

Abstract
HIV-1 transcription is essential for the virus replication cycle. HIV-1 Tat is a viral transactivator that strongly stimulates the processivity of RNA polymerase II (RNAPII) via recruitment of the cyclin T1/CDK9 positive transcription elongation factor, which phosphorylates the C-terminal domain (CTD) of RNAPII. Consistently, HIV-1 replication in transformed cells is very sensitive to direct CDK9 inhibition. Thus, CDK9 could be a potential target for anti-HIV-1 therapy. A clearer understanding of the requirements for CDK9 activity in primary human T cells is needed to assess whether the CDK9-dependent step in HIV-1 transcription can be targeted clinically. We have investigated the effects of limiting CDK9 activity with recombinant lentiviruses expressing a dominant negative form of CDK9 (HA-dnCDK9) in Peripheral Blood Lymphocytes (PBLs) and other cells. Our results show that direct inhibition of CDK9 potently inhibits HIV-1 replication in single-round infection assays with little to undetectable effects on RNAPII transcription, RNA synthesis, proliferation and viability. In PBLs purified from multiple donors, direct inhibition of CDK9 activity blocks HIV-1 replication/transcription but does not prevent T cell activation, as determined via measurement of cell surface and cell cycle entry and progression markers, and DNA synthesis. We have also compared the effects of HA-dnCDK9 to flavopiridol (FVP), a general CDK inhibitor that potently inhibits CDK9. In contrast to HA-dnCDK9, FVP interferes with key cellular processes at concentrations that inhibit HIV-1 replication with potency similar to HA-dnCDK9. In particular, FVP inhibits several T cell activation markers and DNA synthesis in primary PBLs at the minimal concentrations required to inhibit HIV-1 replication. Our results imply that small pharmacological compounds targeting CDK9 with enhanced selectivity could be developed into effective anti-HIV-1 therapeutic drugs.
Keywords: PITALRE (previous designation for CDK9), Transcription, Human T cells, Peripheral Blood Lymphocytes, kinases
1. Introduction
The most common therapeutic method to reduce HIV-1 viral loads is Highly Active Anti-Retroviral Therapy (HAART), which combines drugs that directly target at least two HIV-1 proteins, the reverse transcriptase (RT) and the protease (Pro) (reviewed in Klebl and Choidas, 2006). However, one major problem associated with the current HAART is the appearance of, and selection for novel HIV-1 strains resistant to current antiretroviral drugs. This is because these drugs directly target viral proteins, and thus, become inefficient when HIV-1 mutates. As HIV-1 replicates rapidly, and its RT is prone to errors, mutations in HIV-1 genes encoding for RT or Pro that make these proteins less sensitive to the action of inhibitors are selected rapidly. Thus, there is an urgent need for development of drugs with novel mechanisms of action. In particular, it has become increasingly apparent, that “indirect cellular targets” (non-viral) may represent a solution to this problem (reviewed in Klebl and Choidas, 2006). One potential “indirect target” is the cyclin T1/CDK9 complex.
T-type cyclins (Wei et al., 1998; Peng et al., 1998b) and cyclin K (Fu et al., 1999) interact with CDK9 (Graña et al., 1994) forming distinct complexes termed Positive Transcription Elongation Factor b (P-TEFb) (Marshall and Price, 1995; Peng et al., 1998a). P-TEFb is required for RNAPII transcription in vitro and it has been shown that CDK9 and its associated cyclins are recruited to several promoters in cells. The cyclin T1/CDK9 complex, but not other P-TEFb complexes, is recruited by HIV-1 Tat to the nascent HIV-1 transcript, a step essential for productive transcription of the HIV-1 genome (reviewed in Price, 2000; Garriga and Graña, 2004; Marshall and Graña, 2006).
Previous studies have documented that HIV-1 transcription and replication is very sensitive to inhibition of CDK9 activity using a dominant negative form of CDK9 (dnCDK9) or siRNAs targeted to cyclin T1 and CDK9 (Mancebo et al., 1997; Flores et al., 1999; Chao et al., 2000; Chiu et al., 2004). These studies have been performed by using tumor cell lines such as MAGI and Jurkat cells. It has also been shown that flavopiridol (FVP) inhibits CDK9 activity in vitro with an IC50 of 3 nM (Chao et al., 2000; Chao and Price, 2001), which is significantly lower than its IC50s for other CDKs including CDK4, CDK2, and CDK7 (20-300 nM)(Sedlacek, 2001). FVP inhibits transcription by RNAPII in vitro at the elongation phase and also inhibits HIV-1 Tat transactivation very potently (Chao et al., 2000). Moreover, 300 nM FVP inhibits the rates of RNAPII transcription by approximatelly 70 % in HeLa cells (Chao and Price, 2001). As CDK9 is required for transcription elongation by RNAPII in vitro (Price, 2000) and is so sensitive to FVP inhibition, the potent inhibitory effects of FVP on transcription by RNAPII in cells has lead to the suggestion that CDK9 is required for the transcription of most cellular genes (Chao and Price, 2001). Also, DNA microarray analysis of FVP treated cells showed patterns of altered gene expression similar to those induced by either actinomycin D, an inhibitor of transcriptional initiation, or 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole (DRB), another CDK9 inhibitor, demonstrating that FVP inhibits global transcription (Lam et al., 2001). These data reinforced the suggestion that CDK9 activity is required for transcription of most genes by RNAPII. Importantly, FVP was also shown to inhibit HIV-1 replication in single-round and spread HIV-1 infection assays in Sx22-1 and Jurkat cells, respectively, with IC50s in the low nM range. It was also noted that FVP does not affect the rates of RNAPII transcription in nuclear run on assays of HeLa cells pretreated at these concentration for 1 h (Chao et al., 2000; Chao and Price, 2001).
We and others have previously reported that cyclin T1 expression is upregulated following mitogenic stimulation of human peripheral blood lymphocytes (PBLs), but not transformed T cells (Garriga et al., 1998; Herrmann et al., 1998). Upregulation of cyclin T1 in PBLs correlates with phosphorylation of RNAPII and HIV-1 replication (Garriga et al., 1998) and is coordinated with the expression of other transcriptional regulators (Marshall et al., 2005). T cell activation involves a complex reprogramming of gene expression that leads to the expression of genes required for T cell effector functions, as well as cell cycle entry and proliferation (Huang and Wange, 2004). These coordinated transcriptional programs may require CDK9 activity if CDK9 is required for transcription of most RNAPII genes. Given the low levels of cyclin T1/CDK9 complexes in primary quiescent T cells, it is currently unknown whether inhibition of CDK9 activity in these cells is compatible with T cell activation and how this affects HIV-1 replication. A clearer understanding of the requirements for CDK9 activity in primary human T cells is needed to assess whether the CDK9-dependent step in HIV-1 transcription can be targeted clinically.
With this aim, we have used cell lines to establish conditions to efficiently transduce PBLs with recombinant lentiviruses expressing a dominant negative form of CDK9 (HA-dnCDK9) and determined the effects of directly inhibiting CDK9 activity on T cell activation and HIV-1 replication. We have also compared these effects to those resulting from pharmacologically treating PBLs with FVP. Our results suggest that the effects of dnCDK9 and FVP treatment are not equivalent and suggest that more selective inhibitors of CDK9 could be developed into effective anti-HIV-1 drugs.
2. Materials and Methods
2.1 Cell culture
293T, MAGI and Jurkat cells were grown in Dulbecco’s Modification of Eagle’s Medium (DMEM; Cellgro) supplemented with heat inactivated 10% FBS (Cellgro). PBMCs were isolated as described previously with some modifications (Garriga et al., 1998). PBLs were obtained from PBMCs after monocyte depletion by plastic adherence and resuspended at a concentration of 2 × 106 cells/ml. PBLs were cultured in 24 well cluster plates prior to transduction with lentiviruses or treatment with inhibitors (described below) at 37°C and 5% CO2. Following infection/treatment, PBLs were activated with 1 μg/ml PHA (Sigma-Aldrich) and 1 ng/ml PMA (Sigma-Aldrich). For long-term activation experiments, cells were stimulated with 1 μg/ml PHA, γ-irradiated feeder PBMCs (6 Grays), and 20 U/ml human IL-2 (Roche). For proliferation curves, 293T cells were seeded at 0.5 × 106 cells/10 cm dish and cell number was counted daily using a Neubauer hemocytometer. 293T cell lines stably expressing HA-dnCDK9, HA-CDK9, cyclin T1 or no transgene were generated following transduction with lentiviral constructs expressing each transgene and a puromycin resistance gene. Two days following transduction cells were selected in the presence of puromycin (1 μg/ml).
2.2 Protein assays
Cells were lysed in lysis buffer (50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 250 mM NaCl, 50 mM NaF, 0.1% Triton X-100, 0.1 mM sodium vanadate, 1 mM PMSF, 10 μg/ml leupeptin, 4 μg/ml aprotinin, and 40 μg/ml pepstatin). For Western blot analysis, 10 μg of protein was resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane in 10 mM CAPS (pH 11.0) containing 10% methanol. The following antibodies were used: anti-cyclin T1 (SC-10750), CDK9 (SC-484), p107 (SC-318), Cyclin A (SC-596), and Mcl-1 (SC-819) (Santa Cruz Biotechnology); PARP-1 (51-6639GR) (Becton and Dickinson, BD); ERK1/2 (9102) (Cell signaling); and RNAPII 8wg16, RNAPII H5 (Ser-2 specific) and RNAPII H14 (Ser-5 specific) (Covance).
2.3 Lentiviral constructs, recombinant viral production and cellular transduction
pCEIII and pCPP transfer vectors were derived from pHR’-CMV-LacZ (Naldini et al., 1996) as described previously (Hasham and Tsygankov, 2004). PCEIII-HA-CDK9 and PCEIII-HA-dnCDK9 vectors were generated by blunt-end subcloning the NruI/XbaI fragment from pRC-CMV-HA-CDK9 vectors containing the CDK9 cDNAs under the control of a CMV promoter (Garriga et al., 1996a) into a blunt-ended XhoI site in the PCEIII vector. Similarly, a SpeI-BamHI fragment from pCMV2-flag-HEXIM1 was blunt ended and cloned in the PCEIII vector. pCPP-HA-CDK9 and pCPP-HA-dnCDK9 vectors were generated by subcloning EcoRI digested cDNAs into the EcoRI site of the pCPP vector. Lentiviral particles were generated by transient transfection in 293T cells by CaCl2 method as described in (Marshall et al., 2005) with some significant modifications. Briefly, 15 μg of the transfer vector (directing expression of the gene of interest), 10 μg of pCMVΔ8.2 (packaging construct) and 10 μg of the pCMV-VSV-G (envelope) vector (Sena-Esteves et al., 2004) were cotransfected into 293T cells. HXB2 and VSV-G pseudotyped HIV-1 Luciferase (HIV-1-luc) viruses were generated by cotransfecting 15 μg of pNL4-3.Luc.R-E- (Connor et al., 1995; He et al., 1995) and 10 μg of Env plasmid (HXB2 or VSV-G). Supernatants were harvested at 48 and 60 h after transfection and filtered through 0.45 μm filters. Viruses were pelleted by ultracentrifugation at 50,000 × g for 90 min. at 4°C. Pelleted virus was resuspended in sterile PBS containing 0.1% BSA. Viruses were stored at -80°C prior to use.
MAGI and 293T cells were transduced by addition of viral supernatants to target cells in the presence of 8 μg/ml polybrene (Sigma-Aldrich) to improve transduction efficiency and placed at 37°C and 5% CO2. Sixteen h following transduction, cells were washed once with PBS and then fresh media was added. PBLs were infected in 24 well cluster plates. Concentrated viral supernatant (MOI 250) and polybrene (8 μg/mL) was added to 2 × 106 cells and PBLs were spin-inoculated at 244 × g for 90 minutes at 25°C. Cells were then incubated at 37°C and 5% CO2. Transducing units/ml were determined by infecting 293T cells with serial dilutions of GFP-expressing viruses. Titer was determined by counting the number of EGFP-positive cells from 5 independent fields under a fluorescent microscope.
2.4 HIV-1 single-round replication assays in cell lines and PBLs
0.5 × 106 cells (MAGI/Jurkat) were plated in 60 mm dishes. Twenty four h later the cells were infected with the recombinant lentiviruses indicated in the results section. Eight h later, 1 ml of HIV-1-Luc viral supernatant and 1 ml of complete medium containing polybrene (8 μg/ml) was added to the cells and allowed to infect over night. Following infection, cells were washed twice with medium, refed and collected 48 h later for analysis. 293T stable cell lines (HA-dnCDK9 and puro) were similarly infected with HIV-1-Luc. For FVP (Aventis) experiments, cells were treated with various concentrations (10-300nM) of FVP 3 h post-infection with HIV-1-Luc supernatants, for the duration of the experiment. Luciferase Assays (Promega) were carried out according to manufacturer’s standard protocols.
PBLs were transduced with recombinant lentiviruses as described above. Sixteen h later, 2 × 106 PBLs were infected with 50 μl of concentrated HXB2 pseudotyped HIV-1-Luc viruses via spin inoculation. Twenty four h later, the PBLs concentration was adjusted to 1 × 106 cells/ml with fresh media and stimulated with PMA/PHA. When indicated, cells were treated with FVP (10-300 nM) 3 h prior to stimulation with PMA/PHA. Cells were collected at 24 hours for FACS analysis and at 48 h for luciferase assays, and Western blot analysis. Raw luciferase data was normalized to the protein content of the lysates.
2.5 RNA and DNA synthesis and run-on assays
RNA synthesis experiments were performed as described previously (Sano et al., 2002). Briefly, 1 × 106 cells were incubated for 16 h with 5 μCi/ml of [3H]-uridine (Perkin-Elmer), in the absence or presence of FVP. Total RNA and DNA were purified using Tripure reagents as per manufacture directions (Roche). [3H]-uridine incorporation in RNA was determined 16 h later using a liquid scintillation counter and values were normalized vs. DNA content.
For DNA synthesis assays, 2 × 106 transduced/treated PBLs were seeded in 24 well cluster plates at a concentration of 1 × 106 cells/ml. Cells were stimulated with PMA/PHA as described above. Twenty four h post-activation, 5 μCi/ml [3H]-thymidine was added to the wells. Incorporation of labeled nucleotide was allowed to take place for 24 h. Cells were collected 48 h post-stimulation and lysed in 0.3 M NaOH. After addition of an equal volume of 20% Trichloro acetic acid (TCA), DNA was immobilized on Whatman filter paper (1001-325), washed twice with 10% TCA and once with 95% ethanol using a vacuum flask. Thymidine incorporation was quantitated using a liquid scintillation counter. Values were normalized to cell number.
For nuclear run-on assays, 293T cells were grown to 75% confluency in 15 cm diameter dishes (approx. 45 × 106 cells) and then treated with 300 nM FVP or medium for 1 h. Run on assays were performed essentially as described in (Chao and Price, 2001).
2.6 Flow Cytometry/Flow Assisted Cell Sorting
For flow cytometric analysis of surface markers, 500,000 PBLs were collected 24 h post mitogenic stimulation. PBLs were washed twice with PBS and stained for 20 min with FITC, PE or APC conjugated antibodies to CD25, CD69, or CD62L (BD). PBLs were washed once with PBS and fixed in PBS containing 2% paraformaldehyde (pH 7.4) for 15 min. Following fixation, cells were washed once with PBS and resuspended at a concentration of 1 × 106 cells/ml. Fluorescence profiles were then acquired on a FacsCalibur (BD) flow cytometer. Data was analyzed using CellQuest Pro (BD). For Cell Sorting, transduced PBLs were collected in 15 ml falcon tubes and washed once with RPMI. Cells were resuspended in RPMI containing 5% FBS at a concentration of 30 × 106 cells/ml. Cells were then sorted by GFP fluorescence using a Cytomation MoFlo (Cytomation, Inc., Fort Collins, CO) cell sorter. Sorted PBLs were resuspended in RPMI containing 10% heat-inactivated FBS at a concentration of 1 × 106 cells/ml. Following viral transduction and sorting, cells were activated and analyzed by flow cytometry as described above.
3 Results
With the ultimate goal of transducing PBLs, we generated two series of recombinant lentiviruses. One series directed coexpression of a puromycin resistance gene and either HA-tagged CDK9, dnCDK9, or cyclin T1, while the second series directed coexpression of EGFP and either HA-tagged dnCDK9 or Flag-tagged hexim1 (Fig. 1A). Recombinant lentiviruses were generated in 293T cells via triple transfection of the vectors depicted in Fig. 1A as described in the Methods section. Prior to transduction of human PBLs, these lentiviral vectors were characterized in a variety of tumor cell lines for their ability to affect cell proliferation and viability, cellular transcription as well as HIV-1 transcription/replication via single-round HIV-1 infection assays.
Fig. 1. Generation of recombinant lentiviruses and HIV-1 reporter viruses.
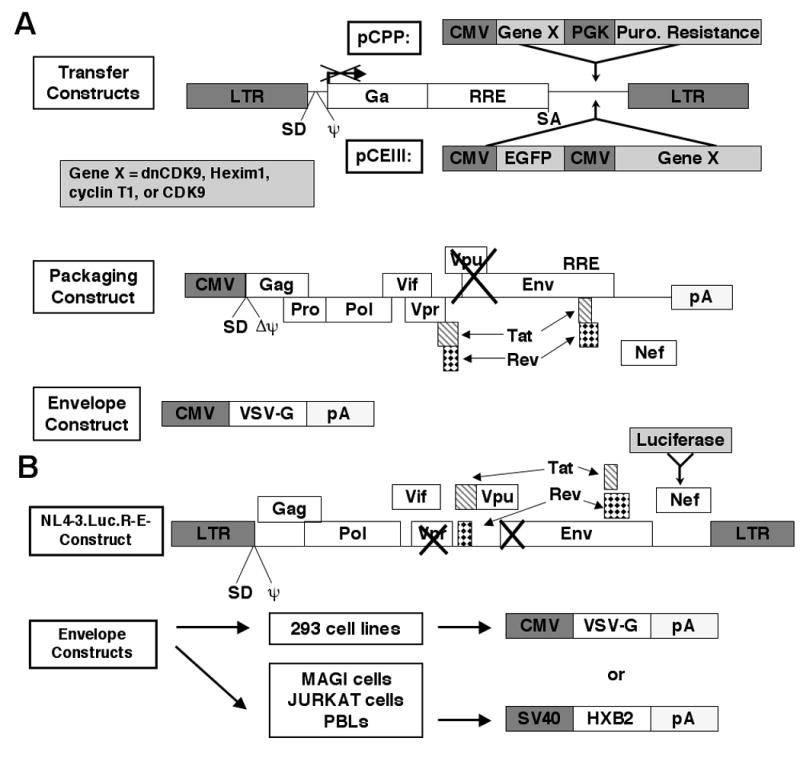
(A) For lentiviral production, 293T cells are transfected via the calcium phosphate method, with a packaging construct that directs the expression of viral proteins minus the Env and Vpu, a plasmid encoding the VSV-G envelope and a transfer plasmid that contains the genes of interest under the control of CMV promoters (expression cassettes) similarly as described previously (Naldini et al., 1996; Hasham and Tsygankov, 2004). In the transfer constructs (pCPP or pCEIII), the indicated expression cassettes are flanked by HIV-1 LTRs and contain the packaging signal, as well as other indicated elements required for efficient lentiviral packaging. Supernatants are collected 48 and 60 hours later and concentrated by ultracentrifugation. cDNAs encoding HA-dnCDK9, HA-CDK9, and HA-cyclin T1 were subcloned in pCPP. Additionally, HA-dnCDK9 and Flag-Hexim1 were subcloned in pCEIII transfer vectors as described in the Methods section. Gene X represents HA-dnCDK9, HA-CDK9, HA-cyclin T1 or Flag-Hexim1. (B) HIV-1-luc viruses pseudotyped with either HXB2 or VSV-G envelopes are generated by cotransfection of the HIV-1 molecular clone mutant NL4-3.Luc.R-E- and the indicated envelope plasmids in 293T cells. These viruses are fully infectious and are replication defective as they exhibit a mutation in the Env and Vpr genes. Luciferase expression is directly proportional to transcription of full-length HIV-1 genome and, thus, reflects replication (Connor et al., 1995; He et al., 1995).
3.1 Stable expression of a dominant-negative form of CDK9 inhibits HIV-1 transcription in 293T cells without effects on cellular RNA synthesis
It has previously been shown that P-TEFb activity is rate-limiting for HIV-1 replication in transformed Jurkat cells expressing a dnCDK9 mutant that inhibited endogenous CDK9 activity by approximately 50% (Flores et al., 1999). Under these conditions, cell viability was reported to be unaffected. Incubation of HeLa cells with flavopiridol (FVP), a potent inhibitor of CDK9, results in potent inhibition of RNAPII transcription at a concentration of 300 nM (Chao and Price, 2001). FVP was also shown to inhibit HIV-1 replication at much lower concentrations in Sx22-1 and Jurkat cells (Chao et al., 2000). To directly compare the effects of inhibiting CDK9 activity in both cellular and HIV-1 transcription, as well as cell viability in a single cell type, we generated 293T cells stably expressing HA-dnCDK9, HA-CDK9, HA-cyclin T1, as well as control puromycin resistant cells. Stable cell clones were generated by lentiviral infection followed by puromycin selection as described in Fig 1 and 2 and named according to the ectopically expressed gene, i.e: HA-dnCDK9, HA-CDK9, HA-cyclin T1 and puro (empty vector) cells. Fig. 2A shows expression of HA-dnCDK9 in one representative clone vs. puro cells (lower panel). Expression of HA-dnCDK9 leads to clear downregulation of endogenous CDK9 protein expression, an effect previously observed when wild type CDK9 is ectopically expressed (Garriga et al., 1996a). Downregulation is due to rapid degradation of monomeric CDK9, which cannot form stable complexes with rate-limiting cyclin T subunits (O’Keeffe et al., 2000; Garriga et al., 2003; Chiu et al., 2004). To determine if HA-dnCDK9 reduces cellular CDK9 activity, anti-CDK9 immunoprecipitates from puro and HA-dnCDK9 whole lysates were incubated with bacterially expressed GST-RNAPII-CTD in in vitro kinase reactions. Fig. 2A shows dramatically reduced RNAPII CTD phosphorylation (upper panel) and CDK9 autophosphorylation (middle panel) in CDK9 complexes immunoprecipitated from HA-dnCDK9 expressing cells as compared to control puro cells. We next determined the effects of HA-dnCDK9 overexpression on the phosphorylation of Ser-2 on the heptad repeats of CTD of RNAPII via Western blot analysis. Fig. 2B shows noticeable downregulation of RNAPII phosphorylation of Ser-2 in vivo without changes on RNAPII expression (Fig 2B, compare middle to upper panel). Thus, inhibition of cellular CDK9 is accompanied by a marked reduction on Ser-2 phosphorylation. Next, we determined the effect of HA-dnCDK9 on cellular proliferation. Fig. 2C shows a comparison of the increase in cell number over time for the indicated cell lines. No significant differences in doubling times were observed between control cells and cells expressing HA-dnCDK9, HA-CDK9 or HA-cyclin T1. Also, HA-dnCDK9 cells have been cultured for several months with no obvious loss of cell viability or proliferative potential. Thus, while overexpression of HA-dnCDK9 in 293T cells leads to decreased phosphorylation on Ser-2 of the CTD of RNAPII, this does not lead to an overall inhibition of cell growth in these cells. Subsequently, we determined the effects of CDK9 inhibition on cellular transcription using both run-on and RNA synthesis assays. In the run-on assay, labeled nucleotide is incorporated into the elongating transcript over a 30 min period. α-amanitin (2 μg/ml), a known RNAPII transcription inhibitor, was used as a control for RNAPII mediated transcription. Interestingly, no differences in transcriptional elongation were observed between parental 293T cells and cells ectopically expressing HA-dnCDK9 (Fig. 2D) or HA-CDK9 (data not shown). In contrast, treatment of control 293T cells with 300 nM FVP for 1 h inhibited global transcriptional elongation by approximately 60 %, in agreement with a previous report using HeLa cells (Chao and Price, 2001). This result was surprising, as the FVP effect on transcription in HeLa cells was attributed to specific inhibition of P-TEFb activity. To confirm these results with a different assay, we determined cellular RNA synthesis in HA-dnCDK9, HA-CDK9 and puro cells, as well as puro cells treated with FVP at the indicated concentrations for 17 hrs. Fig. 2E confirms the results of the run-on assay, showing that FVP, but not the expression of HA-dnCDK9, inhibits cellular RNA synthesis. Obviously, one interpretation of these results is that FVP may inhibit cellular CDK9 more potently than HA-dnCDK9. This is conceivable, as HA-dnCDK9 potently inhibits CDK9 activity but does not fully eliminate it (Fig. 2A). To address this possibility, we compared the effects of HA-dnCDK9 expression and FVP treatment on HIV-1 transcription by using a single-round HIV-1 infection assay with VSV-G pseudotyped HIV-1 reporter virions (HIV-1-luc), which express a luciferase reporter gene. These viruses were generated using a HIV-1 pNL4.3-R-E- molecular clone and a VSV-G envelope (see Fig. 1B), as previously described (Connor et al., 1995; He et al., 1995). The luciferase reporter gene is inserted into the viral Nef gene and, thus, luciferase activity reflects LTR dependent transcription and subsequent transcript processing and expression. However, upon infection no viable viral particles are generated as the HIV-1 Env and Vpr gene products are not expressed (Connor et al., 1995; He et al., 1995). HA-dnCDK9 or puro cells treated with increasing concentrations of FVP were infected with VSV-G pseudotyped HIV-1-luc viruses and collected 48 h later. Luciferase assays demonstrated that HIV-1 transcription was inhibited at comparable levels in HA-dnCDK9 cells and in puro cells treated with 100 nM FVP, but not at lower concentrations of FVP. Thus, in 293T cells the concentration of FVP associated with HIV-1 transcriptional inhibition has effects on cellular RNA synthesis, while HA-dnCDK9 expression inhibits HIV-1 transcription/replication without apparent effects on cellular transcription. This was further explored in MAGI cells as described below.
Fig 2. Direct inhibition of CDK9 activity with HA-dnCDK9 in 293T cells inhibits RNAPII phosphorylation on CTD Ser-2 and HIV-1 transcription, but fails to affect the overall rates of cellular transcription and cell proliferation and viability.
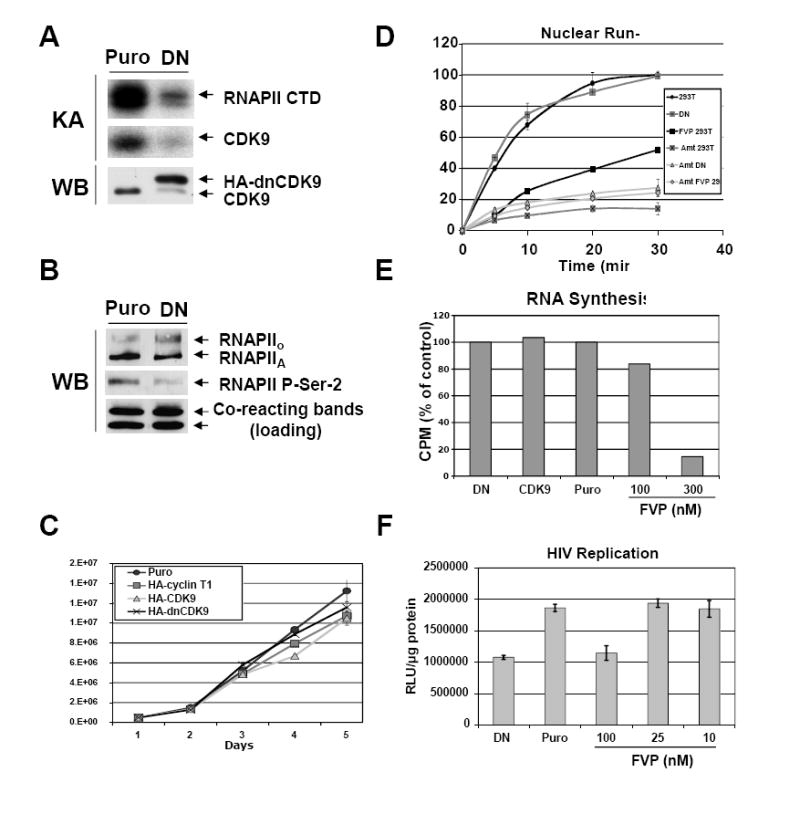
293 cells stably expressing HA-dnCDK9, HA-CDK9 or HA-cyclin T1 and control puromycin resistant (Puro) cells were generated by lentiviral infection followed by selection with puromycin. Lentiviruses were generated as described in Fig 1A and the Methods section using the corresponding pCPP transfer construct for each transgene. Clones expressing the highest transgene levels were selected for further experiments. (A) Ectopic expression of dnCDK9 inhibits CDK9-associated kinase activity. HA-dnCDK9 and endogenous CDK9 expression was determined by Western blot analysis of whole cell lysates of control (Puro) and HA-dnCDK9 (DN) expressing cells (WB). Cellular CDK9 was immunoprecipitated with anti-CDK9 antibodies. Kinase reactions were performed by incubating the immunoprecipitates with GST-RNAPII-CTD (exogenous substrate) as described in the Materials and Methods section. RNAPII-CTD phosphorylation and CDK9 autophosphorylation is shown (KA). (B) Ectopic expression of HA-dnCDK9 inhibits phosphorylation of RNAPII on Ser-2 in vivo. The effects of HA-dnCDK9 expression on RNAPII Ser-2 phosphorylation were determined by Western blot analysis. The expression of total RNAPII is also shown. Cross-reacting bands detected with the anti-Ser-2 antibody are shown as loading control. (C) Cell proliferation and viability of exponentially growing cells was measured by trypan blue staining and cell counting. No significant increase in cell death was detected in cells expressing HA-dnCDK9, HA-CDK9 or HA-cyclin T1. (D) The rates of cellular transcription were determined via run-on assays in 293T and HA-dnCDK9 (DN) cells as described in the Methods section. The effects of FVP (300 nM) and/or α-amanitin (Amt) were also determined where indicated. (E) RNA synthesis in HA-dnCDK9 (DN), HA-CDK9, Puro cells and Puro cells treated with the indicated concentrations of FVP was determined as by measuring [3H]-uridine incorporation into cellular RNA as described in the Methods section. (F) Single-round HIV-1 transcription assays were performed by infecting HA-dnCDK9 (DN), Puro and FVP treated Puro cells with HIV-1-luc viruses pseudotyped with a VSV-G envelope. HIV-1 transcription was measured by determining luciferase activity 48 h post infection in duplicate samples (see Methods section).
3.2 Transient expression of a HA-dnCDK9 inhibits HIV-1 replication in Jurkat and MAGI (CD4+/CCR5+ HeLa) cells with little effect on cellular RNA synthesis
Jurkat and HeLa tumor cell lines have been previously utilized to assess the effects of CDK9 inhibition on HIV-1 transcription, replication and/or cellular transcription (Flores et al., 1999; Chao et al., 2000; Chao and Price, 2001; Chiu et al., 2004). However, the effects on cellular transcription and HIV-1 transcription/replication have not been compared side-by-side using the same cells. Also, both Jurkat and MAGI (Chackerian et al., 1997) cells exhibit receptors that allow infection with HIV-1 viruses carrying an HIV-1 envelope. Thus, these two cell lines were also used as a first step to establish assays for subsequent experiments using primary human PBLs. In the experiments that follow, cells were transduced with EGFP, EGFP/HA-dnCDK9 or EGFP/Flag-Hexim1 lentiviruses for 24 h (lentiviruses generated as described in Fig. 1A), and then challenged with HXB2 envelope pseudotyped HIV-1-luc viruses (generated as described in Fig. 1B). Cells were collected 48 h later to determine HIV-1 transcription/replication, cellular RNA synthesis and/or for analysis of various functional cellular markers via Western blot. Fig. 3A and 3B show the effects of inhibiting CDK9 activity on single-round HIV-1 infection assays in MAGI and Jurkat cells respectively. Compared to EFGP expressing control cells, expression of HA-dnCDK9 lead to inhibition of HIV-1-luc transcription/replication. To corroborate the effects of CDK9 inhibition by an independent mechanism, we expressed Flag-Hexim1. Hexim1 is a physiological inhibitor of CDK9 that forms inactive complexes with CDK9/cyclin T dimers and the 7SK snRNA (reviewed in Garriga and Graña, 2004; Marshall and Graña, 2006). Expression of Flag-Hexim1 potently inhibited HIV-1-luc transcription/replication (Fig 3A and 3B). We next performed a dose-dependent analysis of the effects of both FVP and HA-dnCDK9 on HIV-1 replication and cellular function markers in MAGI cells. MAGI cells were infected with either EGFP or EGFP/HA-dnCDK9 lentiviruses at increasing MOIs (10-100). At an MOI of 10, there was no difference in HIV-1 replication between EGFP and HA-dnCDK9 transduced cells (Fig. 3C). However, at an MOI of 60, expression of HA-dnCDK9, but not EGFP, potently inhibited HIV-1 replication. The effects on HIV-1 replication correlated with the expression of HA-dnCDK9, as the expression of HA-dnCDK9 was below the levels of endogenous CDK9 at an MOI of 10 (Fig. 3D). In contrast, at an MOI of 60, HA-dnCDK9 reached very high levels, which resulted in downregulation of endogenous CDK9 and concomitant inhibition of RNAPII Ser-2 phosphorylation (Fig. 3D). At a 100 MOI, there was further inhibition of HIV-1 replication (Fig. 3C). However, non-specific lentiviral effects started to be noticeable, as some inhibition of HIV-1 replication was observed in EGFP transduced MAGI cells (Fig. 3C). This might be due to transcriptional squelching due to the presence of CMV promoters in the lentiviral constructs. FVP dose-dependent effects were measured in MAGI cells transduced with EGFP expressing lentiviruses at an MOI of 60 prior to treatment, in order to compensate for potential effects due to viral transduction. FVP had no effect on HIV-1 replication at a concentration of 25 nM or less (Fig. 3C). Significant inhibition of HIV-1 replication was observed at a 100 nM, but it was two fold less potent than HA-dnCDK9 (60 MOI). 200 nM FVP potently inhibited HIV-1 replication, but this was accompanied by cell death, as determined by increased detection of poly ADP ribose polymerase (PARP) cleavage (Fig. 3D), which is a hallmark of apoptosis and microscopic observation (data not shown). The comparison of the effects of HA-dnCDK9 at an MOI of 60 and FVP at a concentration of 100 nM allowed us to observe a striking difference on inhibition of Ser-5 phosphorylation of the CTD of RNAPII (Fig. 3D). Ser-5 phosphorylation is thought to be mediated by CDK7 during transcription initiation (Marshall and Graña, 2006). While 100 nM FVP inhibited Ser-5 phosphorylation very potently, HA-dnCDK9 inhibited Ser-2, but not Ser-5, phosphorylation in cells transduced at an MOI of 60. Of note EGFP control lentiviruses did not affect RNAPII phosphorylation at this MOI (at a 100 MOI, we observed a slight reduction on Ser-5 phosphorylation in both EGFP and dn-CDK9 transduced cells indicating non-specific lentiviral effects at high MOIs). As an indirect measurement of general transcription, we determined the expression of Mcl-1. Mcl-1 expression is inducible and rapidly declines when transcription is inhibited (Lam et al., 2001). Fig. 3D shows dose-dependent downreguation of Mcl-1 expression in cells treated with FVP. In contrast, HA-dnCDK9 did not affect Mcl-1 expression, as there is no apparent difference on Mcl-1 proteins levels between dnCDK9 and EGFP expressing cells. Taken together, these results suggest that the potent inhibitory effects of FVP in cellular transcription are due to inhibition of at least one other CTD kinase.
Fig. 3. Direct inhibition of CDK9 with dnCDK9 or Hexim1 is as efficient as FVP in inhibiting HIV-1 replication in single-round infection assays, but dnCDK9 is not a potent inducer of apoptosis and does not inhibit cellular RNA synthesis.
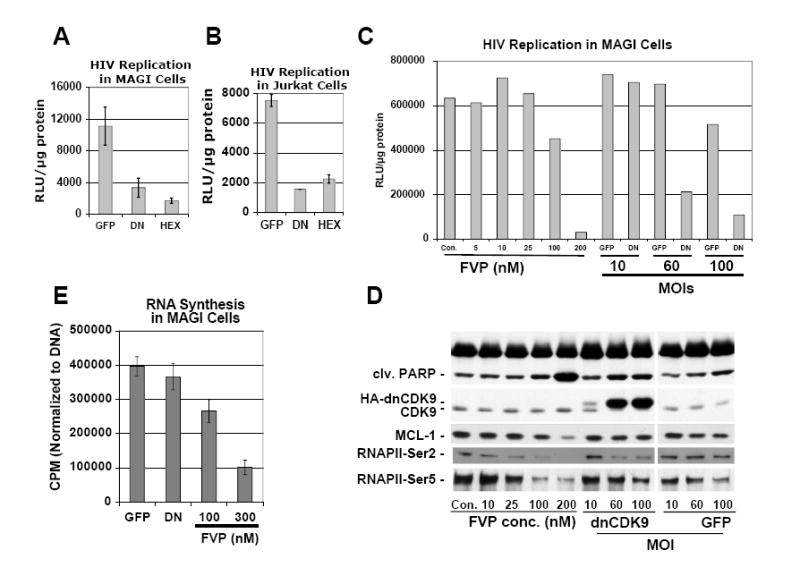
(A and B) Effects of inhibition of CDK9 activity via transduction with HA-dnCDK9 (DN), Flag1-Hexim1 (HEX) or control (EGFP) lentiviruses on HIV-1 replication by single-round HIV-1 infection assays in MAGI (A) and Jurkat (B) cells. C MAGI cells transduced with the indicated lentiviruses were infected with HIV-1-luc viruses pseudotyped with a HXB2 envelope. All cells were harvested 48 hours post-infection. HIV-1 replication was determined by measuring luciferase activity in duplicate samples. (C and D) Comparison of dose-dependent effects of FVP and increased transduction with HA-dnCDK9 (DN) or control (EGFP) lentiviruses (increased MOIs) on (C) HIV-1 replication (as in A) and (D) cellular markers of transcription and apoptosis in MAGI cells via Western blot analysis. (E) Effects of transduction with HA-dnCDK9 lentiviruses or FVP in RNA synthesis in duplicate samples. RNA synthesis was determined as described in Fig. 2E.
Finally, the effects of ectopically expressing HA-dnCDK9 on cellular RNA synthesis were determined in MAGI cells. As in 293T cells, we observed no significant inhibition of RNA synthesis following expression of HA-dnCDK9 as compared to EGFP expression (Fig 3E). In contrast, FVP inhibited RNA synthesis at the concentration needed to inhibit HIV-1 replication (compare Fig. 3C with 3E).
3.3 FVP inhibits mitogen-induced T cell activation in primary human PBLs
CD4+ T cells are a primary target for HIV-1 infection and are central to the pathogenesis of this virus. As the expression of cyclin T1 and CDK9 is low in unstimulated PBLs and increases following mitogenic stimulation, we have suggested that the activity of these complexes might be important for the increased transcriptional needs associated with the process of T cell activation (Marshall et al., 2005). In contrast, in transformed T cells and other cell lines, cyclin T1 levels are comparable to those seen in activated PBLs. Hence, the importance of performing these experiments using PBLs. Thus, to begin examining the effects of inhibiting CDK9 activity during T cell activation we stimulated human PBLs purified from healthy donors with PMA (Phorbol 12-Myristate, 13-Acetate) and PHA (Phytohemagglutinin) as previously described (Garriga et al., 1998; Marshall et al., 2005) in the presence of the indicated concentrations of FVP and measured markers of T cell activation by flow cytometric and Western blot analyses. To allow comparison with transduced PBLs (see below), PBLs used in this experiment were transduced with control lentiviruses expressing EGFP 12 hrs prior to PMA/PHA stimulation. Nevertheless, we obtained similar results using untransduced PBLs (data not shown). Fig. 4A (upper panel) shows a striking decrease in the number of cells expressing the alpha subunit of the IL-2 receptor (CD25 positive cells) in the presence of FVP (24 h post-stimulation). A reduction in the number of positive cells is detectable at concentrations of FVP as low as 30 nM in multiple donors (Fig 4A and data not shown). The lower panel in Fig. 4A shows that mean CD25 fluorescence values, a measurement of its mean surface expression, is gradually downregulated as the concentration of FVP increases. A significant decrease in the expression of the C-type lectin CD69, an early T-cell activation marker, was also observed using concentrations of FVP of 100 nM (Fig. 4B, lower panel), albeit the number of CD69 positive cells was only reduced by 300 nM FVP (Fig. 4B, upper panel). Finally, the expression level of L-selectin (CD62L), a lymphocyte homing surface marker expressed at high levels in quiescent lymphocytes, was also significantly downregulated by FVP at concentrations of 100 nM in quiescent cells (Fig. 4C, lower panel). However, CD62L downregulation following mitogenic stimulation, which is due to early endoprotease-mediated cleavage was not prevented by FVP (Fig. 4C, upper panel). This suggests that at least certain early events associated with T cell activation are not inhibited by FVP. Western blot analysis performed with protein lysates of PBLs harvested 48 h post-mitogenic stimulation, demonstrated that FVP sharply affects the expression of the E2F-dependent p107 gene product, which is upregulated during late G1 and S phase of the cell cycle (cell cycle entry marker), at concentrations as low as 30 nM (Fig. 4D). FVP also potently downregulated Mcl-1, the product of a short-lived transcript (Mcl-1 downregulation reflects inhibition of transcription (Lam et al., 2001)). As compared to mock treated PBLs, FVP increased PARP cleavage (apoptotic marker) in both quiescent and stimulated PBLs even at low concentrations (30-50 nM). Moreover, concentrations of 100-300 nM FVP lead to cell death (microscopically observed, data not shown) and growth inhibition (reduced protein mass, data not shown). Finally, we also determined the direct effects of FVP on DNA synthesis 48 h following PMA/PHA stimulation. Fig. 4E shows that concentrations of FVP as low as 30 nM inhibit thymidine incorporation. Taken together, these results show that FVP potently inhibits T cell activation at high concentrations (100-300 nM). These effects are compatible with a shutdown in general transcription, similar to what is observed in transformed cell lines (Figs. 2 and 3). Surprisingly, FVP also exhibited effects at concentrations that have been previously found to be compatible with cell viability in tumor cell lines (Blagosklonny, 2004).
Fig. 4. Treatment of PBLs with FVP inhibits the expression of markers of T cell activation, cell cycle entry and transcription as well as DNA synthesis.
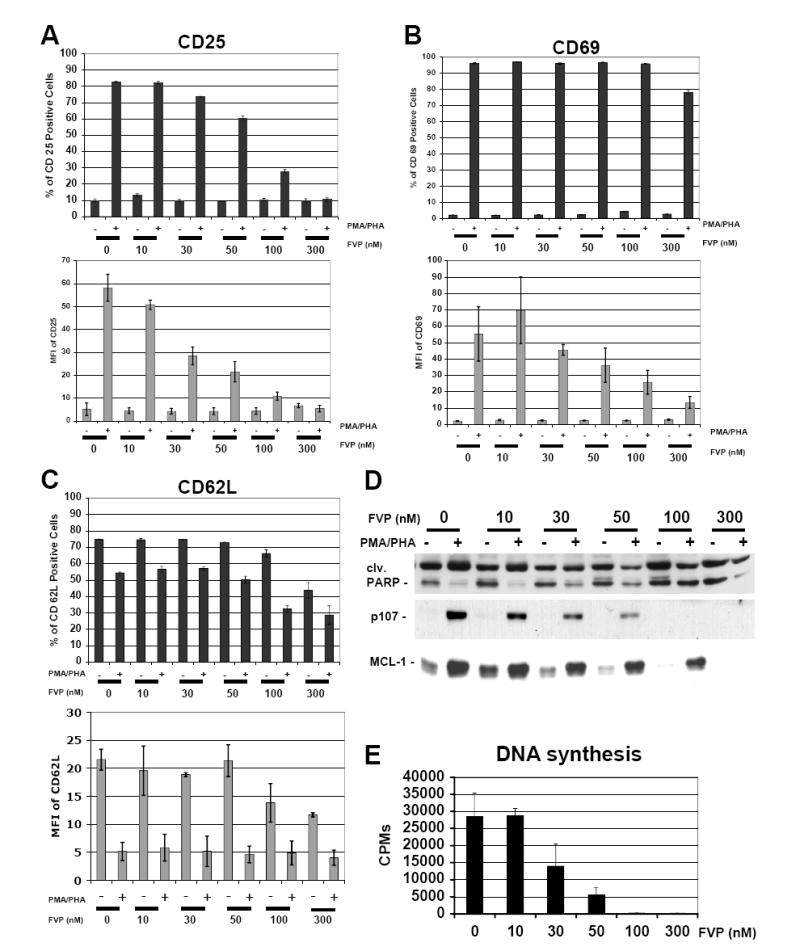
FVP inhibits the expression of the T cell activation markers CD25, CD62L and CD69 (A-C). T cell surface markers were measured 24 hours following mitogenic stimulation by flow cytometric analysis. PBLs were treated as described in the Methods section. All treatments were done in triplicate. Bars on upper panels represent relative numbers of positive cells, whereas lower panels represent the mean fluorescent intensity corresponding to each marker. Experimental variability is represented by error bars. Similar results were obtained using PBLs obtained from other donors. (D) The expression of cell cycle entry (p107), transcription (Mcl1) and apoptosis (PARP cleavage) markers was determined by Western blot analysis of whole cell lysates of PBLs collected 48 h following mitogenic stimulation. (E) FVP inhibits DNA synthesis. DNA synthesis was determined by measuring incorporation of [3H]-thymidine into DNA for 24 hrs. [3H]-thymidine was added 24 hrs post mitogenic stimulation. All treatments were done in duplicate.
3.4 Direct inhibition of CDK9 activity in PBLs by HA-dnCDK9 inhibits HIV-1 replication and appears compatible with T-cell activation
Based on published work linking inhibition of CDK9 by FVP and inhibition of transcription (Chao and Price, 2001; Lam et al., 2001), the effects of FVP on multiple markers associated with T cell activation, transcription and cell viability as well as inhibition of DNA synthesis shown in Fig. 4 could suggest that inhibition of CDK9 activity is incompatible with T cell activation. To test this notion, we investigated whether direct inhibition of CDK9 using HA-dnCDK9 had effects similar to those induced by FVP. Fig. 5 shows a representative experiment in which transduced PBLs were sorted by their associated EGFP fluorescence. Transduction with recombinant lentiviruses coexpressing EGFP and HA-dnCDK9 did not alter CD25 expression, in both sorted and unsorted mitogenically stimulated cells as compared to PBLs transduced with EGFP control lentiviruses. Fig. 5A shows the percent of CD25 positive cells found in each of the indicated cell fractions. In addition, mean fluorescent CD25 expression values were not altered by the expression of HA-dnCDK9 (data not shown). Also, HA-dnCDK9 expression did not affect the number of CD69 positive cells or mean fluorescent CD69 expression values (data not shown). Transduction with HA-dnCDK9/EGFP lentiviruses did not alter the expression of cell cycle entry (p107) and S phase progression (cyclin A) markers in both sorted positive and unsorted cells (Fig. 5B). Similarly, HA-dnCDK9 did not affect the expression of Mcl1, a marker of inhibition of transcription. Importantly, HA-dnCDK9 potently inhibited HIV-1 replication in single-round replication assays (Fig. 5C). In contrast, under the same conditions FVP exhibited effects on the expression of CD25 (Fig 5A), even at concentrations in which it was not as effective as HA-dnCDK9 in inhibiting HIV-1 replication (i.e.: 30 nM, Fig. 5C). With this particular donor, we did not observe clear effects in the expression of cell cycle markers in PBLs treated with 30 nM FVP (Fig. 5B). However, as a consequence of cell death and growth inhibition, protein was limiting for the analysis of PBLs treated with 100 nM FVP, which is consistent with the cellular toxicity observed with PBLs from all donors at this FVP concentration (Figs. 4 and 5, and data not shown). Collectively, these experiments suggest that the toxic effects of FVP are unlikely to be due to the sole inhibition of CDK9. These data have been confirmed by using PBLs obtained from multiple donors with very similar results. Fig 6A shows that HA-dnCDK9 expression inhibits HIV-1 replication in PBLs from multiple donors. We have also compared the effects of FVP and HA-dnCDK9 on HIV-1 replication over a 5-day time course (Fig. 6B). PBLs were infected with the indicated lentiviruses and/or treated with FVP (10-300nM) and then stimulated with irradiated PBMCs, IL-2 (20U/ml) and PHA (1 ug/ml). Samples were collected at 2 and 5 days post activation. HIV-1 replication was dramatically lower in HA-dnCDK9 transduced cells as compared to controls (Fig. 6B). FVP treatment also inhibited HIV-1 replication. However, lower concentrations of FVP (10-30 nM) were unable to inhibit HIV-1 replication as effectively as HA-dnCDK9. Moreover, to rule out the possibility that inhibition of CDK9 with HA-dnCDK9 was inhibiting HIV-1 replication indirectly by limiting a non-obvious step during T cell activation, we stimulated PBLs with PMA/PHA 24 hrs prior to transduction with HA-dnCDK9 or EGFP lentiviruses. This timing allows for expression of T cell surface markers and entry into the G1 phase of the cell cycle. In fact, considering the time needed for expression of the transgenes, T cell activation has progressed further before any effects due to HA-dnCDK9 are possible. Forty eight hrs following PMA/PHA stimulation, the transduced PBLs were challenged with HIV-1-luc and PBLs were collected 96 hrs post mitogenic stimulation. Fig. 6C shows clear inhibition of HIV-1 replication with the expression of HA-dnCDK9 in preactivated PBLs. The results shown in Figs 6B and 6C suggest that expression of HA-dnCDK9 is effective at inhibiting HIV-1 replication in cycling PBLs. An additional control experiment was performed to rule out the possibility that transduction with recombinant lentiviral vectors could have an inhibitory effect on HIV-1 infection as well as on other early events associated with the HIV-1 cycle. PBLs were first infected with HIV-1-luc overnight, and subsequently infected with EGFP or HA-dnCDK9 lentiviruses. 16 h later, PBLs were activated with PMA/PHA. PBLs were collected and examined by luciferase assay analysis 48 h following stimulation. As expected, HIV-1 replication was inhibited in PBLs transduced with HA-dnCDK9 as opposed to cells transduced with EGFP lentivirus (Fig. 6D). Thus, the inhibition of HIV-1 replication observed in cells expressing HA-dnCDK9 is not due to non-specific inhibition of HIV-1 infection by the recombinant lentiviruses. Finally, to determine the effects of direct inhibition of CDK9 on DNA synthesis, we performed thymidine incorporation assays. Fig. 6E shows that DNA synthesis was comparable in both EGFP and HA-dnCDK9 transduced PBLs stimulated with PMA/PHA. In contrast, FVP clearly inhibited DNA synthesis even at 30 nM (Fig. 6E) in agreement with the result shown in Fig. 4E.
Fig. 5. Expression of dnCDK9 in PBLs inhibits HIV-1 replication in single round assays, but does not affect T cell activation as determined by the expression of T cell activation and cell cycle markers.
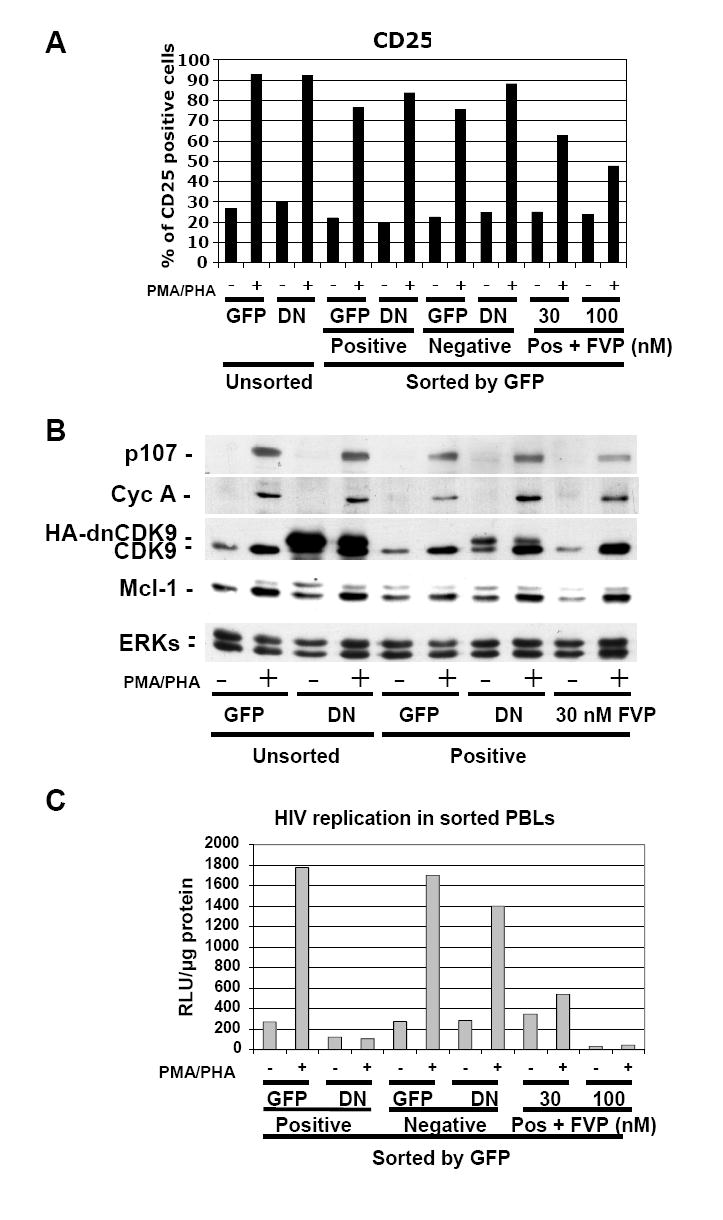
PBLs were transduced with HA-dnCDK9/EGFP (DN) or EGFP (GFP) lentiviruses and sorted 48 hrs later to separate a population of cells enriched with EGFP expressing cells (Positive) from a population with no detectable EGFP expression (Negative). Sorted EGFP-transduced control PBLs were also treated with FVP as indicated (Pos + FVP). Sorted and unsorted PBLs were then stimulated with PMA/PHA and processed for analysis of CD25 surface marker expression (A) or WB (B) analysis as in Fig. 4. In contrast to FVP, HA-dnCDK9 does not affect CD25 expression at the T cell surface (A) or cell cycle markers (B). (C) The cellular fractions described above were infected with HXB2 pseudotyped HIV-luc viruses and HIV-1 transcription/replication was determined at 48 hrs post-stimulation by measuring luciferase activity. HA-dnCDK9 inhibits HIV-1 replication more potently that 30 nM FVP without the cellular effects associated with FVP.
Fig. 6. Direct CDK9 activity downregulation inhibits HIV-1 replication in multiple donors, over extended time periods and independently of the T cell activation stage.
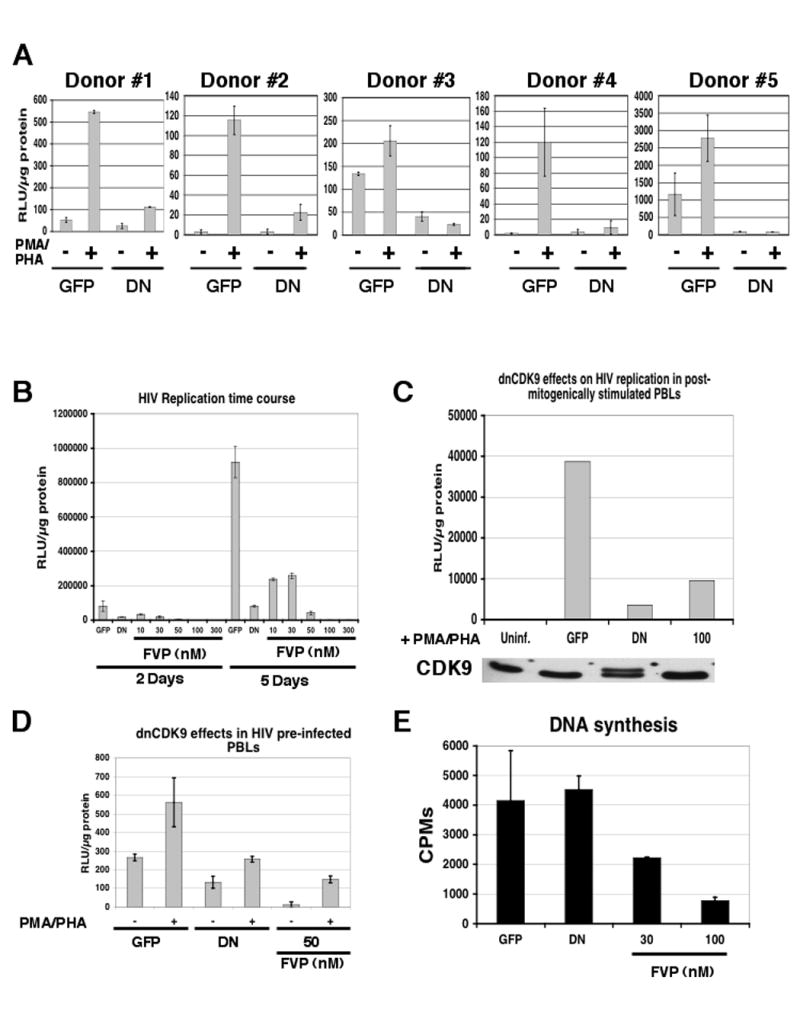
(A) dnCDK9 inhibits HIV-1 replication in PBLs from multiple donors (duplicate samples). (B) comparison of the effects of FVP and inhibition of CDK9 activity via transduction with HA-dnCDK9 lentiviruses on HIV-1 replication by single-round HIV-1 infection assays in PBLs up to 5 days post-mitogenic stimulation. Cells were transduced as described above. PBLs were stimulated with irradiated PBMCs, IL-2 (20 U/ml) and PHA (1 μg/ml) and collected at 2 and 5 days and HIV-1 replication was determined via luciferase assays in duplicate samples. (C) HA-dnCDK9 inhibits HIV-1 replication when expressed in post-stimulated PBLs (D) HA-dnCDK9 expression inhibits HIV-1 replication in PBLs previously infected with HIV-1-luc viruses. Cells were first infected with HIV-1-luc overnight, and subsequently infected with EGFP or HA-dnCDK9 lentiviruses. Sixteen h later, PBLs were activated with PMA/PHA. PBLs were collected and prepared for luciferase assays 48 h following stimulation (duplicate samples). (E) HA-dnCDK9 expression does not inhibit DNA synthesis in PBLs. DNA synthesis was determined by measuring incorporation of [3H]-thymidine into DNA for 24 hrs. [3H]-thymidine was added 24 hrs post activation in duplicate samples.
Collectively, these results show that CDK9 can be inhibited in primary human T cells in a manner that blocks HIV-1 replication without affecting T cell activation, proliferation and viability. In contrast, FVP affects the expression of markers of T cell activation and proliferation as well as DNA synthesis at the concentrations that exhibit an anti-HIV-1 activity comparable to that of HA-dnCDK9. These results imply that FVP has targets other than CDK9 that mediate its potent cellular effects.
4. Discussion
The activity of the cyclin T1/CDK9 complex is essential for Tat dependent stimulation of HIV-1 transcription. Previous work performed using transformed cell lines suggested that CDK9 could be a potential novel therapeutic cellular target to inhibit HIV-1 replication (Mancebo et al., 1997; Flores et al., 1999; Chao et al., 2000; Chiu et al., 2004). However, these results should be interpreted with caution, as CDK9 has also been implicated in the control of cellular transcription both in vitro (Price, 2000) and in experiments using cells treated with FVP and DRB, which potently inhibit cellular transcription and CDK9 activity (Chao and Price, 2001; Lam et al., 2001).
T cell activation involves a complex coordination of gene expression programs that lead to the expression of gene products required for T cell effector functions and cell proliferation. One of the most rapid steps during T cell activation is de novo expression of IL-2, which is accompanied and further enhanced by expression of the high affinity IL-2 receptor and allows for rapid expansion of specialized T cell populations (Gaffen and Liu, 2004). Given the potential requirement of CDK9 for general transcription, and the low levels of cyclin T1/CDK9 complexes in quiescent T cells, it is currently unknown whether inhibition of CDK9 activity in these cells is compatible with T cell activation and how this affects HIV-1 replication. Moreover, the direct effects of inhibiting CDK9 activity in primary cells are unknown. In this report we have investigated the effects of directly limiting CDK9 activity in a variety of cells including primary human lymphocytes. Moreover, we have compared the effect of direct inhibition of CDK9 with the effects of FVP on proliferation, viability, transcription/RNA synthesis and, more importantly, on T cell activation and HIV-1 replication. Our results show that limiting CDK9 activity in primary human PBLs potently inhibits HIV-1 replication without preventing T cell activation as determined by measuring a variety of T cell activation markers and DNA synthesis. Notably, IL-2 receptor alpha expression levels and p107 were not affected by direct inhibition of CDK9 activity using HA-dnCDK9. In contrast, the concentrations of FVP required to achieve a similar block in HIV-1 replication, inhibit T cell activation. Our results also suggest that FVP inhibits other RNAPII kinases in addition to CDK9 even at the low nanomolar range.
The first suggestion that CDK9 could be a potential cellular, as opposed to viral, target for anti-HIV-1 therapy came from a in vitro transcriptional screen of a library of small molecules for inhibitors of Tat-dependent HIV-1 transcription (Mancebo et al., 1997). This screen identified various related compounds with the ability to inhibit Tat dependent transcription, which were found to be inhibitors of CDK9. Subsequent studies using a dominant negative mutant form of CDK9 demonstrated that HIV-1 replication is very sensitive to inhibition of CDK9 activity in transformed Jurkat cells without loss of cell viability or effects on transcription of a group of genes analyzed (Flores et al., 1999). Others found that the CDK inhibitor FVP is a very potent inhibitor of CDK9 activity in vitro (IC50 of ~3 nM; (Chao and Price, 2001), which is lower than the IC50 reported for cyclin D1/CDK4 (20 nM) and cyclin B/CDC2 (30 nM) (Sedlacek, 2001). Interestingly, at low concentrations, FVP was found to inhibit HIV-1 replication potently in transformed cells, without apparent effects on cellular transcription (Chao et al., 2000). However, we have found that FVP affects cellular function at concentrations which are effective at inhibiting HIV-1 replication in a variety of cell types. Specifically, at FVP concentrations of 100 nM we have observed inhibition of cellular RNA synthesis in transformed 293 and MAGI cells. At this concentration, FVP inhibits HIV-1 replication comparably or less potently than dnCDK9 or the CDK9 inhibitor Hexim1, but dnCDK9 does not inhibit RNA synthesis. The effects of FVP are even more dramatic in primary T cells whose activation following mitogenic stimulation is affected by FVP concentrations as low as 10-30 nM with different degrees of severity. The effects of FVP on T cell activation are consistent with the requirement of increased transcription to generate factors essential for T cell effector functions and proliferation. However, we were surprised to detect effects even at low concentrations of FVP, as tumor cell lines proliferate and are viable under these conditions. Given the requirement of IL-2 synthesis for optimal T-cell activation and increased expression of the high affinity IL-2 receptor in vitro (Reem and Yeh, 1984; Welte et al., 1984; Depper et al., 1985; Reem et al., 1985; Schorle et al., 1991), it is likely that transcriptionally regulated elements within these pathways are very sensitive to FVP treatment. In contrast, HA-dnCDK9 is as effective as 30-50 nM FVP in inhibiting HIV-1 replication in PBLs from multiple donors. But it does not prevent T cell activation as judged from the lack of perturbations in the expression of a variety of T cell activation and cell cycle markers as well as the absence of effects on DNA synthesis.
Others have recently reported that siRNA directed to either CDK9 or cyclin T1 was effective in inhibiting HIV-1 replication in transformed HeLa cells without obvious effects on cell viability (Chiu et al., 2004). Our results in MAGI and 293 cells agree with these data. However, in this previous study siRNA mediated downregulation of cyclin T1/CDK9 complexes had little effect on the overall cellular CDK9-associated activity. It was suggested that the cell maintains CDK9 activity by reducing the pools of inactive cyclin T1/CDK9 complexes associated with 7sk snRNA by converting them to the active form, as the expression of the cyclin T1/CDK9 complex is knocked-down. It was also suggested that the dynamic equilibrium between active and inactive cyclin T1/CDK9 complexes allow cells to maintain a threshold of kinase activity, which might be essential for viability (Chiu et al., 2004). However, we clearly see inhibition of RNAPII CTD phosphorylation on Ser-2 following expression of dnCDK9 in a variety of cells, including MAGI cells, which are derived form HeLa cells, suggesting that under our experimental conditions cellular CDK9 activity is effectively downregulated. In MAGI cells, inhibition of HIV1 replication correlates with effective expression of dnCDK9 and inhibition of Ser-2 phosphorylation, with no effects on Ser-5 phosphorylation, as compared to EGFP transduced cells. This was accomplished at an MOI of 60. At lower MOIs the expression of the dnCDK9 is insufficient to affect Ser-2 phosphorylation and consequently no effects on HIV-1 replication were noted. Conversely, FVP inhibits phosphorylation of Ser-5, in addition to Ser-2 in MAGI cells (Fig. 3) and other cell types (data not shown), suggesting that FVP targets a second CTD kinase, which may explain its potent effects on inhibition of RNAPII transcription. In this regard, a recent report has shown that DRB, a drug that inhibits transcriptional elongation by RNAPII and CDK9, inhibits phosphorylation of both CTD Ser residues during transcriptional elongation at particular genes and that phosphorylation of these two residues exhibits different kinetics (Gomes et al., 2006). Moreover, our results do not rule out the possibility that some of the effects of FVP on T cell activation may be caused by inhibition of CDKs that play roles in the regulation of the cell cycle, i.e CDK4, and/or other G1/S CDKs, whose inhibition could result in cell cycle entry delays in mitogenicaly stimulated PBLs.
It is also important to note that expression of dnCDK9 in PBLs does not prevent scheduled upregulation of endogenous CDK9 upon mitogenic stimulation, which together with a decrease in the expression of dnCDK9 that is consistently observed during T cell activation, results in an increase in the CDK9/dnCDK9 ratio. This may lead to delayed activation/upregulation of CDK9 activity at some point post-mitogenic stimulation. Although this is conceivable, inhibition of CDK9 in quiescent or stimulated cells (Fig. 5C and Fig. 6C) is sufficient to potently inhibit HIV replication without affecting the expression of T cell activation markers and DNA synthesis (Fig. 5A and B and Fig. 6C). Thus, our results do not signify that CDK9 activity is not required for T cell activation, but rather that CDK9 activity can be downregulated in primary T lymphocytes in a manner that potently inhibits HIV-1 replication without interfering with T cell activation. Moreover, it is possible that other functionally redundant CDKs are sufficient to sustain essential cellular transcription when CDK9 activity is significantly reduced. In this regard, gene knockout of several CDKs in mice has revealed broad compensation and redundancy (Santamaria and Ortega, 2006). Gene profiling experiments will be important to determine whether direct CDK9 inhibition affects the expression of key cellular genes under these conditions.
Our results in human primary peripheral blood lymphocytes suggest that highly selective inhibition of CDK9 activity is a feasible strategy to inhibit HIV-1 replication and imply that small pharmacological compounds targeting CDK9 with enhanced selectivity could be developed into effective anti-HIV-1 therapeutic drugs.
Acknowledgments
We thank Qiang Zhou, Miguel Sena-Esteves and David Price for cDNA constructs and Jose-Ramon Suarez (Aventis) for providing flavopiridol. PNL4-3.Luc.R-E- plasmid was obtained through the NIH AIDS Research Reference Reagent Program (NIAID) from Dr. Nathaniel Landau. We thank Alison Kurimchak for technical assistance and Dr. Gunther Boden, May Truongcao and the Temple University General Clinical Research Center staff for assisting in the blood collection and Betty Moran and Norm Nagl for technical advice. Grant Support: R. M. was supported by an award from the Department of Defense, Breast cancer research Program (DAMD 17-02-1-0576). This work was supported in part by grants to X.G. including an NIH R01 (AI45450) and a Career Development Award (K02 AI01823) from the National Institute of Allergy and Infectious Diseases and a W. W. Smith grant (A9802/9901) and to A.Y.T. (R01 CA78499, National Cancer Institute). Facilities used for this work were supported in part by a Shared Resources for Cancer Research grant R24 (CA88261-01) and a General Clinical Research Grant NIH (M01 RR00349).
Abbreviations
- PMA
13-Acetate
- DRB
5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole
- AIDS
acquired immune deficiency syndrome
- BD
Becton and Dickinson
- CTD
C-terminal domain
- CDK9
cyclin-dependent kinase 9
- DMEM
DNA, Dulbecco’s modification of Eagle’s medium
- EGFP
enhanced green fluorescent protein
- FVP
flavopiridol
- FACS
flow activated cell sorting
- HAART
highly active antiretroviral therapy
- MOI
HIV-1, IL, multiplicity of infection
- NIAID
National Institute of Allergy and Infectious Diseases
- NIH
National Institutes of Health
- PBMC
peripheral blood mononuclear cells
- PBLs
peripheral blood lymphocytes
- PHA
phorbol 12-myristate, phytohemagglutinin
- PARP
poly ADP ribose polymerase
- PVDF
polyvinylidene difluoride
- P-TEFb
positive transcription elongation factor b
- Pro
protease
- RT
reverse transcriptase
- RNAPII
RNA, RNA polymerase II
- Ser
serine
- TCA
trichloro acetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blagosklonny MV. Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle. 2004;3:1537–42. doi: 10.4161/cc.3.12.1278. [DOI] [PubMed] [Google Scholar]
- Chackerian B, Long EM, Luciw PA, Overbaugh J. Human immunodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian immunodeficiency virus infection. J Virol. 1997;71:3932–9. doi: 10.1128/jvi.71.5.3932-3939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem. 2000;275:28345–8. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–9. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Cao H, Jacque JM, Stevenson M, Rana TM. Inhibition of human immunodeficiency virus type 1 replication by RNA interference directed against human transcription elongation factor P-TEFb (CDK9/CyclinT1) J Virol. 2004;78:2517–29. doi: 10.1128/JVI.78.5.2517-2529.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virol. 1995;206:935–44. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Depper JM, Leonard WJ, Drogula C, Kronke M, Waldmann TA, Greene WC. Interleukin 2 (IL-2) augments transcription of the IL-2 receptor gene. Proc Natl Acad Sci U S A. 1985;82:4230–4. doi: 10.1073/pnas.82.12.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Lee G, Kessler J, Miller M, Schlief W, Tomassini J, Hazuda D. Host-cell positive transcription elongation factor b kinase activity is essential and limiting for HIV type 1 replication. Proc Natl Acad Sci U S A. 1999;96:7208–13. doi: 10.1073/pnas.96.13.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu TJ, Peng J, Lee G, Price DH, Flores O. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J Biol Chem. 1999;274:34527–30. doi: 10.1074/jbc.274.49.34527. [DOI] [PubMed] [Google Scholar]
- Gaffen SL, Liu KD. Overview of interleukin-2 function, production and clinical applications. Cytokine. 2004;28:109–23. doi: 10.1016/j.cyto.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Garriga J, Bhattacharya S, Calbo J, Marshall RM, Truongcao M, Haines DS, Grana X. CDK9 is constitutively expressed throughout the cell cycle, and its steady-state expression is independent of SKP2. Mol Cell Biol. 2003;23:5165–73. doi: 10.1128/MCB.23.15.5165-5173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga J, Graña X. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene. 2004;337:15–23. doi: 10.1016/j.gene.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Garriga J, Mayol X, Graña X. The CDC2-related kinase PITALRE is the catalytic subunit of active multimeric protein complexes. Biochem J. 1996a;319:293–298. doi: 10.1042/bj3190293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga J, Peng J, Parreno M, Price DH, Henderson EE, Graña X. Upregulation of cyclin T1/CDK9 complexes during T cell activation. Oncogene. 1998;17:3093–102. doi: 10.1038/sj.onc.1202548. [DOI] [PubMed] [Google Scholar]
- Gomes NP, Bjerke G, Llorente B, Szostek SA, Emerson BM, Espinosa JM. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006;20:601–12. doi: 10.1101/gad.1398206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graña X, De Luca A, Sang N, Fu Y, Claudio PP, Rosenblatt J, Morgan DO, Giordano A. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc Natl Acad Sci USA. 1994;91:3834–8. doi: 10.1073/pnas.91.9.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasham MG, Tsygankov AY. Tip, an Lck-interacting protein of Herpesvirus saimiri, causes Fas- and Lck-dependent apoptosis of T lymphocytes. Virol. 2004;320:313–29. doi: 10.1016/j.virol.2003.11.032. [DOI] [PubMed] [Google Scholar]
- He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–11. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J Virol. 1998;72:9881–8. doi: 10.1128/jvi.72.12.9881-9888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wange RL. T cell receptor signaling: beyond complex complexes. J Biol Chem. 2004;279:28827–30. doi: 10.1074/jbc.R400012200. [DOI] [PubMed] [Google Scholar]
- Klebl B, Choidas A. CDK9/cyclin T1: a host cell target for antiretroviral therapy. Future Virology. 2006;1:317–330. [Google Scholar]
- Lam LT, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:RESEARCH0041. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11:2633–44. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–8. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- Marshall RM, Graña X. Mechanisms controlling CDK9 activity. Front Biosci. 2006;11:2598–2613. doi: 10.2741/1994. [DOI] [PubMed] [Google Scholar]
- Marshall RM, Salerno D, Garriga J, Graña X. Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J Immunol. 2005;175:6402–11. doi: 10.4049/jimmunol.175.10.6402. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- O’Keeffe B, Fong Y, Chen D, Zhou S, Zhou Q. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem. 2000;275:279–87. doi: 10.1074/jbc.275.1.279. [DOI] [PubMed] [Google Scholar]
- Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem. 1998a;273:13855–60. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998b;12:755–62. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;20:2629–34. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reem GH, Yeh NH. Interleukin 2 regulates expression of its receptor and synthesis of gamma interferon by human T lymphocytes. Science. 1984;225:429–30. doi: 10.1126/science.6429853. [DOI] [PubMed] [Google Scholar]
- Reem GH, Yeh NH, Urdal DL, Kilian PL, Farrar JJ. Induction and upregulation by interleukin 2 of high-affinity interleukin 2 receptors on thymocytes and T cells. Proc Natl Acad Sci U S A. 1985;82:8663–6. doi: 10.1073/pnas.82.24.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med. 2002;8:1310–7. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- Santamaria D, Ortega S. Cyclins and CDKS in development and cancer: lessons from genetically modified mice. Front Biosci. 2006;11:1164–88. doi: 10.2741/1871. [DOI] [PubMed] [Google Scholar]
- Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–4. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- Sedlacek HH. Mechanisms of action of flavopiridol. Crit Rev Oncol Hematol. 2001;38:139–70. doi: 10.1016/s1040-8428(00)00124-4. [DOI] [PubMed] [Google Scholar]
- Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122:131–9. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–62. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Welte K, Andreeff M, Platzer E, Holloway K, Rubin BY, Moore MA, Mertelsmann R. Interleukin 2 regulates the expression of Tac antigen on peripheral blood T lymphocytes. J Exp Med. 1984;160:1390–403. doi: 10.1084/jem.160.5.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]