;8:485–496. doi: 10.1002/jbmr.5650080413. [DOI] [PubMed] [Google Scholar]
- 26.Rittling S R, Matsumoto H N, McKee M D, Nanci A, An X -R, Novick K E, Kowalski A J, Noda M, Denhardt D T. J Bone Miner Res. 1998;13:1101–1111. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- 27.Liaw L, Birk D E, Ballas C B, Whitsitt J S, Davidson J M, Hogan B L M. J Clin Invest. 1998;101:1468–1478. doi: 10.1172/JCI1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller S C, Jee W S. Anat Rec. 1979;193:439–462. doi: 10.1002/ar.1091930309. [DOI] [PubMed] [Google Scholar]
- 29.Simonet W S, Lacey D L, Dunstan C R, Kelley M, Chang M-S, Luthy R, Nguyen H Q, Wooden S, Bennett L, Boone T, et al. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 30.Lacey D L, Timms E, Tan H-L, Kelley M J, Dunstan C R, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 31.Bucay N, Sarosi I, Dunstan C R, Morony S, Tarpley J, Capparelli C, Scully S, Tan H L, Xu W, Lacey D L, et al. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soriano P, Montgomery C, Geske R, Bradley A. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 34.Boyce B F, Yoneda T, Lowe C, Soriano P, Mundy G R. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McHugh K P, Hodivala-Dilke K, Cheng S -L, Zheng M H, Avioli L V, Hynes R O, Ross F P, Teitelbaum S L. Bone. 1998;23:S190. [Google Scholar]
- 36.Dodds R A, Cristiano F, Feild J, Kapadia R, Liang X, Debouck C, Kola I, Gowen M. Bone. 1998;23:S164. [Google Scholar]
- 37.Grigoriadis A E, Wang Z Q, Cecchini M G, Hofstetter W, Felix R, Fleisch H A, Wagner E F. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 38.Felix R, Cecchini M G, Fleisch H. Endocrinology. 1990;127:2592–2594. doi: 10.1210/endo-127-5-2592. [DOI] [PubMed] [Google Scholar]
- 39.Kimble R B, Matayoshi A B, Vannice J L, Kung V T, Williams C, Pacifici R. Endocrinology. 1995;136:3054–3061. doi: 10.1210/endo.136.7.7789332. [DOI] [PubMed] [Google Scholar]
- 40.Kimble R B, Bain S, Pacifici R. J Bone Miner Res. 1997;12(6):935–941. doi: 10.1359/jbmr.1997.12.6.935. [DOI] [PubMed] [Google Scholar]
- 41.Ammann P, Rizzoli R, Bonjour J P, Bourrin S, Meyer J M, Vassalli P, Garcia I. J Clin Invest. 1997;99:1699–1703. doi: 10.1172/JCI119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorenzo J A, Naprta A, Rao Y, Alander C, Glaccum M, Widmer M, Gronowicz G, Kalinowski J, Pilbeam C C. Endocrinology. 1998;139:3022–3025. doi: 10.1210/endo.139.6.6128. [DOI] [PubMed] [Google Scholar]
- 43.Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan G A. EMBO J. 1994;13:1189–1196. doi: 10.1002/j.1460-2075.1994.tb06368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jilka R L, Hangoc G, Girasole G, Passeri G, Williams D C, Abrams J S, Boyce B, Broxmeyer H, Manolagas S C. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- 45.Kitamura H, Kawata H, Takahashi F, Higuchi Y, Furuichi T, Ohkawa H. Am J Pathol. 1995;147:1682–1692. [PMC free article] [PubMed] [Google Scholar]