

The crystal structure of dimerized radixin FERM domain has been determined. It was found that the adhesion molecule-binding site of one molecule is masked by the C-terminal residues of the other molecule.
Keywords: radixin, ERM, FERM domain, masking
Abstract
ERM (ezrin/radixin/moesin) proteins bind to the cytoplasmic tail of adhesion molecules in the formation of the membrane-associated cytoskeleton. The binding site is located in the FERM (4.1 and ERM) domain, a domain that is masked in the inactive form. A conventional masking motif, strand 1 (residues 494–500 in radixin), has previously been identified in the C-terminal tail domain. Here, the crystal structure of dimerized radixin FERM domains (residues 1–310) is presented in which the binding site of one molecule is occupied by the C-terminal residues (residues 295–304, strand 2) of the other molecule. The residues contain a conserved motif that is compatible with that identified in the adhesion molecules. The residues might serve as a second masking region in the inactive form of ERM proteins.
1. Introduction
ERM (ezrin/radixin/moesin) proteins mediate the association of actin filaments with plasma membranes by binding to both membrane-associated adhesion molecules and actin filaments. The ERM-family proteins are involved in cell adhesion, motility and morphogenesis in addition to participating in signal transduction pathways (Tsukita & Yonemura, 1999 ▶; Sun et al., 2002 ▶; Bretscher et al., 2002 ▶). ERM proteins can be divided into three regions: an N-terminal FERM (band 4.1 protein and ERM homology) domain consisting of ∼300 residues (radixin residues 1–297), a central α-domain of ∼200 residues (radixin residues 310–470) and a C-terminal tail domain of ∼100 residues (radixin residues 477–583) (Edwards & Keep, 2001 ▶; Hoeflich & Ikura, 2004 ▶).
When inactive, ERM proteins are present in the cytosol and the adhesion molecule-binding sites on their FERM domains are masked (Gary & Bretscher, 1995 ▶; Takahashi et al., 1997 ▶; Andreoli et al., 1994 ▶; Magendantz et al., 1995 ▶; Hirao et al., 1996 ▶; Bretscher et al., 2002 ▶). A conventional masking motif, strand 1 (residues 494–500 in radixin), has previously been identified in the C-terminal tail domain (Pearson et al., 2000 ▶) and was proposed to interfere directly with the binding of adhesion molecules to the FERM domain (Hamada et al., 2003 ▶). Proper inactivation by masking is important in order to avoid unnecessary or inappropriate interactions with adhesion molecules and actin filaments. Indeed, transfected cells that express the mutant ezrin T567D, a phosphomimetic mutant used to explore the function of activated ERM proteins, appear to form inappropriate structures such as lamellipodia, membrane ruffles and microvilli tufts (Oshiro et al., 1998 ▶; Gautreau et al., 2000 ▶). In addition, ERM proteins and the closely related protein merlin, the gene product of neurofibromatosis type 2 (NF2) tumour suppressor, are thought to play a role in cancer development (McClatchey, 2003 ▶). Here, the crystal structure of a dimerized pair of radixin FERM domains is presented. It was found that the binding site of one molecule was unexpectedly masked by the C-terminal residues 295–304 of the other.
2. Materials and methods
2.1. Crystallization
The C-terminal extended FERM domain (radixin residues 1–310) was cloned from mouse radixin that exhibits 100% amino-acid sequence identity to that of human radixin. The protein was purified as described previously (Hamada, Matsui et al., 2000 ▶) and concentrated to 23 mg ml−1 in a solution containing 300 mM NaCl, 10 mM Na MES pH 6.8 and 1 mM dithiothreitol (DTT). Clusters of needle-shaped crystals were initially obtained using the PEG 6000 Grid Screening kit (Hampton Research). After refining the crystallization conditions, the largest crystal (20 × 50 × 500 µm) grew in 18% PEG 3350 and 0.1 M Na HEPES pH 7.5 at 293 K.
2.2. X-ray data collection
For X-ray data collection, crystals were transferred to a cryoprotectant solution containing 25–30%(v/v) glycerol and subsequently frozen in liquid nitrogen. X-ray data were collected on beamline BL44XU at SPring-8. The mounted crystal was maintained at 100 K in the cryonitrogen stream and exposed to the X-ray beam with a wavelength of 0.90 Å. The diffraction data were recorded on a DIP2040B detector system and were indexed using HKL2000 (Otwinowski & Minor, 1997 ▶). Statistical data are listed in Table 1 ▶.
Table 1. Crystallographic data for the dimerized radixin FERM domains.
Values in parentheses are for the outer resolution shell.
| X-ray data | |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 57.2, b = 70.2, c = 110.8, β = 99.0 |
| Resolution (Å) | 30–2.8 (2.9–2.8) |
| Mosaicity (°) | 0.2–0.5 |
| Reflections (total/unique) | 68724/19703 |
| Completeness (%) | 96.4 (76.9) |
| 〈I/σ(I)〉 | 11.0 (1.5) |
| Rmerge (%) | 8.7 (56.4) |
| Refinement | |
| No. of residues included | 297 (Mol-1), 302 (Mol-2) (of 310) |
| No. of atoms | 5018 |
| Rwork/Rfree† (%) | 22.2/26.9 |
| Ramachandran plot (%) | |
| Most favoured | 83.7 |
| Additional allowed | 14.4 |
| Generously allowed | 1.3 |
| Disallowed | 0.6 |
| Average B factor (Å2) | |
| Mol-1 | 69.5 |
| Mol-2 (FERM/C-term 295–304) | 81.1/88.3 |
| R.m.s. bond length (Å) | 0.008 |
| R.m.s. bond angles (°) | 1.3 |
R
work = 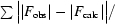
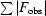 . R
free is the same as R
work except that it was calculated for a 5% subset of all reflections that were never used in crystallographic refinement.
. R
free is the same as R
work except that it was calculated for a 5% subset of all reflections that were never used in crystallographic refinement.
2.3. Structure determination
Molecular replacement (MR) was performed using CNS (Brünger et al., 1998 ▶) with the previously determined FERM domain (residues 3–295; Hamada, Shimizu et al., 2000 ▶) as a search model. Two molecules were found in the asymmetric unit of the crystal. The calculated MR maps showed clear residual electron densities (in both 2F o − F c and F o − F c maps) at the ICAM-2-binding site of molecule 1 (Mol-1). The densities were continuous to the C-terminus of molecule 2 (Mol-2). Omit density maps confirmed that the densities represent the C-terminal residues 296–304 of Mol-2 that were missing in the search model. These residues were modelled manually using O (Jones et al., 1991 ▶). Finally, the overall structure was refined using CNS in the absence of NCS (non-crystallographic symmetry) restraints or constraints. Three residues (His161 and Lys262 of Mol-1 and Asp252 of Mol-2) were refined into the disallowed region of the Ramachandran plot (Table 1 ▶). Electron-density map was poor for His161 and Lys262, but clear for Asp252, which was found in the same region in the FERM–ICAM-2 structure (Hamada et al., 2003 ▶).
3. Results and discussion
3.1. Overall structure
The crystal structure of the dimerized radixin FERM domain was determined by molecular replacement and refined to 2.8 Å resolution (Table 1 ▶). The final model includes residues 3–299 and 3–304 for molecule 1 (Mol-1) and molecule 2 (Mol-2), respectively. As previously reported, the FERM domain consists of three subdomains A, B and C (Figs. 1 ▶ a and 1 ▶ b; Pearson et al., 2000 ▶; Hamada, Shimizu et al., 2000 ▶). Subdomain A possesses a typical ubiquitin fold, whereas subdomain B possesses an α-helix bundle structure. Subdomain C, possessing a standard seven-stranded β-sandwich (strands β1C–β7C) with a long capping α-helix (α1C) at the C-terminus, was grouped into the phosphotyrosine-binding (PTB) domain superfamily. In the present crystal structure, the FERM-domain structure of Mol-1 and Mol-2 is essentially the same except for small conformational changes observed in the side chains of β5C of Mol-1 (residues 245–251 shown in Fig. 1 ▶ c). These changes result from binding of the C-terminal extended residues of Mol-2.
Figure 1.

Crystal structure of dimerized radixin FERM domains. (a) Overall structure as a ribbon model. Mol-1 is shown in light blue and Mol-2 in pink, with its C-terminal extended residues (Arg295–Gln304) in red. The radixin FERM domain consists of subdomains A (the N-terminal 82 residues), B (residues 96–119) and C (residues 204–297). (b) A side view shows that Mol-1 and Mol-2 are aligned in a similar orientation. The blue dashed line represents the membrane-interaction surface of a dimer and blue arrows indicate binding sites to phosphatidylinositol 4,5-bisphosphate (PIP2; Hamada, Shimizu et al., 2000 ▶). Figures were prepared using PyMOL (http://pymol.sourceforge.net). (c) Schematic representation of the interactions between the C-terminal extended residues (strand 2 in pink) and subdomain C (light blue). Hydrogen bonds are shown as dashed lines and distances are indicated.
3.2. C-terminal extended residues of Mol-2 bind to subdomain C of Mol-1
Surprisingly, the structure revealed that the C-terminal region of Mol-2 (residues 295–304) binds the groove of the PTB-like subdomain C of Mol-1, an interaction mediated by an antiparallel β–β association. Five residues of the extended region (Asp298–Val302) form a short β-strand (strand 2) associated with strand β5C from subdomain C of Mol-1, with six hydrogen bonds between the main chains (<3.1 Å; Fig. 1 ▶ c). Many of the side chains are also involved in intermolecular interactions, which together contribute to the binding specificity. The nonpolar side chain of Val302 (Mol-2) is buried in a hydrophobic pocket formed by the side chains of Ile245, Ile248, Trp242 and His288 of Mol-1. The C-terminal residues beyond Gln304 of Mol-2 were disordered.
As shown in Fig. 1 ▶(b), although the orientation of the two FERM domains differs by ∼70° from each other, Mol-1 and Mol-2 are aligned parallel; subdomains A and C are aligned above and subdomain B below. The FERM domain of Mol-2 can be made completely parallel to that of Mol-1 by a single rotation of ∼70° (ignoring steric hindrance). It is structurally impossible for the C-terminal region to bind subdomain C (β5C) of the same molecule given the short nature of the linker present between strand 2 and subdomain C (α1C). In the present structure, interactions were only observed between subdomain C of Mol-1 and the C-terminus of Mol-2, while the C-terminal residues in Mol-1 are disordered and the binding site in subdomain C of Mol-2 remains open. There are no direct domain–domain contacts between Mol-1 and Mol-2 except for the single hydrogen bond formed between the last FERM residue of Mol-2 (Arg295) and Asn251 of subdomain C of Mol-1 (Fig. 1 ▶ c); the O atom of the main chain of Arg295 on helix α1C accepts a hydrogen bond from the NH2 group of the side chain of Asn251 (Mol-1). A single hydrogen bond at the edge of the domain is insufficient to keep one FERM domain associated with the other; thus, the FERM domains would remain flexible with respect to each other in solution. Considering that the strand 2–β5C association is not mediated by the rigid interactions between the two FERM domains, it seems unlikely that the interaction results from direct crystal packing between the two molecules. Gel-filtration chromatography of radixin 1–310 gave an estimated molecular weight of 42 kDa (data not shown), which is slightly larger than the calculated weight of a monomer (37 kDa) but smaller than that of a dimer.
3.3. Comparison of strand 2 with ICAM-2 and strand 1
Several adhesion molecules have been proposed to interact with ERM proteins, including CD44 (Tsukita et al., 1994 ▶; Serrador et al., 1997 ▶; Yonemura et al., 1998 ▶), CD43 (Yonemura et al., 1993 ▶, 1998 ▶; Serrador et al., 1998 ▶), ICAM-1 (Heiska et al., 1998 ▶), ICAM-2 (Yonemura et al., 1998 ▶; Helander et al., 1996 ▶; Heiska et al., 1998 ▶) and PSGL-1 (Snapp et al., 2002 ▶). A crystal structure of a radixin FERM domain complexed with the cytoplasmic tail of ICAM-2 revealed that the FERM domain recognizes the signature sequence RxxTYxVxxA (Motif 1, where x represents any amino acid; Hamada et al., 2003 ▶). The two hydrophobic residues (Tyr and Val in ICAM-2) are especially important and play a critical role in the hydrophobic β–β association with subdomain C. Both the hydrophobic residues and the homologous sequence motif can be found in other FERM-binding adhesion molecules (Fig. 2 ▶ a).
Figure 2.

The second masking motif in comparison with the conventional masking motif and adhesion molecules. (a) Sequence alignment of the second and conventional masking motifs in human ERM and merlin proteins and the juxtamembrane cytoplasmic region of adhesion molecules that bind ERM. The red arrow at the top indicates β-strand formation of the second masking motif in the present structure (strand 2). Conserved residues in the motif are highlighted in the following colours: basic residues in blue, acidic residues in red, hydrophobic residues in yellow, serine and threonine in magenta and glutamine and asparagine in green. The numbers on the right-hand side of the column indicate C-terminal residue numbers in the sequences. The residues for which structures are available in the PDB (β-strands and loop regions shown in Fig. 2 ▶ b) are boxed. Asterisks (*) indicate the key residues in the FERM-binding motif in mouse ICAM-2 (Hamada et al., 2003 ▶). The percentage amino-acid similarity between the full-length radixin and moesin, ezrin and merlin are 81, 75 and 44%, respectively. (b) Strand 2 (second masking motif, shown in red) binds to subdomain C (in light blue) in the same conformation as strand 1 of the moesin C-tail domain (Pearson et al., 2000 ▶; conventional masking motif, in green) and the ICAM-2 cytoplasmic peptide (Hamada et al., 2003 ▶; in blue). The present structures were superimposed on the other two structures at subdomain C with r.m.s. deviations of 0.70 and 1.20 Å (for 93 Cα atoms), respectively. The N- and C-terminal residue numbers of the peptides are labelled. Superimposition of the molecules was performed using LSQMAN (Kleywegt & Jones, 1997 ▶). The β3 tail of integrin was also shown to bind to the same site of the talin FERM domain (Garcia-Alvarez et al., 2003 ▶; not shown). (c) Structural variability in residues connecting the FERM and α-domains. Sequences of the deposited FERM structures [PDB codes 1gc7 and 1gc6 (Hamada, Shimizu et al., 2000 ▶), 1j19 (Hamada et al., 2003 ▶), 1e5w (Edwards & Keep, 2001 ▶) and 1isn (Shimizu et al., 2002 ▶)] around the loop region connecting helix α1C and the α-domain are aligned. The α-helices are shown as light-green boxes and the β-strand in Mol-1 (strand 2) as a red arrow. Residues that were included in the crystallization sample but for which electron density was not observed are shown as dashed lines. The two conserved hydrophobic residues essential for intermolecular β-sheet formation are highlighted in yellow. (d) Superimposition of the present structure, ICAM-2-bound form of the radixin FERM domain (PDB code 1j19, shown in blue), free-form moesin (1e5w, green) and free-form merlin (PDB code 1isn, yellow). The orientation of Mol-1 (light grey) and Mol-2 (red) are the same as in Fig. 1 ▶(b). Structures were superimposed at helix α1C of Mol-2. Each C-terminus is labelled with its last residue number and PDB code.
Interestingly, the C-terminal region of ERMs was found to possess a similar motif (KxxTIxVxxM) and was shown to bind to the same site on subdomain C with the same β-strand formation (second masking motif, shown in red in Fig. 2 ▶ b). Moreover, the motif is highly conserved in merlin and all ERM-protein family members (Fig. 2 ▶ a). The two hydrophobic residues are fully conserved, with only Ile300 being replaced with leucine in ezrin and merlin. In contrast, the motif in the conventional masking region within the C-terminal tail domain (strand 1, 488–494 in moesin and 494–500 in radixin; Pearson et al., 2000 ▶) is poorly conserved in ERM and merlin. Especially in ERMs, the first hydrophobic residue of strand 1 is replaced with alanine. Since the side chain of alanine is smaller, its hydrophobic interaction with residue Ile248 of subdomain C (Fig. 1 ▶ c) would be considerably weaker than the interaction between adhesion molecules and strand 2.
The two masking regions, strand 1 (green in Fig. 2 ▶ b) and residues 295–304 (strand 2, red), directly compete with adhesion molecules (blue) for binding to subdomain C. In the FERM–ICAM-2 complex (Hamada et al., 2003 ▶), the 11 residues of the ICAM-2 peptide bury as much as 1370.9 Å2 of the total accessible surface area of the complex and eight main-chain hydrogen bonds stabilize the complex. These relatively larger values explain the high binding affinity of ICAM-2 for the FERM domain, which is in the nanomolar range (K d = 16 nM in the case of radixin; Hamada et al., 2003 ▶). In contrast, the binding affinity of the conventional masking motif, strand 1, to subdomain C is expected to be lower; in the moesin FERM–C-tail complex, the β–β interaction was unstable and easily affected by crystal packing, inducing domain swapping (Pearson et al., 2000 ▶). The swapped structure showed that strand 1 buries only 868.2 Å2 of the surface, with six hydrogen bonds between the main chains. In summary, it is likely that masking of the FERM domain by strand 1 itself is insufficient to compete with and block the strong binding of several adhesion molecules such as ICAM-2. This seems to be inconsistent with the previously proposed masking mechanism by which full-length proteins in the inactive form restrict the binding of adhesion molecules (Bretscher et al., 2002 ▶).
On the other hand, in the present structure, nine residues (residues Arg295 and Pro297–Gln304) of the C-terminal extended region (strand 2) bury 1003.1 Å2 of the surface, which is larger than that of strand 1. Thus, residues 295–304 might serve as a second masking region in the inactive form of ERM and merlin proteins. Such a property of the duplicative masking motifs within a protein could compensate for their low binding affinity by increasing the number of motifs available near the FERM domain. Recently, the structure of the FERM domain of focal adhesion kinase was determined (Ceccarelli et al., 2006 ▶). In this structure, the residues just C-terminal to the FERM domain also mask the same site (β5C), but an intramolecular parallel β-sheet was formed rather than an intermolecular antiparallel interaction.
3.4. Structural variability in the residues connecting FERM and α-domains
A list of crystal structures of FERM domains that were obtained from crystallization of samples including C-terminal extended residues is shown in Fig. 2 ▶(c). In the three structures [PDB codes 1j19 (Hamada et al., 2003 ▶), 1e5w (Edwards & Keep, 2001 ▶) and 1isn (Shimizu et al., 2002 ▶)], the C-terminal residues were observed as a helix that might be the N-terminal part of the long α-helix (α-domain). In moesin, which has the longest extended C-tail (PDB code 1e5w, shown in green in Fig. 2 ▶ d), helix formation of residues 299–308 was stabilized through several interactions with the FERM domain (Edwards & Keep, 2001 ▶). Similar interactions between the helix and FERM domains were also observed in radixin (PDB code 1j19, in blue; Hamada et al., 2003 ▶) and merlin (PDB code 1isn, in yellow; Shimizu et al., 2002 ▶), yielding almost the same orientation of the helix. Thus, this region has the capacity to form two different secondary structures, a β-strand and an α-helix, acting as a so-called ‘chameleon’ sequence that adopts context-dependent secondary structures (Minor & Kim, 1996 ▶; Tan & Richmond, 1998 ▶; Mezei, 1998 ▶). In the other three radixin structures [the free form 1gc7, the IP3-bound form 1gc6 (Hamada, Shimizu et al., 2000 ▶) and Mol-1 of the present work], electron densities corresponding to the C-terminal residues were not observed owing to disorder, which suggests intrinsic structural flexibility in these residues (shown as dashed lines in Fig. 2 ▶ c). Flexibility was also suggested by the results of a proteolysis assay (Hoeflich et al., 2003 ▶).
3.5. Second actin-binding site
In merlin, although the C-terminal actin-binding region is missing, the protein can still bind to actin filaments (Brault et al., 2001 ▶; Xu & Gutmann, 1998 ▶; James et al., 2001 ▶). Interestingly, there are many reports that map the additional but independent F-actin-binding sites in ERM and merlin to a region that overlaps with the present second masking motif; that is, ezrin residues 281–310 (Martin et al., 1997 ▶), 281–333 (Roy et al., 1997 ▶; corresponding to the same residue numbers in radixin, reviewed in Tsukita & Yonemura, 1999 ▶) and merlin residues 280–323 (Brault et al., 2001 ▶; corresponding to radixin residues 264–308). The present structure suggests the possibility that ERM and merlin proteins might utilize interaction of the second masking motif with the FERM domain to compete with and suppress the binding of this region to actin.
Supplementary Material
PDB reference: radixin FERM domain, 2d2q, r2d2qsf
Acknowledgments
We thank Drs Sa. Tsukita and Sh. Tsukita for providing the radixin FERM domain cDNA, S. Terawaki and N. Yoshihara for their assistance in protein purification and Drs A. Nakagawa, E. Yamashita and M. Yoshimura at SPring-8 for their help in the synchrotron experiments. This work was supported by a Protein 3000 project on Signal Transduction from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan to TH and by a Grant-in-Aid for Young Scientists and a Grant-in-Aid for Scientific Research on Priority Areas from the Japan Society for the Promotion of Science (JSPS) to KK.
References
- Andreoli, C., Martin, M., Le Borgne, R., Reggio, H. & Mangeat, P. (1994). J. Cell Sci.107, 2509–2521. [DOI] [PubMed] [Google Scholar]
- Brault, E., Gautreau, A., Lamarine, M., Callebaut, I., Thomas, G. & Goutebroze, L. (2001). J. Cell Sci.114, 1901–1912. [DOI] [PubMed] [Google Scholar]
- Bretscher, A., Edwards, K. & Fehon, R. G. (2002). Nature Rev. Mol. Cell Biol.3, 586–599. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Ceccarelli, D. F., Song, H. K., Poy, F., Schaller, M. D. & Eck, M. J. (2006). J. Biol. Chem.281, 252–259. [DOI] [PubMed] [Google Scholar]
- Edwards, S. D. & Keep, N. H. (2001). Biochemistry, 40, 7061–7068. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez, B., de Pereda, J. M., Calderwood, D. A., Ulmer, T. S., Critchley, D., Campbell, I. D., Ginsberg, M. H. & Liddington, R. C. (2003). Mol. Cell, 11, 49–58. [DOI] [PubMed] [Google Scholar]
- Gary, R. & Bretscher, A. (1995). Mol. Biol. Cell, 6, 1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau, A., Louvard, D. & Arpin, M. (2000). J. Cell Biol.150, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada, K., Matsui, T., Tsukita, S. & Hakoshima, T. (2000). Acta Cryst. D56, 922–923. [DOI] [PubMed] [Google Scholar]
- Hamada, K., Shimizu, T., Matsui, T., Tsukita, S. & Hakoshima, T. (2000). EMBO J.19, 4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada, K., Shimizu, T., Yonemura, S., Tsukita, S. & Hakoshima, T. (2003). EMBO J.22, 502–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiska, L., Alfthan, K., Gronholm, M., Vilja, P., Vaheri, A. & Carpen, O. (1998). J. Biol. Chem.273, 21893–21900. [DOI] [PubMed] [Google Scholar]
- Helander, T. S., Carpen, O., Turunen, O., Kovanen, P. E., Vaheri, A. & Timonen, T. (1996). Nature (London), 382, 265–268. [DOI] [PubMed] [Google Scholar]
- Hirao, M., Sato, N., Kondo, T., Yonemura, S., Monden, M., Sasaki, T., Takai, Y. & Tsukita, S. (1996). J. Cell Biol.135, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich, K. P. & Ikura, M. (2004). Int. J. Biochem. Cell Biol.36, 2131–2136. [DOI] [PubMed] [Google Scholar]
- Hoeflich, K. P., Tsukita, S., Hicks, L., Kay, C. M., Tsukita, S. & Ikura, M. (2003). Biochemistry, 42, 11634–11641. [DOI] [PubMed] [Google Scholar]
- James, M. F., Manchanda, N., Gonzalez-Agosti, C., Hartwig, J. H. & Ramesh, V. (2001). Biochem. J.356, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J. & Jones, T. A. (1997). Methods Enzymol.277, 525–545. [DOI] [PubMed]
- McClatchey, A. I. (2003). Nature Rev. Cancer, 3, 877–883. [DOI] [PubMed]
- Magendantz, M., Henry, M. D., Lander, A. & Solomon, F. (1995). J. Biol. Chem.270, 25324–25327. [DOI] [PubMed] [Google Scholar]
- Martin, M., Roy, C., Montcourrier, P., Sahuquet, A. & Mangeat, P. (1997). Mol. Biol. Cell, 8, 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezei, M. (1998). Protein Eng.11, 411–414. [DOI] [PubMed] [Google Scholar]
- Minor, D. L. Jr & Kim, P. S. (1996). Nature (London), 380, 730–734. [DOI] [PubMed] [Google Scholar]
- Oshiro, N., Fukata, Y. & Kaibuchi, K. (1998). J. Biol. Chem.273, 34663–34666. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pearson, M. A., Reczek, D., Bretscher, A. & Karplus, P. A. (2000). Cell, 101, 259–270. [DOI] [PubMed] [Google Scholar]
- Roy, C., Martin, M. & Mangeat, P. (1997). J. Biol. Chem.272, 20088–20095. [DOI] [PubMed] [Google Scholar]
- Serrador, J. M., Alonso-Lebrero, J. L., del Pozo, M. A., Furthmayr, H., Schwartz-Albiez, R., Calvo, J., Lozano, F. & Sanchez-Madrid, F. (1997). J. Cell Biol.138, 1409–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador, J. M., Nieto, M., Alonso-Lebrero, J. L., del Pozo, M. A., Calvo, J., Furthmayr, H., Schwartz-Albiez, R., Lozano, F., Gonzalez-Amaro, R., Sanchez-Mateos, P. & Sanchez-Madrid, F. (1998). Blood, 91, 4632–4644. [PubMed] [Google Scholar]
- Shimizu, T., Seto, A., Maita, N., Hamada, K., Tsukita, S., Tsukita, S. & Hakoshima, T. (2002). J. Biol. Chem.277, 10332–10336. [DOI] [PubMed] [Google Scholar]
- Snapp, K. R., Heitzig, C. E. & Kansas, G. S. (2002). Blood, 99, 4494–4502. [DOI] [PubMed] [Google Scholar]
- Sun, C. X., Robb, V. A. & Gutmann, D. H. (2002). J. Cell Sci.115, 3991–4000. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., Sasaki, T., Mammoto, A., Takaishi, K., Kameyama, T., Tsukita, S. & Takai, Y. (1997). J. Biol. Chem.272, 23371–23375. [DOI] [PubMed] [Google Scholar]
- Tan, S. & Richmond, T. J. (1998). Nature (London), 391, 660–666. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., Oishi, K., Sato, N., Sagara, J. & Kawai, A. (1994). J. Cell Biol.126, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita, S. & Yonemura, S. (1999). J. Biol. Chem.274, 34507–34510. [DOI] [PubMed] [Google Scholar]
- Xu, H. M. & Gutmann, D. H. (1998). J. Neurosci. Res.51, 403–415. [DOI] [PubMed] [Google Scholar]
- Yonemura, S., Hirao, M., Doi, Y., Takahashi, N., Kondo, T. & Tsukita, S. (1998). J. Cell Biol.140, 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura, S., Nagafuchi, A., Sato, N. & Tsukita, S. (1993). J. Cell Biol.120, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: radixin FERM domain, 2d2q, r2d2qsf