

Abstract
The obligate intracellular parasite Toxoplasma gondii is an important pathogen of humans and animals. Some of the devastating consequences of toxoplasmosis are in part due to the lysis of the host cell during parasite egress. The process of egress is poorly understood and since it is asynchronous in tissue culture its study has been limited to those conditions that induce it, such as artificial permeabilization of the host cell and induction of calcium fluxes with ionophores. Given that permeabilization leads to egress by the activation of motility upon a drop in host cell potassium concentration, we investigated whether the ionophore nigericin, which selectively causes efflux of potassium from the cell without the need for permeabilization, would cause egress. Nigericin effectively causes intracellular parasites to exit their host cell within 30 min of treatment with the drug. Our results show that nigericin-induced egress depends on an efflux of potassium from the cell and requires phospholipase C function and parasite motility. This novel method of inducing and synchronizing egress mimics the effect of artificial permeabilization in all respects. Nevertheless, since the membrane remains intact during the treatment, in our nigericin-induced egress we are able to detect parasite-dependent permeabilization of the host cell, a known step in induced egress. In addition, consistent with the model that loss of host cell potassium leads to egress through the activation of intraparasitic calcium fluxes, a previously isolated Toxoplasma mutant lacking a sodium hydrogen exchanger and defective in responding to calcium fluxes does not undergo nigericin-induced egress. Thus, the discovery that nigericin induces egress presents a novel assay that allows for the genetic and biochemical analysis of the signaling mechanisms that lead to the induction of motility and egress.
Keywords: Toxoplasma, Egress, Potassium, Calcium, Nigericin, TgNHE1
1. Introduction
Toxoplasma gondii is an obligate intracellular parasite capable of entering and replicating within virtually any nucleated cell from a broad range of warm-blooded animals. In addition, this protozoan parasite is an important pathogen of both humans and animals. In immunocompromised individuals such as acquired immunodeficiency syndrome (AIDS) patients, new infections or reactivation of pre-existing Toxoplasma cysts can lead to encephalitis and death (Luft and Remington, 1992; Israelski and Remington, 1993; Slavin et al., 1994). Additionally, in congenital infections, the disease can lead to fetal death or severe neurological problems (Wong and Remington, 1994).
Some of the devastating and life-threatening effects of an uncontrolled Toxoplasma infection are a direct consequence of its lytic cycle, which consists of attachment to the host cell, invasion, intracellular replication and egress. Egress by Toxoplasma is rapid and results in lysis of the host cell. Not withstanding its importance in the pathogenesis of Toxoplasma, very little is known about the mechanisms and parasite proteins involved in egress. Nonetheless, it is known that egress can be induced by activating the motility machinery of the parasite with the induction of calcium fluxes within the host cell and the intracellular parasite (Endo et al., 1982; Stommel et al., 1997; Black et al., 2000). Accordingly, most of our current understanding about egress comes from studies utilizing the calcium ionophore A23187, which efficiently induces intracellular parasites to undergo egress.
During calcium ionophore-induced egress (Ca2+IIE) parasites become motile and extend their conoid before they egress, suggesting that this is an active process involving the cytoskeleton and motility machinery. Furthermore, the characterization of mutants with a pronounced delay in Ca2+IIE revealed that upon exposure to A23187 the host plasma membrane and the parasitophorous vacuole are actively permeabilized by an unidentified parasite- and calcium-dependent mechanism (Black et al., 2000). Time-lapse video microscopy of parasites leaving their host cell upon ionophore-induced egress show that, instead of rupturing the host cell during egress, the parasites appear to penetrate the vacuolar membrane and exit the host cell at discrete sites, constricting their bodies through the plasma membrane as they do during invasion (Black and Boothroyd, 2000). Interestingly, it has been shown that a parasite protein, RON4, which localizes to the constriction ring during invasion, is specifically seen around the constriction of the parasite during calcium ionophore-induced egress (Alexander et al., 2005). These observations have led to the hypothesis that induced egress parallels invasion and utilizes some of the same signaling mechanisms and proteins (Hoff and Carruthers, 2002).
Recently, it was reported that permeabilization of the host cell membrane with Staphylococcus aureus α-toxin effectively induces intracellular parasites to exit their parasitophorous vacuole and host cell (Moudy et al., 2001). Interestingly, this permeabilization-induced egress is blocked if the K+ concentration outside the host cell is approximately the same as inside the cell, which presumably stops the loss of K+ once the cell is permeabilized. Indeed, it is the decrease in K+ concentration alone (and not that of other ions) that induces the parasite to exit upon permeabilization (Moudy et al., 2001). Additionally, the experiments described by Moudy et al. show that upon efflux of K+ from the host cell the parasites become motile. This occurs as a result of a signaling mechanism that involves a parasite phospholipase C (PLC) as well as Ca2+ fluxes within the parasite (Moudy et al., 2001). Accordingly, from their work a possible stepwise model for induced egress arises in which first, permeabilization of the host cell results in a loss of K+ from the host cell cytoplasm. Next, the parasite detects these changes and a cell signaling cascade is activated, resulting in release of Ca2+ from a parasite intracellular compartment, the ensuing activation of parasite motility and finally parasite exit from the host cell.
The ability of the parasite to respond to changes in [K+] in the environment is also evident with extracellular parasites. When incubated in [K+] similar to what the parasite encounters when inside a host cell, the parasites are not motile and therefore cannot invade host cells (Endo et al., 1987). This paralyzing effect is reversible in that the parasites recover and invade host cells when the [K+] is returned to normal extracellular levels (Kafsack et al., 2004). This ability of the parasite to perceive changes in its environment would allow it to exit compromised cells (the result of active permeabilization or cell death) and, since the outside environment is low in K+, to maintain its motile state and quickly invade new cells.
Based on these results, it is likely that the parasites actively detect fluctuations in [K+]. Since the particular parasite proteins involved in this process are not known, further microscopic, genetic and molecular analyses of how K+ efflux induces egress are needed. Using S. aureus α-toxin-driven permeabilization to induce K+ efflux and egress has several disadvantages when attempting to study this phenomenon. Foremost, by artificially permeabilizing the cell numerous ions and small molecules will move in and out and thus any observed effect might be caused by factors other than the loss of K+. In addition, it is known that during egress the parasites themselves induce permeabilization of the host cell (Black et al., 2000). Thus the use of S. aureus α-toxin driven permeabilization to induce egress would preclude the analysis of this step during the egress process. All these factors together underscore the need for a new way of inducing K+ efflux while keeping the host cell membrane intact. Here we report a novel method for inducing and synchronizing egress of T. gondii based on the effects of the K+ ionophore nigericin. In addition, we provide evidence that nigericin-induced egress mimics permeabilization-induced egress in all respects and that it can be used as a tool in the genetic dissection of the egress process.
2. Materials and methods
2.1. Parasite and host cell maintenance and reagents
Parasites were maintained by passage through human foreskin fibroblasts (HFFs) at 37°C and 5% CO2. Normal culture medium was Dubelco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine and 100 units penicillin/100 µg streptomycin per ml. Ionophore assays were performed using a standard extracellular buffered solution (EC buffer), which consisted of 141.8 mM NaCl, 5.8 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, 25 mM N-2-Hydroxyehtylpiperazine-N'-2-ethanesulfonic acid (HEPES)-NaOH, pH 7.2 (Kafsack et al., 2004). Nigericin sodium salt (Calbiochem) and U-73122 (Sigma) were dissolved in dimethyl sulphoxide (DMSO) at 1 mM to make stock solutions.
2.2. Parasite strains
For ease of monitoring parasite replication and egress, we engineered strains of Toxoplasma expressing both the green fluorescent protein (GFP) and β-galactosidase (β gal). For this purpose, a vector carrying GFP under the control of the Toxoplasma Gra1 promoter and βgal under the control of the Toxoplasma Tubulin promoter was created by cloning a gra1GFP SalI fragment from pGraGFP (Seeber and Boothroyd, 1996; Kim et al., 2001) into the XhoI site of ptubβgal (Seeber and Boothroyd, 1996; Kim et al., 2001). The resulting construct with a head-to-head configuration of the Tubulin and Gra1 promoters was linearized with BamHI and introduced into two different parasite strains, RH and Prugniaud (Zenner et al., 1993), which had previously been manipulated to lack the Toxoplasma hypoxanthine/xanthine guanine phosphoribosyl transferase (HPT) gene product (RHΔhpt and PruΔhpt, respectively) (Donald et al., 1996). Transfected parasites stably expressing GFP were isolated by flow cytometry and GFP+ clones were subsequently tested for βgal expression by growing them in the presence of the substrate chlorophenol red-βD-galactopyranoside (CPRG) as previously described (Seeber and Boothroyd, 1996). The resulting PruΔhpt+GFP+βgal and RHΔhpt+GFP+βgal strains were confirmed to have normal growth, egress and Ca2+IIE phenotypes compared with PruΔhpt and RHΔhpt, respectively. Of these two strains, RHΔhpt+GFP+βgal was utilized in the experiments described here.
2.3. Nigericin-induced egress assays
For this assay 105 parasites were allowed to infect HFFs grown in 24-well tissue culture plates in normal medium and grown at 37°C for 24 h. Intracellular parasites were then washed three times with PBS pre-warmed to 37°C. These parasites were then exposed to either 10 µM nigericin or DMSO carrier alone for varying amounts of times at 37°C in either EC buffer or EC buffer with varying amounts of KCl. After this incubation, media was removed and the cells were fixed with 3.5% formaldehyde in PBS for 20 min. Using fluorescence microscopy to detect the GFP parasites, intact vacuoles in nigericin and DMSO-treated cells were counted in 10 randomly selected fields. The percentage of vacuoles that remained intact upon nigericin treatment was calculated by dividing the number of intact vacuoles in nigericin-treated cells over that of intact vacuoles in DMSO-treated cells. For ease of interpretation the resulting percentage was subtracted from 100 to represent the percentage of vacuoles lysed (i.e. egress) upon nigericin treatment. In experiments using parasites that do not express GFP, parasites and intact vacuoles were visualized by staining methanol-fixed cells with Diff-quik (Dade-Behring) as previously described (Arrizabalaga et al., 2004).
2.4. Inhibitor assays
For nigericin egress assays using the inhibitor U-73122, 105 parasites were allowed to infect HFFs in 24-well tissue culture plates in DMEM, at 37°C, for 24 h. Intracellular parasites were then pre-incubated with 0, 2 or 10 µM U-73122 in EC buffer at 37°C for 30 min. After pre-incubation, the solution was removed and replaced with EC buffer containing the same amount of U-73122 plus either 10 µM nigericin or an equivalent amount of DMSO as a solvent control at 37°C for 30 min. After this treatment cultures were fixed with 3.5% formaldehyde and egress was quantified as described above. To test whether PLC inhibition of nigericin-induced egress could be reversed by directly inducing calcium fluxes, intracellular parasites were exposed to 1 µM A23187 for 10 min after pre-incubation with U-73122 and treatment with nigericin.
To test the effect of disrupting the actin-dependent parasite motility on nigericin-induced egress, intracellular parasites were treated for 30 min with 10 µM nigericin and 10 µM cytochalasin D (Sigma) in EC buffer. Egress efficiency was determined as described for the nigericin-induced egress assay.
2.5. Host cell permeabilization assay
The integrity of host plasma and vacuolar membranes was examined using immunofluorescence as previously described (Black et al., 2000). In brief, parasites were grown for 24 h in HFFs on glass coverslips and then treated with 10 µM nigericin for 15 min. The nigericin-treated cells were then fixed with 3.5% formaldehyde for 20 min and the parasites that were within disrupted host cells were stained with a rabbit anti-SAG1 (a parasite surface antigen) and an Alexa fluor 594 conjugated goat anti-rabbit secondary antibody (Molecular Probes). When using parasite strains that express GFP, this reporter was used to detect all parasites regardless of the state of their host cell. Thus, parasites within permeabilized cells appear both green and red when inspected using a Zeiss Axiovert 40 CFL microscope at 400× magnification. The percentage of pearmibilization was determined by dividing the number of vacuoles in permeabilized cells (number of red vacuoles) over the total number of vacuoles inspected (number of green vacuoles).
For those parasite strains that do not express GFP the permeabilization assay was modified according to established protocols (Black et al., 2000). After treatment with 10 µM nigericin for 15 min, cultures were fixed with 3.5% formaldehyde and stained with a rabbit anti-SAG1 antibody as described above, to detect vacuoles within disrupted host cells. To see all parasites regardless of the status of the host cell, this was followed by permeabilization with 0.2% Triton X-100 in PBS, and all parasites were labeled with a second SAG1 antibody that has been generated in mice. The two primary antibodies were visualized with an anti-rabbit IgG secondary antibody with a red fluorescent tag (Alexa Fluor 594 - Molecular Probes) and an anti-mouse IgG secondary antibody with a green fluorescent tag (Alexa Fluor 488 - Molecular Probes), respectively. Thus parasites in cells that were disrupted following nigericin treatment were co-labeled red and green, while those in cells that remained intact after nigericin treatment were labeled green only. The percentage of vacuoles within permeabilized cells was calculated by dividing the number of red vacuoles over the number of green ones.
3. Results
3.1. Nigericin induces intracellular parasites to undergo egress
To explore whether the selective efflux of K+ can induce egress without having to artificially permeabilize the host cell with S. aureus α-toxin, we studied the effect of the K+ ionophore nigericin on intracellular parasites. For this purpose parasites, expressing GFP (RHΔhpt+GFP+βgal) to ease visualization, were treated with varying concentrations of nigericin for 30 min 24 h p.i. From these studies it was determined that treatment with 2 µM nigericin resulted in a low number of vacuoles being lysed (15%) while 10 µM nigericin induced > 96% of parasites to exit host cells (Fig. 1A). The effect of 10 µM nigericin on intracellular parasites is detectable by 15 min, at which time point 22% of vacuoles are lysed, whereas by 30 min approximately 100% of parasites have exited host cells (Fig. 1B).
Fig. 1.

Effect of nigericin on intracellular parasites. Intracellular RHΔhpt+GFP+βgal parasites were treated with nigericin for: A) 30 min at either 2 or 10 µM nigericin and B) at different time points for 15 or 30 min with 10 µM nigericin. The percentage egress was determined by comparing the total remaining intact vacuoles in the treated culture with those of an untreated culture. Each data point represents the average of at least three independent experiments and the error bars represent the S.D. For each experiment at least 100 vacuoles were inspected.
3.2. Nigericin-induced egress is dependent on efflux of K+
While nigericin is known to induce efflux of K+ in eukaryotic cells, it can also have other effects such as the stimulation of the mitochondrial ATPase (Estrada et al., 1967; Graven et al., 1967). To investigate whether the induction of egress by nigericin is due to an efflux of K+, we exposed intracellular parasites to nigericin in the presence of different concentrations of KCl. Nigericin effectively induces egress when extracellular [K+] is greatly lower than the expected intracellular [K+] (Fig. 2). By contrast, it was observed that the efficiency of egress was greatly diminished when [KCl] was increased to levels that mimic intracellular [K+]: 52.7% egress at 120 mM KCl, 14.1% in 140 mM KCl and 0% at 160 mM KCl (Fig. 2). These results indicate that efficient nigericin-induced egress requires [K+] outside the host cell to be lower than inside, strongly suggesting that nigericin promotes egress by inducing an efflux of K+.
Fig. 2.

Effect of extarcellular K+ on nigericin-induced egress. Intracellular RHΔhpt+GFP+βgal parasites were treated with 10 µM nigericin in extracellular buffer containing increasing concentrations of KCl. The percentage egress was determined as in Fig. 1. Each data point represents the average of at least three independent experiments and the error bars represent the S.D. For each experiment at least 100 vacuoles were inspected.
It is possible that the lack of an effect by nigericin in high extracellular [K+] is due to a general inability of the parasite to undergo egress under these conditions and not because nigericin can’t extrude K+ from the host cell against a K+ gradient. To explore this possibility we took advantage of the effect of A23187, a calcium ionophore that effectively induces egress of intracellular parasites. After being treated with 10 µM nigericin for 30 min in the presence of 150 mM KCl, parasites were exposed to either 1 µM A23187 or DMSO as a solvent control. As shown in Fig. 3, parasites can effectively undergo egress when treated with A23187 in [K+] that normally blocks nigericin-induced egress. This suggests that high [K+] is not inhibitory by itself and that its effect on nigericin-induced egress is due to the fact that nigericin induces egress by causing a K+ efflux from the host cell. Moreover, this result indicates that the effect of nigericin is upstream of the intraparasitic calcium fluxes known to be involved in the induction of egress.
Fig. 3.

Effect of A23187 on intracellular parasites after exposure to nigericin in 150 mM KCl. Intracellular RHΔhpt+GFP+βgal parasites were exposed to 10 µM nigericin in the presence of 150mM KCl. Media was then changed to either nigericin in 150 mM KCl or nigericin and 1 µM A23187 in 150 mM KCl. After a 10 min incubation, the percentage egress was determined for each experiment. Each data point represents the average of at least three independent experiments and the error bars represent the S.D.
3.3. Nigericin-induced egress is dependent on calcium signaling
Loss of host cell K+ after permeabilization causes egress by inducing a transient increase in [Ca2+] within the parasite which is dependent on the signaling protein PLC (Moudy et al., 2001). To investigate whether the same signaling pathway is involved in inducing egress upon exposure to nigericin, intracellular parasites were treated with varying concentrations of U-73122, an inhibitor of PLC, before being exposed to nigericin. When parasites are treated for 30 min with 10 µM of U-73122, they become resistant to the effect of nigericin (Fig. 4). Interestingly, the inhibition of nigericin-induced egress by U-73122 can be overcome by artificially causing calcium fluxes with the calcium ionophore A23187 (data not shown). Together, these results suggest that, just as in the case of permeabilization-induced egress, nigericin-induced egress depends on PLC and it is likely to require calcium fluxes within the parasite.
Fig. 4.

Effect of phospholipase C (PLC) inhibition on nigericin-induced egress. Intracellular RHΔhpt+GFP+βgal parasites were treated with the indicated concentrations of U-73122 for 30 min, after which time point 10 µM nigericin was added for another 30 min of incubation. The percentage egress was determined as in Fig. 1. Each data point represents the average of at least three independent experiments and the error bars represent the S.D. For each experiment at least 10 fields of view were inspected.
3.4. Mutants delayed in responding to Ca2+ fluxes do not respond to nigericin
Previous research, as well as our experiments presented here, suggests that a loss of K+ from the host cell induces egress of Toxoplasma by inducing a flux of Ca2+ within the parasite. This hypothesis predicts that nigericin-induced egress depends on an efficient response by the parasite to calcium fluxes. To test this hypothesis we used various Toxoplasma mutants resistant to the effect of the calcium ionophore A23187, which induces egress by causing calcium fluxes (Black et al., 2000; Arrizabalaga et al., 2004). One of these mutants, which lacks the sodium/hydrogen exchanger (NHE) TgNHE1, has a defect in maintaining calcium homeostasis and thus has an abnormally high cytosolic level of calcium (Arrizabalaga et al., 2004). Given this defect, the Tgnhel knock-out strain cannot efficiently detect and respond to the changes in calcium concentration induced by A23187, which is manifested as a delay in A23187-induced egress. If, as predicted, nigericin induces egress by the downstream induction of calcium fluxes within the parasite, the Tgnhe1 knock-out strain (RHΔnhe1) should have an altered response to nigericin. Fig. 5 shows the percentage of egress based on the number of vacuoles lysed after 30 min exposure to nigericin for the parental strain RHΔhpt and the RHΔnhe1 knockout strain. While the parental strain efficiently responds to nigericin, exhibiting 96% egress after 30-min exposure to 10 µM nigericin, the Tgnhe1 knockout strain is resistant to this effect, showing only 9% egress at the same time point (Fig. 5). Besides confirming that the Tgnhe1 knockout strain cannot respond to nigericin, this result indicates that the disruption of calcium homeostasis caused by the lack of TgNHE1 results in an inability to respond not only to the large calcium fluxes induced by the calcium ionophore but also to the more biologically relevant fluxes caused by the calcium signaling events downstream of K+ loss from the cell.
Fig. 5.

Effect of nigericin on mutants with a delay in responding to calcium ionophores. Intracellular parasites of the parental strain (RHΔhpt), the Tgnhe1 knockout strain (RHΔnhe1) and the Tgnhe2 knockout strain (RHΔnhe2) were treated with 10 µM nigericin for 30 min. Of these strains only RHΔnhe1 has an altered response to Ca2+ fluxes. The percentage egress was determined as in Fig. 1. Each data point represents the average of at least three independent experiments and the error bars represent the S.D. For each experiment at least 10 fields of view were inspected which represent a total of over 100 vacuoles.
As a control for our experiment with the RHΔnhe1 knockout strain, we also tested a strain with a knockout of Tgnhe2, an NHE that does not influence Ca2+ or calcium ionophore-induced egress (Karasov et al., 2005). As expected, this mutant strain, RHΔnhe2, responds normally to nigericin-induced egress (Fig. 5). In addition to the RHΔnhe1 knockout strain, we tested the previously characterized chemical mutant MBE1.1 (Black et al., 2000), which also exhibits a delay to the effects of the calcium fluxes induced by A23187. Similar to what we observed with the RHΔnhe1 knockout strain, MBE1.1 does not undergo egress when exposed to nigericin (data not shown). Taken together, these results indicate that mutants with defects in responding to calcium fluxes cannot respond to nigericin, which is consistent with the observation that nigericin-induced egress is dependent on the induction of a calcium flux within the parasite.
3.5. Permeabilization is a step in nigericin-induced egress
Previous work using the calcium ionophore A23187 to artificially induce egress indicated that prior to parasites exiting the host cell, the plasma membrane is permeabilized in a parasite-dependent manner (Black et al., 2000). Given that, just as in the case of A23187-induced egress, calcium fluxes within the parasite are a step in nigericin’s effect, one would predict host cell permeabilization to be a step in nigericin-induced egress. To test this hypothesis, we treated GFP-expressing intracellular parasites with nigericin for 15 min, at which time point most parasites remain inside the cell, and tested the integrity of the host-cell membrane using antibodies directed to the parasite and visualized with a red fluorescent labeled secondary antibody see (Materials and methods). This antibody will only reach the parasites when the host plasma membrane and the parasitophorous vacuolar membranes are permeabilized. Thus, only those parasites within permeabilized cells will appear red and all parasites will be green (due to the GFP marker). Using this assay we determined that after 15 min of treatment with nigericin, 18% of infected host cells were permeabilized (Fig. 6A). This level of permeabilization is considerably higher that what is seen in the absence of nigericin (2% of host cells permeabilized, Fig. 6A). The permeabilization of host cells seen after nigericin treatment is eliminated when the extracellular [K+] is increased to intracellular levels (Fig. 6A). This result shows that permeabilization is due to an event downstream of K+ efflux and not a direct effect of nigericin.
Fig. 6.

Permeabilization of host cells during nigericin treatment. A) RHΔhpt+GFP+βgal parasites were treated with 10 µM nigericin for 15 min 24 h post-invasion in extracellular (EC) buffer or EC buffer with 142 mM KCl. B) Cells infected with either the RHΔhpt strain or the nigericin-resistantstrain RHΔnhe1 were treated with 10 µM nigericin or DMSO carrier alone for 15 min. In both A and B parasites in permeabilized cells were detected using an antibody specific to the surface of the parasite as described in Material and methods. For each experiment at least 100 vacuoles were inspected, each bar represents the average of at least three independent experiments and the error bars represent the S.D.
To assess whether the permeabilization event was parasite-dependent we tested the effect of nigericin on cells infected with the RHΔnhe1 knockout strain, which is non-responsive to nigericin. Given that neither the RHΔnhe1 parasites nor the parental strain RHΔhpt express GFP, we modified our permeabilization assay with the use of two antibodies directed at the parasite, one used after nigercin treatment and fixation (to see parasites in cells that were permeabilized upon nigericin treatment) and the other one used after subsequent detergent permeabilization (to see all parasites, see Materials and methods). When cells infected with the nigericin-resistant RHΔnhe1 strain are treated with nigericin we detect only 5% of host cells being permeabilized, which is equal to the background permeabilization levels seen in the untreated controls (Fig. 6B). This level of permeabilization is significantly less than what is detected in cells infected with the parental strain RHΔhpt after nigericin treatment (Fig. 6B). These results confirm that permeabilization is a parasite-dependent event, since if nigericin caused the permeabilization on its own we would observe permeabilization in the nigericin-treated RHΔnhe1-infected cells. In conjunction, these assays show that host cell permeabilization is a step in induced egress downstream of calcium signaling and indicate that nigericin induces egress by the same mechanisms as the calcium ionophores.
3.6. Parasite motility is required for nigericin-induced egress
Our results suggest that loss of K+ from the host cell resulting from nigericin treatment induces parasites to undergo egress by inducing their motility. Thus, it is expected that the effect of this ionophore should be dependent on the motility machinery of the parasite. To test this hypothesis we studied the effect of the actin inhibitor cytochalasin D on nigericin-induced egress. Toxoplasma moves by a gliding motility mechanism that depends on its actin microfilaments and this motility is completely blocked by cytochalasin D (Dobrowolski and Sibley, 1996). When intracellular parasites are exposed to nigericin in the presence of cytochalasin D the parasites remain inside the cell (Fig. 7). This result indicates that nigericin cannot induce egress of non-motile parasites and that motility activation is therefore a likely step in nigericin-induced egress.
Fig. 7.
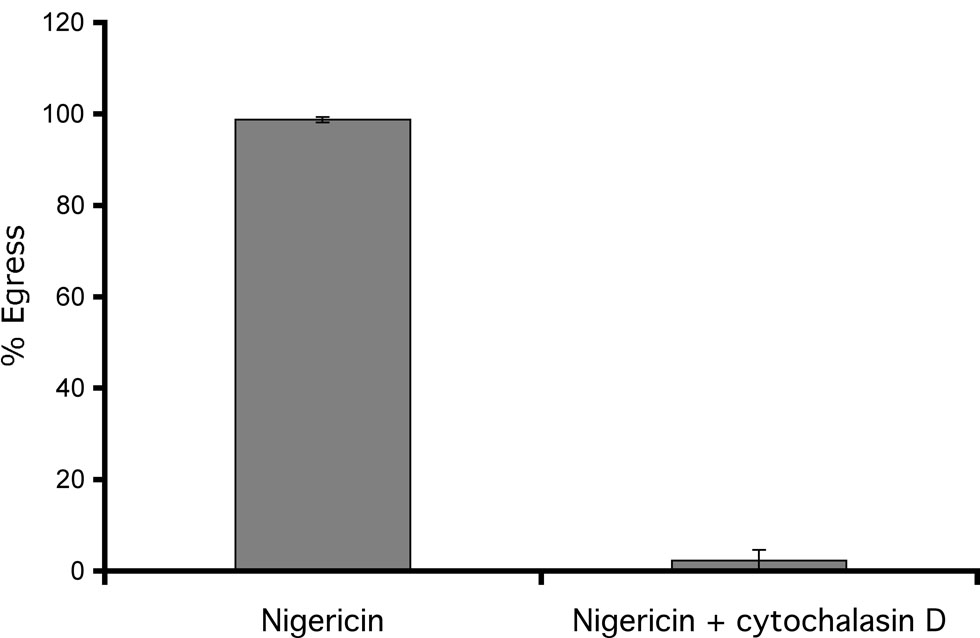
Effect of cytochalasin D on nigericin-induced egress. Intracellular RHΔhpt+GFP+βgal parasites were treated with 10 µM nigericin either in the presence or absence of 10 µM cytochalasin D for 30 min. The percentage egress was determined as in Fig. 1. Each data point represents the average of at least three independent experiments and the error bars represent the S.D. For each experiment at least 100 vacuoles were inspected.
4. Discussion
When [K+] in a Toxoplasma infected cell is reduced by permeabilizing the host membrane, the parasite quickly exits the cell. We have found a novel way to induce motility and egress by specifically inducing K+ efflux without the need for permeabilization with detergents or bacterial toxins. This method is based on the effects of nigericin, a polyether antibiotic originally isolated from Streptomyces hygroscopicus and known to promote K+/H+ exchange across membranes (Graven et al., 1967; Henderson et al., 1969). By disrupting H+ and K+ concentration within a cell or an organelle, nigericin can change cell pH (Shavit et al., 1968; Henderson et al., 1969), disrupt membrane potential (Markin et al., 1975) and change cell volume (Dise et al., 1980). Our results presented here show that nigericin effectively induces egress of T. gondii. During treatment, nigericin can probably affect both the host cell and the intracellular parasites. Nevertheless, given that this egress effect is blocked by high extracellular [K+], nigericin-induced egress is likely due to the effect of K+ efflux from the host cell, which is consistent with experiments that use permeabilization to induce K+ efflux and egress (Moudy et al., 2001). In addition, just as in the case of permeabilization-induced egress, our assay confirms that loss of host cell K+ is upstream of intraparasitic calcium fluxes in the pathway leading to egress. This is evidenced by the fact that calcium ionophores can overcome a block in nigericin-induced egress and that mutants with a defect in detecting calcium fluxes cannot react to nigericin.
While we observe that nigericin effectively induces Toxoplasma egress, it has been previously reported that exposing intracellular Toxoplasma parasites to nigericin results in parasite death (Couzinet et al., 1994). Interestingly, we consistently observe egress when using nigericin at concentrations apparently higher than those used to kill the parasite in the previous report (Couzinet et al., 1994) and under our conditions we only observe lethality when using nigericin at concentrations higher than 20 µM. Given this contradiction we explored the reasons for the conflicting results. The experiments showing that nigericin kills intracellular parasites were performed by exposing these parasites to nigericin for prolonged periods of time in the presence of normal growth medium, which included 5% inactivated calf serum (Couzinet et al., 1994). By contrast, all our studies in which we observe nigericin-induced egress were performed using extracellular (EC) buffer in the absence of serum. Interestingly, we have previously observed that calcium ionophore-induced egress is inhibited by serum (Arrizabalaga, Black and Boothroyd, unpublished data). This led us to hypothesize that the effect of nigericin on intracellular parasites is influenced by the medium used in the experiment. To explore this possibility, we treated intracellular parasites with 10 µM nigericin in either DMEM with 5% inactivated calf serum or in EC buffer with no serum. As expected, no egress was observed with serum containing media (data not shown). This is in contrast to nearly 100% egress efficiency when nigericin exposure is performed in the absence of serum. Moreover, when we exposed intracellular parasites to nigericin in the presence of serum for more than 24 h we observed the same morphological signs of parasite death described by Couzinet and colleagues (Couzinet et al., 1994) (data not shown). These results easily explain the conflict between our data and published results.
Another reported effect of nigericin on T. gondii is the inhibition of motility of extracellular parasites (Endo et al., 1987). Indeed, when we expose extracellular parasites to 10 µM of nigericin, invasion efficiency is considerably reduced (by approximately 50%), presumably due to an inhibition of the motility machinery (data not shown). Nonetheless, our results suggest that motility is not affected by nigericin when the parasites are within the cell. As described above, after exposure with nigericin under high [K+] (which blocks nigericin-induced egress) the parasites can be induced to undergo egress with the calcium ionophore A23187. Given that A23187 cannot induce non-motile parasites to undergo egress (Black et al., 2000), this indicates that intracellular parasites exposed to nigericin retain their motility. Furthermore, when parasite motility is directly inhibited by the actin inhibitor cytochalasin D, nigericin does not induce egress, confirming that parasite motility is required for nigericin-induced egress. Thus, while we cannot exclude the possibility that nigericin affects the intracellular parasites in some way, it appears that it does not affect their ability to move.
Given that nigericin-induced egress is dependent on calcium homeostasis, calcium signaling and motility, it can be used as a system for the genetic dissection of the pathways leading to egress. A possible approach to identifying the proteins required during induced egress would be to isolate and characterize mutants that cannot undergo egress after nigericin treatment. Providing evidence for the effectiveness of such a method is the fact that a previously isolated Ca2+IIE mutant disrupted in TgNHE1 is non-responsive to nigericin, which indicates that disruption of one gene alone can result in a disruption of nigericin-induced egress. While in some respects nigericin-induced egress recapitulates the phenomenon seen with calcium ionophores, nigericin-induced egress depends on events that are upstream of the events induced by the calcium ionophores and thus allows for a broader look at the mechanisms leading to egress.
One of the most noticeable differences between nigericin-induced egress and Ca2+IIE is the kinetics of the effect. The induction of egress by nigericin is considerably slower than that seen with calcium ionophores (A23187 induces > 90% of parasites to exit within 2 min) (Black et al., 2000). The reason behind this observation is not known at this point, but it might be due to either a slow rate of K+ loss across the host cell membrane or a need for signaling mechanisms not required when calcium fluxes are directly induced. Nonetheless, the slower rate of egress with nigericin allows for the observation of steps in the pathway that normally occur too rapidly to be detected. For example, just as with calcium ionophores, nigericin treatment results in a parasite-dependent host cell permeabilization step. While we can effectively detect this permeabilization event with nigericin without any further manipulation, when using a calcium ionophore the parasite-dependent permeabilization of the host cell membrane can only be detected if the parasites are paralyzed with cytochalasin D to avoid egress and the ensuing membrane rupture (Black et al., 2000). Therefore, nigericin-induced egress might constitute a good assay to determine the mechanism behind this permeabilization event, which could be the result of either a secreted parasite factor or the activation of a host factor in a parasite-dependent manner.
Detection of changes in its environment is of crucial importance to an intracellular parasite such as T. gondii, especially as it moves from the outside to the inside of the host cell. Given the vast differences in ionic composition between the extra- and intracellular environments it is not surprising that Toxoplasma has the ability to respond to changes in the concentration of ions such as K+. The connection between activation of motility and extracellular [K+] is not exclusive to this parasite. Salmonid sperm, for instance, is not motile in high [K+] but becomes motile if [K+] is reduced to normal extracellular levels (Morisawa et al., 1983), a phenomenon that mimics what is observed with T. gondii. The nigericin-induced egress assay that we have developed will allow us to study detection of, and response to, K+ changes within the context of a normal process of the parasite, i.e. egress, and therefore it will aid the study of events that are part of the response to those changes including motility, calcium signaling and egress.
Acknowledgements
This work was supported by NIH grants from the NCRR Center of Biomedical Research Centers P20 (RR15587) and the NIAID K22 program (AI061293-01). M.D.L. is supported by the Idaho IDEA Network of Biomedical Research Excellence grant from the NIH/NCRR (P20 RR016454). The creation of the GFP-expressing parasite lines was performed by GA while a postdoctoral fellow at Stanford University in the laboratory of Dr. John Boothroyd. The SAG1 antibodies used in our work were a gift from Dr. John Boothroyd.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander DL, Mital J, Ward GE, Bradley P, Boothroyd JC. Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog. 2005;1:e17. doi: 10.1371/journal.ppat.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrizabalaga G, Ruiz F, Moreno S, Boothroyd JC. Ionophore-resistant mutant of Toxoplasma gondii reveals involvement of a sodium/hydrogen exchanger in calcium regulation. J Cell Biol. 2004;165:653–662. doi: 10.1083/jcb.200309097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MW, Arrizabalaga G, Boothroyd JC. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol Cell Biol. 2000;20:9399–9408. doi: 10.1128/mcb.20.24.9399-9408.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MW, Boothroyd JC. Lytic cycle of Toxoplasma gondii. Microbiol Mol Biol Rev. 2000;64:607–623. doi: 10.1128/mmbr.64.3.607-623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzinet S, Dubremetz JF, David L, Prensier G. Toxoplasma gondii: activity of the polyether ionophorous antibiotic nigericin on tachyzoites in cell culture. Exp Parasitol. 1994;78:341–351. doi: 10.1006/expr.1994.1037. [DOI] [PubMed] [Google Scholar]
- Dise CA, Goodman DB, Rasmussen H. Selective stimulation of erythrocyte membrane phospholipid fatty acid turnover associated with decreased cell volume. J Biol Chem. 1980;255:5201–5207. [PubMed] [Google Scholar]
- Dobrowolski JM, Sibley LD. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 1996;84:933–939. doi: 10.1016/s0092-8674(00)81071-5. [DOI] [PubMed] [Google Scholar]
- Donald RG, Carter D, Ullman B, Roos DS. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J Biol Chem. 1996;271:14010–14019. doi: 10.1074/jbc.271.24.14010. [DOI] [PubMed] [Google Scholar]
- Endo T, Sethi KK, Piekarski G. Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp Parasitol. 1982;53:179–188. doi: 10.1016/0014-4894(82)90059-5. [DOI] [PubMed] [Google Scholar]
- Endo T, Tokuda H, Yagita K, Koyama T. Effects of extracellular potassium on acid release and motility initiation in Toxoplasma gondii. J Protozool. 1987;34:291–295. doi: 10.1111/j.1550-7408.1987.tb03177.x. [DOI] [PubMed] [Google Scholar]
- Estrada OS, Graven SN, Lardy HA. Potassium Ion-dependent hydrolysis of adenosine triphosphate induced by nigericin in mito chondria. J Biol Chem. 1967;242:2925–2932. [PubMed] [Google Scholar]
- Graven SN, Lardy HA, Estrada OS. Antibiotics as tools for metabolic studies. 8. Effect of nonactin homologs on alkali metal cation transport and rate of respiration in mitochondria. Biochemistry. 1967;6:365–371. doi: 10.1021/bi00854a001. [DOI] [PubMed] [Google Scholar]
- Henderson PJ, McGivan JD, Chappell JB. The action of certain antibiotics on mitochondrial, erythrocyte and artificial phospholipid membranes. The role of induced proton permeability. Biochem J. 1969;111:521–535. doi: 10.1042/bj1110521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff EF, Carruthers VB. Is Toxoplasma egress the first step in invasion? Trends Parasitol. 2002;18:251–255. doi: 10.1016/s1471-4922(02)02240-7. [DOI] [PubMed] [Google Scholar]
- Israelski DM, Remington JS. Toxoplasmosis in patients with cancer. Clin Infect Dis. 1993;17 Suppl 2:S423–S435. doi: 10.1093/clinids/17.supplement_2.s423. [DOI] [PubMed] [Google Scholar]
- Kafsack BF, Beckers C, Carruthers VB. Synchronous invasion of host cells by Toxoplasma gondii. Mol Biochem Parasitol. 2004;136:309–311. doi: 10.1016/j.molbiopara.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Karasov AO, Boothroyd JC, Arrizabalaga G. Identification and disruption of a rhoptry-localized homologue of sodium hydrogen exchangers in Toxoplasma gondii. Int J Parasitol. 2005;35:285–291. doi: 10.1016/j.ijpara.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Kim K, Eaton MS, Schubert W, Wu S, Tang J. Optimized expression of green fluorescent protein in Toxoplasma gondii using thermostable green fluorescent protein mutants. Mol Biochem Parasitol. 2001;113:309–313. doi: 10.1016/s0166-6851(01)00212-2. [DOI] [PubMed] [Google Scholar]
- Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clin Infect Dis. 1992;15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- Markin VS, Sokolov VS, Bogulavsky LI, Jaguzhinsky LS. Nigericin-induced charge transfer across membranes. J Membr Biol. 1975;25:23–45. doi: 10.1007/BF01868566. [DOI] [PubMed] [Google Scholar]
- Morisawa M, Suzuki K, Morisawa S. Effects of potassium and osmolality on spermatozoan motility of salmonid fishes. J Exp Biol. 1983;107:105–113. doi: 10.1242/jeb.107.1.105. [DOI] [PubMed] [Google Scholar]
- Moudy R, Manning TJ, Beckers CJ. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J Biol Chem. 2001;276:41492–41501. doi: 10.1074/jbc.M106154200. [DOI] [PubMed] [Google Scholar]
- Seeber F, Boothroyd JC. Escherichia coli beta-galactosidase as an in vitro and in vivo reporter enzyme and stable transfection marker in the intracellular protozoan parasite Toxoplasma gondii. Gene. 1996;169:39–45. doi: 10.1016/0378-1119(95)00786-5. [DOI] [PubMed] [Google Scholar]
- Shavit N, Dilley RA, San Pietro A. Ion translocation in isolated chloroplasts. Uncoupling of photophosphorylation and translocation of K+ and H+ ions induced by Nigericin. Biochemistry. 1968;7:2356–2363. doi: 10.1021/bi00846a043. [DOI] [PubMed] [Google Scholar]
- Slavin MA, Meyers JD, Remington JS, Hackman RC. Toxoplasma gondii infection in marrow transplant recipients: a 20 year experience. Bone Marrow Transplant. 1994;13:549–557. [PubMed] [Google Scholar]
- Stommel EW, Ely KH, Schwartzman JD, Kasper LH. Toxoplasma gondii: dithiolinduced Ca2+ flux causes egress of parasites from the parasitophorous vacuole. Exp Parasitol. 1997;87:88–97. doi: 10.1006/expr.1997.4187. [DOI] [PubMed] [Google Scholar]
- Wong SY, Remington JS. Toxoplasmosis in pregnancy. Clin Infect Dis. 1994;18:853–861. doi: 10.1093/clinids/18.6.853. [DOI] [PubMed] [Google Scholar]
- Zenner L, Darcy F, Cesbron-Delauw MF, Capron A. Rat model of congenital toxoplasmosis: rate of transmission of three Toxoplasma gondii strains to fetuses and protective effect of a chronic infection. Infect Immun. 1993;61:360–363. doi: 10.1128/iai.61.1.360-363.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]