

Abstract
In Xenopus somatic cell nuclear cloning, the nucleoli of donor nuclei rapidly and almost completely disappear in egg cytoplasm. We previously showed that the germ cell-specific proteins FRGY2a and FRGY2b were responsible for this unusually drastic nucleolar disassembly. The nucleolar disassembly occurs without inhibition of pre-rRNA transcription, a well known trigger for nucleolar segregation, and the mechanism for the nucleolar disassembly by FRGY2a and FRGY2b remains largely unknown. In this study, we searched for FRGY2a-interacting proteins and investigated the functional consequences of their interactions through a series of experiments. We showed that during the nucleolar disassembly, FRGY2a localized to the nucleoli of isolated nuclei and was capable of disassembling purified nucleoli, suggesting a direct interaction between FRGY2a and nucleolar components. Using a His tag pull-down approach, we identified the abundant and multifunctional nucleolar protein B23 as a potential target of FRGY2a and its related human protein YB1. A specific interaction between FRGY2a/YB1 and B23 was confirmed by co-immunoprecipitation. Finally, B23 knockdown using short interfering RNA and a subsequent add-back experiment confirmed that B23 was necessary for nucleolar disassembly by YB1. We propose that FRGY2a and YB1 disassemble nucleoli by sequestering B23, which is associated with pre-ribosomes and other structurally important nucleolar components.
Reversible disassembly of nucleoli is one of the most striking phenomena that donor nuclei exhibit in Xenopus somatic cell nuclear cloning. Physiologically, Xenopus eggs and early embryos do not contain nucleoli or transcribe rRNA until the midblastula transition, when zygotic transcription initiates and nucleoli are assembled (1). When somatic nuclei are injected into eggs, the nucleoli of the donor nuclei disappear within 40 min and reappear during the midblastula transition, exactly recapitulating physiological nucleolar disassembly and reassembly (2). To understand the molecular mechanism of nucleolar disassembly in the donor nuclei and gain new insight into the physiological dynamics of nucleoli in early Xenopus embryos, we established an in vitro nucleolar disassembly assay by combining Xenopus egg extract and somatic nuclei. Using this assay, we have found that the Xenopus germ cell-specific proteins FRGY2a and FRGY2b (collectively called FRGY2a/b) are responsible for the reversible disassembly of somatic nucleoli in egg cytoplasm (3).
FRGY2a/b were initially isolated as key proteins responsible for masking maternally derived mRNA within eggs and early embryos to prevent premature translation of mRNA (4). Later, they were additionally discovered to be transcription factors for several germ cell-specific genes (5). FRGY2a (336 amino acids) and FRGY2b (324 amino acids) share 83.0% identity at the amino acid level, and each recombinant protein can disassemble nucleoli on its own (3). The N-terminal domains of these proteins contain the evolutionarily conserved cold shock domain, which is responsible for their binding to the Y-box DNA sequence 5′-CTGATTGG-3′ of germ cell-specific gene promoters during transcriptional activation or repression (6, 7). The N-terminal domains also contain two RNA-binding motifs, RNP1-like and RNP2-like, within the cold shock domain, providing binding sites for mRNA. The C-terminal domains of these proteins are about 200 amino acids long, including four basic/aromatic amino acid islands, and are capable of disassembling nucleoli in isolated nuclei and in transfected cells, without the N-terminal domains. When Xenopus somatic nuclei are treated with the C-terminal domain of FRGY2a for 2 h, the nucleoli are almost completely disassembled, leaving only tiny remnants containing rDNA and associated RNA polymerase I transcription machinery. Surprisingly, the nucleolar remnants can continue pre-rRNA transcription as efficiently as intact nucleoli, indicating that nucleolar disassembly mediated by FRGY2a/b is not due to the inhibition of pre-rRNA transcription, which is the most common trigger for nucleolar segregation (8, 9). Until now, virtually nothing was known about how FRGY2a/b disassemble nucleoli. Since FRGY2a/b are the first identified proteins that have the capability of disassembling nucleoli, determining the molecular basis by which they promote this process will yield new insights into the structural organization of the nucleolus in a broader context than that of nuclear cloning. In this study, we isolated proteins that interact with FRGY2a during nucleolar disassembly and found that the multifunctional nucleolar protein B23 was necessary for nucleolar disassembly by FRGY2a.
EXPERIMENTAL PROCEDURES
Preparation of Recombinant Proteins
cDNAs of FRGY2a, FRGY2a-C (amino acids 120–336; -N and -C after the protein names indicate the N- and C-terminal domains, respectively), FRGY1, FRGY1-C-(113–303), YB1, YB1-N-(1–134), YB1-C-(135–325), enhanced green fluorescence protein-tagged FRGY2a (EGFP4-FRGY2a), EGFP-FRGY2a-N-(1–119), and EGFP-FRGY2a-C-(120–336) were subcloned into the pET-21c(+) vector with a His6 tag at the C terminus (Novagen). The accuracy of the constructs was confirmed by DNA sequencing. These plasmids were introduced into Escherichia coli BL21 (DE3), and production of the recombinant proteins was induced by treatment with 0.5 mm isopropyl thiogalactoside for 3 h. E. coli pellets obtained from an 8-liter culture were resuspended in 160 ml of recombinant binding buffer (20 mm Tris HCl, pH 8.0, 500 mm NaCl, 10 mm imidazole, 0.1% Triton X-100, 10% glycerol, 8 m urea, 0.2 mm phenylmethylsulfonyl fluoride, 2 mm leupeptin, and 1.5 mm pepstatin A) and sonicated with a Sonifier 450 (Branson) for 1 min six times with 50% duty cycle at output 6. The sonicated E. coli suspension was centrifuged at 12,000 × g for 20 min at 4 °C. The supernatant was gently rocked with 40 ml of nickel resin nickel-nitrilotriacetic acid (Qiagen) for 1 h at 4 °C. The nickel-nitrilotriacetic acid was packed into an Econo-column (Bio-Rad) and washed with 480 ml of recombinant binding buffer, and the bound protein was eluted with recombinant elution buffer (the same as recombinant binding buffer except for 500 mm imidazole, instead of 10 mm). Eluted recombinant protein (about 10 ml) was renatured by dialysis against a series of buffers at 4 °C as follows. First, 1 liter of 1 m urea, 2 m NaCl buffer (10 mm HEPES, pH 7.8, 2 m NaCl, 1 m urea, 1 mm EDTA, 0.003% Triton X-100, 0.2 mm phenylmethylsulfonyl fluoride, 2 mm leupeptin, and 1.5mm pepstatin A) was used for 1 h. Second, 1 liter of 0.5 m urea, 2 m NaCl buffer (the same as 1 m urea, 2 m NaCl buffer, except for 0.5 m urea) was used for 1 h. Third, 1 liter of 2 m NaCl buffer (the same as 1 m urea, 2 m NaCl buffer, except for no urea) was used for 12 h. Finally, 2 liters of Buffer A (10 mm HEPES, pH 7.8, 150 mm NaCl, 4 mm MgCl2, 1 mm dithiothreitol, 0.003% Triton X-100, 10% glycerol, 0.2mm phenylmethylsulfonyl fluoride, 2 mm leupeptin, and 1.5 mm pepstatin A) was used for 4 h.
Purification of YB1 from HeLa Cells
We prepared whole cell extract from a 10-liter culture of HeLa S3 cells (National Cell Culture Center) as described previously with some modifications (10). Briefly, we added 100 µl of cytoplasmic buffer (10 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 0.5 mm dithiothreitol, 0.2 mm phenylmethylsulfonyl fluoride, 2 mm leupeptin, 1.5 mm pepstatin A, and 0.025% Nonidet P-40) to 107 cells and incubated on ice for 20 min. After centrifugation at 6,000 × g for 10 min at 4 °C, we recovered supernatant as the cytoplasmic extract. We then added 50 µl of nuclear buffer (20 mm HEPES, pH 7.4, 420 mm NaCl, 1.5 mm MgCl2, 0.5 mm dithiothreitol, 0.2 mm EDTA, 25% glycerol, 0.2 mm phenylmethylsulfonyl fluoride, 2 mm leupeptin, and 1.5 mm pepstatin A) to the pellet and incubated it on ice for 20 min. We centrifuged the sample at 15,000 × g for 15 min at 4 °C and collected supernatant as the nuclear extract. The cytoplasmic and nuclear extracts were combined and kept in aliquot at −70 °C.
The extract was first applied to an SP-Sepharose cation exchange column, and bound protein was eluted by a linear gradient of NaCl from 150 mm to 1 m in Buffer A. The fractions that showed nucleolar disassembly activity, eluted by 700 – 800mm NaCl, were pooled and dialyzed against Buffer A. The dialyzed sample was applied to a HiTrap heparin column, and bound protein was eluted by an NaCl gradient from 150 mm to 1 m in Buffer A. Pooled active fractions, eluted by 650 – 750 mm NaCl, were dialyzed against Buffer A, applied to a Mono Q 5/5 anion exchange column, and fractionated by a linear gradient of NaCl from 150 mm to 1 m in Buffer A. Finally, the pooled active fractions, eluted by 450 – 550 mm NaCl in Buffer A, were fractionated by a Superdex 200 gel filtration column equilibrated with Buffer A. All columns were purchased from Amersham Biosciences. Throughout this purification process, the nucleolar disassembly activity co-fractionated with YB1, as monitored by immunoblotting using anti-YB1 antibody (gift from V. Evdokimova) (11). Although YB1 was widely distributed in fractions obtained from the SP-Sepharose column, only the fractions that demonstrated nucleolar disassembly activity were further fractionated as described above.
Reverse Transcription-PCR of FRGY1 from XL2 Cells
Total RNA isolated from XL2 cells with TRIzol (Invitrogen) was reverse transcribed using SuperScript II (Invitrogen). cDNA of FRGY1 was amplified with the primers 5′-GGTAGCAGCGAGGTTGAAACAC-3′ and 5′-CTCAGCCCCGCCCTGCTCAG-3′ using Platinum Taq polymerase (Invitrogen). The amplified cDNA was sequenced for verification.
In Vitro Nucleolar Disassembly Assay
HeLa and Xenopus embryonic fibroblasts XL2 cells at 2 × 106 cell/ml were treated with digitonin (50 µg/ml) to permeabilize both the plasma membrane and the nuclear envelope (12). Permeabilized cells are hereafter called nuclei. The nucleolar disassembly reaction consisted of 15 µm FRGY2a-related proteins (FRGY2a, FRGY1, YB1, and their N- and C-terminal domains), 1 × 105 nuclei, and the energy regenerating system (1 mm ATP, 1 mm GTP, 20 mm phosphocreatine, and 1 µm creatine phosphokinase) in Buffer A. After incubation of the reaction mix for 2 h at 25 °C, the nuclei were fixed with 4% paraformaldehyde and spun onto coverslips by centrifugation at 1,000 × g for 7 min for immunofluorescence microscopy as described below. For Fig. 2C, the nuclei on coverslips were treated with 0.1 u/µl (final concentration) of DNase I (Invitrogen) in 100 µl of Buffer A for 30 min at 25 °C.
FIGURE 2. Nucleolar localization of EGFP-FRGY2a.
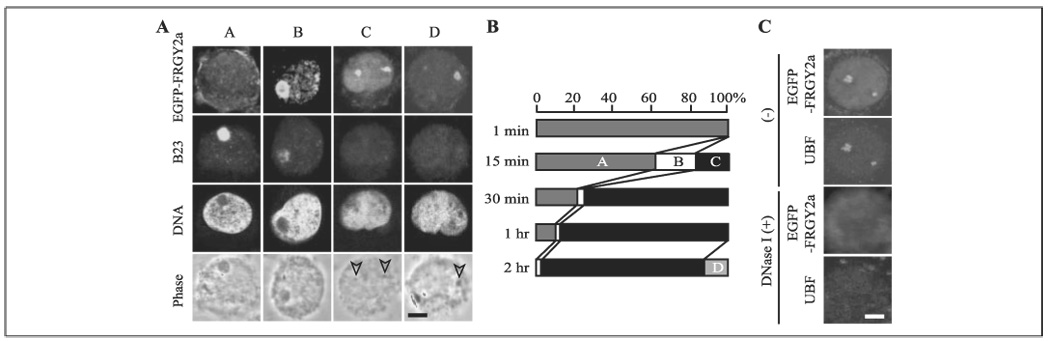
A, four localization patterns (A–D) of localization of EGFP-FRGY2a and B23 in XL2 nuclei during nucleolar disassembly. Arrowheads indicate nucleolar remnants. Bar, 4 µm in A and C. B, proportion of the patterns A–D during nucleolar disassembly based on the observation of 200 nuclei at each time point. C, disappearance of nucleolar EGFP-FRGY2a and UBF by DNase I treatment after incubation of XL2 nuclei with EGFP-FRGY2a for 2 h.
HeLa cell nucleoli were prepared based on a published protocol (13). Briefly, 2 × 107 HeLa cells were resuspended in 500 µl of Buffer 1 (10 mm HEPES, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, and 0.5 mm dithiothreitol) and were ground 10 times with a Micro Duall tissue grinder (Kontes, 0.15-mm clearance) followed by centrifugation at 230 × g for 5 min at 4 °C. The pellet was resuspended in 500 µl of Buffer 2 (10 mm HEPES, pH 7.9, 10 mm MgCl2, and 250 mm sucrose) and overlaid onto 3 ml of Buffer 3 (10 mm HEPES, pH 7.9, 0.5 mm MgCl2, and 350 mm sucrose) in a 5-ml test tube. After centrifugation at 1,400 × g for 5 min at 4 °C, the pellet was resuspended in 1 ml of Buffer 3 and sonicated with a Sonifier 450 for 1 min 5 five times with 50% duty cycle at output 3. After confirmation under a phase contrast microscope that grossly intact nucleoli were isolated, the nucleolar suspension was overlaid onto 3 ml of Buffer 4 (10 mm HEPES, pH 7.9, 0.5 mm MgCl2 and 880 mm sucrose) in a 5-ml tube and centrifuged at 2,800 × g for 10 min at 4 °C. The nucleolar pellet was resuspended in 100 µl of Buffer 3 and incubated with Buffer A or FRGY2a as described above for the nucleolar disassembly assay.
Transfection of FRGY1, YB1, and YB1-C into HeLa Cells
We subcloned cDNAs of FRGY1, YB1, and YB1-C into pcDNA3.1/Myc-His(−)A with a His6 tag at the C terminus (Invitrogen). For transfection of the plasmids, we added 1 µg of plasmid and 1 µl of Lipofectamine 2000 (Invitrogen) to 1 × 105 HeLa cells cultured on cover slides in 48-well plates following the instruction of Lipofectamine 2000. Vector alone was used for the mock transfection. Protein expression was detected by immunofluorescence microscopy using anti-His antibody (Santa Cruz Biotechnology, diluted at 1:1,000) and Cy3-labeled secondary antibody (Amersham Biosciences, 1:1,000) 24 h after transfection.
Immunofluorescence Microscopy
Nuclei were stained with primary antibodies against Xenopus B23 (provided by M. Schmidt-Zachmann, diluted at 1:30,000) (14), mouse B23 (Santa Cruz Biotechnology, 1:1,000), UBF (a gift from B. McStay, 1:1,000) (15) and fibrillarin (Cytoskeleton, 1:1,000), and secondary antibodies labeled with Cy2, Cy3, or Cy5 (Amersham Biosciences, all 1:1,000). Topro 3 (Molecular Probes) was used for DNA counterstaining. The mounting solution for the cover slides contained 1,4-diazabicyclo(2.2.2)octane (Sigma), 50% glycerol, and phosphate-buffered saline. Images were captured with an Axioskop 2 fluorescence microscope equipped with a Radiance 2100 confocal system (Zeiss). The results were quantified with 200 randomly selected nuclei or cells in each experiment.
Electron Microscopy
This procedure was performed as described previously (1). The nuclei were fixed with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4. They were post-fixed in 1% osmium tetroxide, dehydrated in graded ethanol, and embedded in Polybed 812 (Polysciences). Ultrathin sections (80 nm) were stained with uranyl acetate and lead citrate and examined with the Joel 1200-EX electron microscope.
His Tag Pull-down
After incubation of HeLa or XL2 nuclei with 15 µM YB1, YB1-N, YB1-C, and FRGY2a-C for 1.5 h, extract was prepared from the nuclei as described above. Thirty µl of extract prepared from 2 × 106 nuclei was incubated with 150 µl of binding buffer (50 mm NaH2PO4, pH 8.0, 350 mm NaCl, 10 mm imidazole, 0.1% Triton X-100, and 10% glycerol) and 50 µl of nickel resin at 4 °C for 2 h. The resin was washed with 800 µl of binding buffer, and the bound proteins (input and associated proteins) were eluted by elution buffer (the same as binding buffer except for 250 mm imidazole). The input and associated proteins were compared by silver staining of SDS-PAGE gels.
Mass Spectrometry
Protein bands were cut out from the SDS protein gel and digested with trypsin. The peptide fragments were applied to Biflex III (Bruker Daltonics) for matrix-assisted laser desorption ionization-time of flight and analyzed with the MASCOT software (Matrix Science). Protein identities were confirmed by LCQ Classic ion trap mass spectrometry (ThermoFinnigan) equipped with the Pico View nanoelectrospray source (New Objective).
Immunoprecipitation
HeLa cells transfected with YB1-C-His6 using Lipofectamine 2000 were incubated for 24 h. Fifty µl of extract prepared from these cells as described above was precleared with GammaBind G-Sepharose (Amersham Biosciences) and then incubated with 4 µg of anti-His antibody or control IgG (both from Santa Cruz Biotechnology) for 1 h at 4 °C with gentle rocking. Following the addition of 30 µl of GammaBind G, the extract was incubated for an additional 1 h at 4 °C with gentle rocking. The proteins bound to GammaBind G were eluted with SDS-PAGE sample buffer, applied to SDS-PAGE, and immunoblotted with anti-His antibody (diluted at 1:1,000) and anti-B23 antibody (Zymed Laboratories Inc., 1:1,000).
For immunoprecipitation of radiolabeled proteins, cDNAs encoding YB1-C and B23 were subcloned into the pSP64 poly(A) expression plasmid. These two proteins were separately prepared in vitro in the presence of [35S]Met using the TnT quick coupled transcription/translation system (Promega). Fifty µl of the reaction mix containing 35S-labeled YB1-C was precleared with 40 µl of GammaBind G-Sepharose and then incubated with 4 µg of recombinant B23-His6 for 1 h at 4 °C. After another incubation for 1 h at 4 °C with 6 µg of anti-His antibody or control IgG, 20 µl of GammaBind G-Sepharose was added and further incubated for 1 h at 4 °C. Finally, the proteins bound to the GammaBind G were eluted with SDS-PAGE sample buffer and applied to gel electrophoresis and autoradiography. Immunoprecipitation from the combination of YB1-C-His6 and 35S-labeled B23 was performed by the same procedure.
B23 Knockdown by siRNA
The sense strand sequences of short interfering RNA (siRNA) are as follows: WT-1 (5′-AAGUAUAUCUGGAAAGCGGUC-3′), MT-1 (5′-AAGUAUAUCUCCAAAGCGGUC-3′, underlined are mutated ribonucleotides), WT-2 (5′-AGUGGAAGCCAAAUUCAUCTT-3′), and CTL (5′-ACUACCGUUGUUAUAGGUGTT-3′). These siRNA were fluorescein-labeled at the 5′ end. HeLa cells (3 × 106) were seeded in a 10-cm tissue culture plate and transfected with 100 nm siRNA on day 1 and day 4 using 70 µl of Lipofectamine 2000 each. The cells were used for the nucleolar disassembly assay on day 8. It was necessary to transfect siRNA twice to knock down the B23 level below the threshold of detection by immunostaining. To add back B23, we subcloned B23 cDNA into the pDsRed-C1 vector (Clontech). We introduced 50 µg of the plasmid with 50 µl of Lipofectamine 2000 into HeLa cells in a 10-cm dish that had already been treated with B23 siRNA. The DsRed-positive cells were sorted with a FACSAria fluorescence-activated cell sorter (BD Biosciences) and used for the nucleolar disassembly assay with 15 µM YB1-C.
RESULTS
Nucleolar Disassembly by FRGY2a-related Proteins
We first studied nucleolar disassembly with FRGY1, a Xenopus FRGY2a-related protein, and its human ortholog, YB1. FRGY1 is a ubiquitously expressed somatic cell protein that is 46.7% identical to FRGY2a at the amino acid level (5). FRGY1 is expressed in Xenopus XL2 cells as demonstrated by reverse transcription-PCR (Fig. 1A). In human cells, YB1 plays multiple roles in transcriptional regulation of a wide variety of genes, masking of mRNA in many cell types, and mediation of stress responses (7). In immunofluorescence staining, YB1 is primarily detectable in the cytoplasm without any clear signal in nucleoli (16). To examine the roles of these proteins in nucleolar disassembly, we prepared recombinant full-length, N-terminal, and C-terminal domains of FRGY2a, FRGY1, YB1, and EGFP-FRGY2a (Fig. 1B). In addition, we purified endogenous YB1 from HeLa cell extract based on its ability to disassemble HeLa cell nucleoli in isolated nuclei (Fig. 1B). Since HeLa cells contain 2.6 × 106 YB1 molecules/cell (17) and 1 ml of HeLa cell pellet, which was depleted with culture medium as much as possible, contains 0.7−1.2 × 109 cells, we estimated the YB1 concentration within HeLa cells to be 3.0−5.2 µm. (2.6 × 106 molecules/cell × 0.7 × 109 cells/ml × 10³ ml/l)/(6.02 × 1023 molecules/mole) = 3.0 µm. (6.02 × 1023 is Avogadro’s number.) Endogenous YB1 was undetectable in the isolated nuclei (data not shown). All of these proteins, except for the N-terminal domains, could disassemble nucleoli in over 90% of the nuclei from HeLa and XL2 cells, as monitored by loss of definitive nucleoli with phase contrast microscopy and the disappearance of the abundant nucleolar protein B23 (also called numatrin, NO38, and nucleophosmin) from nuclei in the in vitro nucleolar disassembly assay (Fig. 1C). These proteins were effective above 15 µM, which was slightly higher than the estimated YB1 concentration within the cells. This difference can be explained by potential problems with the recombinant proteins, such as aggregation and aberrant three-dimensional structures. Although FRGY2a purified from Xenopus egg extract could disassemble Xenopus and mammalian nucleoli, crude Xenopus egg extract could disassemble only Xenopus nucleoli (3). The reason for this species specificity of egg extract is currently unknown.
FIGURE 1. Nucleolar disassembly by FRGY2a, FRGY1, and YB1.
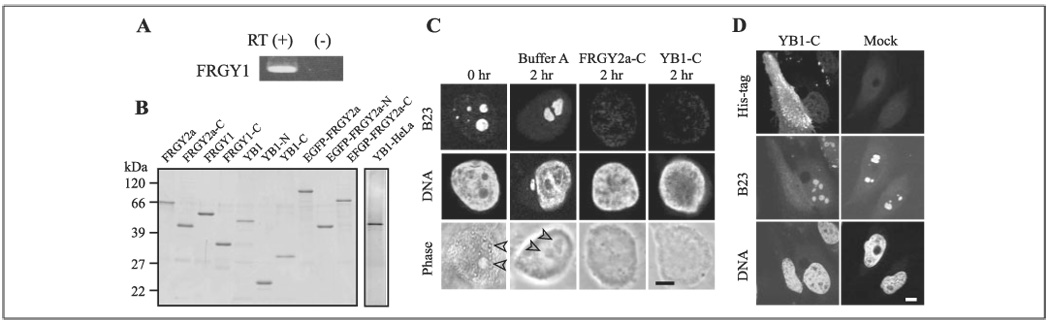
A, reverse transcription-PCR of FRGY1 from total RNA isolated from XL2 cells. The results with (+) and without (−) reverse transcription are shown. B, silver-stained gels loaded with YB1 purified from HeLa cell extract (YB1-HeLa), recombinant FRGY2a, and its related proteins. C, disassembly of HeLa cell nucleoli by recombinant FRGY2a-C and YB1-C. HeLa nuclei were incubated with Buffer A and these recombinant proteins at 15 µm for 2 h followed by immunostaining with anti-B23 antibody. A nucleus before incubation (0 h) is shown for comparison. DNA was counterstained with Topro 3. Arrowheads indicate nucleoli. Scale bars, 4 µm for C and D. D, dispersal of B23 in HeLa cells by transfected His-tagged YB1-C. The cells were co-stained with anti-His antibody, anti-B23 antibody, and Topro 3. Note that the YB1-C expressing cell does not show black spots corresponding to nucleoli in the Topro 3 staining. Mock, mock transfection.
Transfected full-length FRGY2a is sequestered in the cytoplasm of XL2 cells, presumably by association with mRNA through the RNP1-like and RNP2-like motifs in the N-terminal domain, and does not disassemble nucleoli (3). In contrast, transfected FRGY2a-C can enter nuclei and disassemble nucleoli (3). Likewise, although transfected full-length FRGY1 and YB1 did not enter nuclei or disassemble nucleoli (data not shown), FRGY1-C and YB1-C could enter nuclei and disassemble nucleoli in 25–40% of the expressing HeLa and XL2 cells (Fig. 1D). The nuclei with dispersed nucleoli displayed irregular shapes 48 h after transfection, and most of these cells died by 72–96 h after transfection (not shown).
Nucleolar Accumulation of FRGY2a during Nucleolar Disassembly
Next we investigated the subnuclear localization of EGFP-FRGY2a during nucleolar disassembly in XL2 nuclei. Although video microscopy of a single cell would have been ideal for this purpose, the high background level of the EGFP signal in the reaction mix made it difficult to interpret. As an alternative, we incubated XL2 nuclei with EGFP-FRGY2a and aborted the reaction at several time points by paraformaldehyde fixation to observe the temporal profile of the EGFP-FRGY2a localization during nucleolar disassembly. A preliminary study confirmed that EGFP alone did not localize to nucleoli (data not shown). The combination of EGFP-FRGY2a localization and nucleolar disassembly could roughly be divided into four patterns (Fig. 2, A and B). Pattern A showed negligible nuclear uptake of EGFP-FRGY2a and no dispersal of B23. Fifteen minutes into the reaction, pattern B, in which EGFP-FRGY2a primarily accumulated in the nucleoli and was weakly distributed in the nucleoplasm, appeared in ~20% of the nuclei. Although the nucleoli remained grossly intact in the phase contrast image of this pattern, the B23 level was significantly decreased, suggesting that nucleolar disassembly was already taking place at the molecular level. After 30 min of incubation, EGFP-FRGY2a localized to small nucleolar remnants accompanied by significant nucleoplasmic distribution in about 80% of the nuclei (pattern C). B23 was undetectable in the nucleoli in this pattern. Pattern C remained until the end of the course, except that EGFP-FRGY2a was no longer detectable in the nucleoplasm in ~10% of the nuclei after 2 h (pattern D). In pattern D, EGFP-FRGY2a colocalized with the RNA polymerase I transcription factor UBF, which remained bound to rDNA during nucleolar disassembly (3), implying binding of EGFP-FRGY2a to rDNA or associated molecules (Fig. 2C). This was confirmed by the disappearance of the EGFP signal with DNase I treatment of the nuclei. EGFP-FRGY2a-C showed essentially the same four distribution patterns during nucleolar disassembly (data not shown). The nucleolar localization indicates direct interactions between FRGY2a and an unidentified nucleolar component(s). Because pre-rRNA transcription is not grossly affected by FRGY2a (3), we do not know the functional implications of the localization of EGFP-FRGY2a on rDNA or associated molecules.
FRGY2a Directly Disassembles Nucleoli
To further examine whether FRGY2a disassembles nucleoli as a result of direct interactions with nucleolar components or it requires other nuclear or cytoplasmic components during this process, we incubated isolated HeLa cell nucleoli with FRGY2a. We washed the isolated nucleoli extensively with Buffer A prior to incubation to eliminate other nuclear and cytoplasmic components from the nucleolar suspension. Double staining of the nucleoli with antibodies against B23 and UBF (as an indicator of rDNA) was used to monitor nucleolar disassembly. The nucleoli incubated in Buffer A remained as large as those in intact cells (B23 stain) and contained both B23 (green) and UBF (red), as shown by the yellow signals in the merged image (Fig. 3). By contrast, the nucleoli incubated with FRGY2a were smaller (B23 stain), and their B23 was separated from UBF as demonstrated by the many red particles in the merged image. This result shows that FRGY2a can directly disassemble nucleoli without significant contributions from other cellular components.
FIGURE 3. Disassembly of isolated nucleoli by FRGY2a.
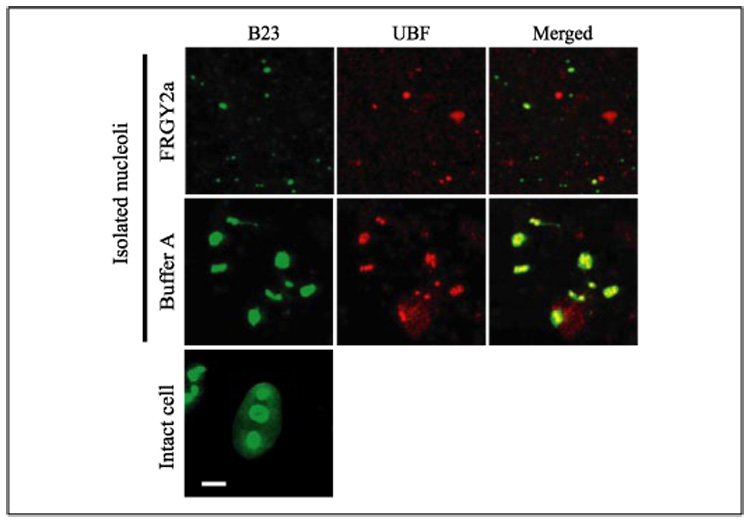
Isolated HeLa cell nucleoli double-stained with anti-B23 (green) and anti-UBF (red) antibodies following incubation with FRGY2a and Buffer A for 2 h are shown. Intact HeLa cell nuclei are also shown for size comparison of the nucleoli. Bar, 4 µm.
Isolation of FRGY2a-interacting Proteins
To identify molecules that interact with FRGY2a during nucleolar disassembly, we employed the His tag pull-down approach. After incubation of HeLa nuclei with YB1-N, YB1-C, and FRGY2a-C (all tagged with His6) separately for 90 min, the His-tagged bait and associated proteins were isolated using nickel resin and applied to SDS-PAGE. Since only YB1-C and FRGY2a-C could disassemble nucleoli, we focused on proteins that were pulled down with both YB1-C and FRGY2a-C (Fig. 4A, second gel from the left, YB1-C HPD and FRGY2a-C HPD) but not with YB1-N (YB1-N HPD). Only one band at about 40 kDa fulfilled this criterion (Fig. 4A, arrowheads). Mass spectrometry identified this protein as B23. When this experiment was repeated with the combination of XL2 nuclei and FRGY2a-C, B23 was again recovered with FRGY2a-C (Fig. 4A, a doublet indicated by an arrow). Because B23 is involved in ribosome biogenesis by interacting with 40 S and 65 S pre-ribosomal particles (18), we suspected that B23 might be indirectly bound to YB1-C and FRGY2a-C via nucleolar RNA. However, RNase A treatment of the pulled down sample did not alter the protein pattern, indicating that binding of FRGY2a-C and B23 was not mediated by RNA (Fig. 4A, RNase A − and +). The His tag pull-down approach also recovered other pre-ribosomal proteins, depending on the combination of the recombinant proteins and cell types. They included proteins S20 and S25 and the nucleolar protein NO 29, which forms a complex with B23 and is also a component of pre-ribosomes (19) (Fig. 4B). Because these three proteins do not contain a His stretch, which is necessary for binding to nickel resin, it is tempting to hypothesize that they were recovered as pre-ribosomal fragments indirectly bound to nickel resin via B23. B23 release has been used in this study as an indicator of nucleolar disassembly solely because the anti-B23 antibody provided a strong signal in immunostaining. It is a coincidence that B23 turned out to be a binding protein for FRGY2a and YB1.
FIGURE 4. Binding of YB1-C and FRGY2a-C to B23.
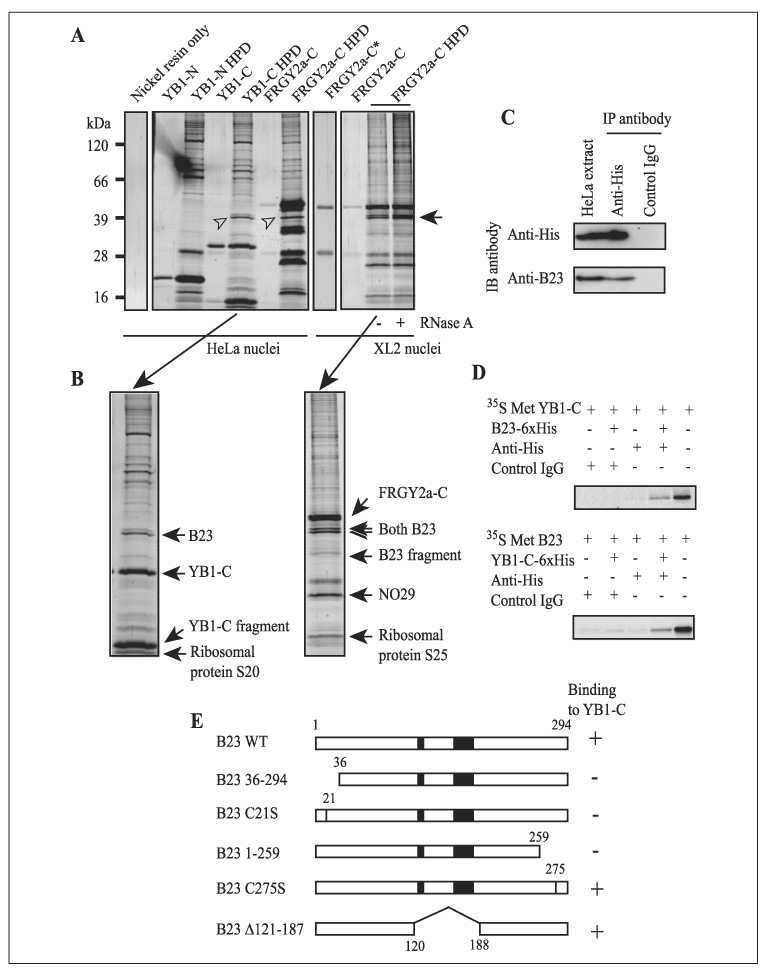
A, His tag pull-down of proteins bound to YB1-N, YB1-C, and FRGY2a-C from extract of HeLa nuclei (first and second from the left) and XL2 nuclei (first and second from the right) after nucleolar disassembly. Input recombinant protein and pulled-down proteins (HPD) were loaded onto the gels and were detected by silver staining. B23 is indicated by arrowheads and an arrow (doublet). The relative ratio between input and the HPD on the gels was 1:10 for each pair, except for FRGY2a-C*, which contained four times as much FRGY2a-C as other FRGY2a-C wells. B, identification of the proteins detected in A by mass spectrometry. C, immunoblotting of proteins immunoprecipitated with anti-His antibody from extract prepared from HeLa cells that were transfected with YB1-C-His6. Antibodies used for immunoprecipitation (IP anti-body) and for detection by immunoblotting (IB antibody) are shown. Normal rabbit IgG was used as control IgG. The far left well contains the input HeLa cell extract. The relative ratio between the input HeLa extract and the precipitated protein applied to the protein gel was 1:5 (1 × 105 cells equivalent for HeLa extract and 5 × 105 cells equivalent for Anti-His). D, autoradiography to examine co-immunoprecipitation of YB1-C and B23. The far right wells contain input radiolabeled proteins with the relative ratio between input and precipitated protein loaded on the gels at 3:1. E, mutation analysis of B23 domains required for binding to YB1-C. The number represents amino acid number. Cys-21 was replaced by Ser in B23 C21S and Cys-275 by Ser in B23 C275S. Black boxes indicate acidic amino acid clusters.
Co-immunoprecipitation of YB1-C and B23
Binding of YB1-C and B23 was confirmed by two co-immunoprecipitation experiments, one with proteins isolated from transfected cells and the other with proteins translated in vitro. In the first approach, we transfected HeLa cells with YB1-C-His6 and cultured the cells for 24 h. We then prepared extract from the transfected cells and immunoprecipitated YB1-C-His6 with anti-His antibody. Immunoblotting of the precipitated proteins showed that B23 was co-precipitated by anti-His antibody, but not by control IgG (Fig. 4C), demonstrating that YB1-C and B23 interact under endogenous conditions.
In the second approach, we radiolabeled YB1-C with [35S]Met by in vitro transcription/translation and incubated the protein with human B23-His6. Anti-His antibody co-precipitated [35S]Met YB1-C only in the presence of B23-His6 and anti-His antibody (Fig. 4D). The combination of [35S]Met B23 and YB1-C-His6 produced the same result. Further co-immunoprecipitation studies of a series of B23 mutants showed that amino acids located within the 35 residues in the N terminus and another 35 residues in the C terminus of B23 were crucial for binding to YB1-C in vitro (Fig. 4E). Among the N-terminal 35 amino acids, Cys-21 is crucial for binding to YB1-C, as demonstrated by the lack of binding in the replacement mutant of Cys-21 to Ser. It is somewhat surprising that residues 121–187 of B23, which contain two clusters of acidic amino acids, were unnecessary for binding to YB1-C (Fig. 4E) because our previous study showed that four basic/aromatic amino acid clusters in FRGY2a-C were necessary for nucleolar disassembly. These four positively charged amino acid clusters presumably bind to other negatively charged regions in B23. Overall, these findings demonstrate that B23, the only commonly recovered protein from both HeLa and XL2 cells by the His tag pull-down experiment, is capable of binding to YB1-C, depending on both its N termini and its C termini.
Requirement of B23 for Nucleolar Disassembly by FRGY2a
To clarify whether binding of YB1-C and FRGY2a-C to B23 was the cause or the result of nucleolar disassembly, we depleted B23 in HeLa cells using siRNA and incubated the cells with YB1-C. Two independent 21-mer wild type RNA duplexes, WT-1 and WT-2, efficiently depleted more than 80% of B23 as shown by immunostaining and immunoblotting (Fig. 5, A and B). Importantly, the nucleoli in these cells were grossly intact, as shown by phase contrast images, retention of another nucleolar protein fibrillarin (Fig. 5A), and electron microscopy (Fig. 5D). B23 depletion is known to partially inhibit DNA replication, but the cells remain alive (20, 21). As negative controls, we prepared two siRNAs: MT-1, in which two ribonucleotides were replaced in WT-1, and CTL, which had no homology with known sequences in BLAST search. Neither of these control siRNAs induced B23 loss upon transfection into HeLa cells (Fig. 5, A and B). Results for CRL are not shown).
FIGURE 5. Prevention of YB1-mediated nucleolar disassembly by B23 depletion.
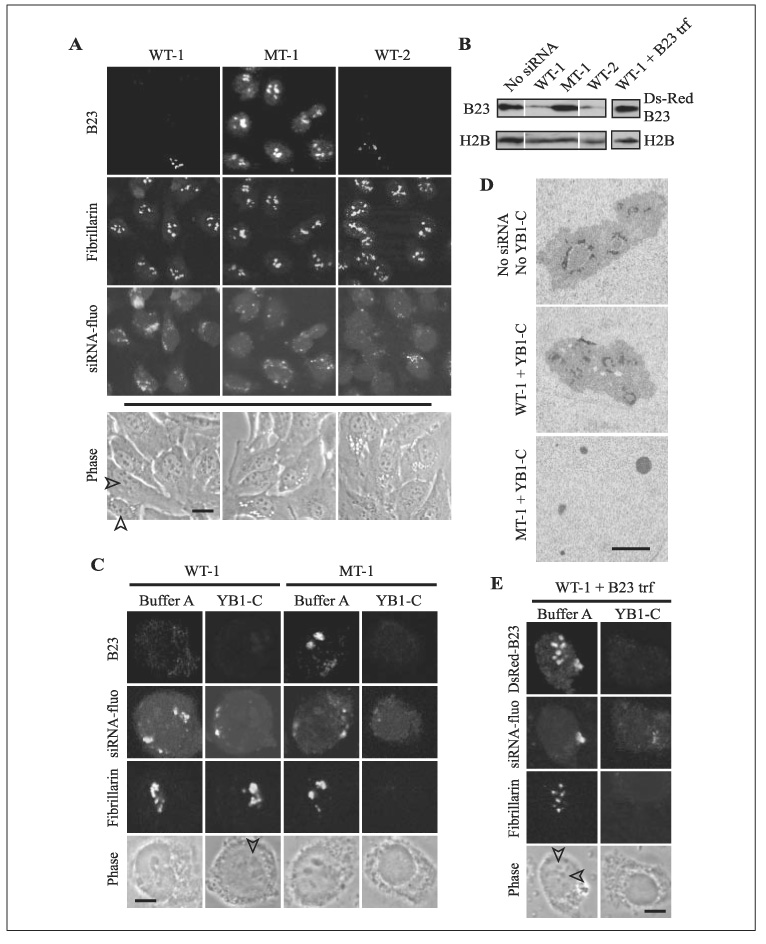
A, the specific loss of B23 in HeLa cells treated with siRNAs WT-1 and WT-2, but not by MT-1, is shown. The cells were double-stained with antibodies against B23 and fibrillarin. Incorporation of siRNA into the cells was detected by fluorescein tagged to siRNA (siRNA-fluo). Phase contrast images were captured from different cells in culture medium before immunostaining since the mounting solution obscured the phase contrast images. Arrowheads indicate nucleoli. B, immunoblotting of B23 and histone H2B (loading control) in the cells treated with siRNA. WT-1 + B23 trf indicates that the cells were treated with WT-1 siRNA first and then transfected with DsRed-B23. Each lane contained 1 × 105 cells. C, YB1-C could not disassemble the nucleoli of HeLa cells in which B23 was depleted by WT-1 siRNA. Note that fibrillarin remained in large nucleoli (arrowhead) in the WT-1-treated nucleus when incubated with YB1-C. In contrast, fibrillarin was lost, and the nucleoli became invisible in the MT-1-treated nucleus incubated with YB1-C. D, electron micrographs of the nuclei treated with siRNA and YB1-C. E, When WT-1- treated HeLa cells were transfected with DsRed- B23, the cells regained sensitivity to the nucleolar disassembly activity of YB1-C. Bar, 20 µm(A), 4 µm (C and E), and 1 µm (D).
When B23-depleted nuclei were incubated with YB1-C for 2 h, more than 75% of the nuclei retained intact nucleoli as judged by phase contrast images and fibrillarin staining (Fig. 5C, arrowhead), indicating resistance to YB1-C-mediated nucleolar disassembly. WT-2 showed the same results (data not shown). However, when nuclei treated with MT-1 and CTL were incubated with YB1-C, the nucleoli disappeared and fibrillarin became undetectable. Resistance of nucleoli to YB-1-C mediated nucleolar disassembly after B23 depletion by WT-1 was confirmed by electron microscopy. There was no evidence of fragmented nucleoli in the cells transfected with WT-1 and subsequently treated with YB1-C (Fig. 5D, WT-1 + YB1-C) in more than 50 nuclear sections examined. In contrast, only small nucleolar fragments were detectable in the nuclei treated with MT-1 and YB1-C.
Finally, we confirmed the specific requirement of B23 for nucleolar disassembly by adding back B23 into the B23-depleted HeLa cells. First, we transfected the DsRed-tagged B23 into HeLa cells whose B23 had already been depleted by WT-1. Although the expression level of the transfected B23 was variable among the cells, the average expression level was similar to that of endogenous B23 (Fig. 5B). After collecting the cells expressing DsRed-B23 by fluorescence-activated cell sorting, we isolated the nuclei and incubated them with YB1-C for the nucleolar disassembly assay. As shown in Fig. 5E, DsRed-B23 and fibrillarin disappeared from the nuclei together with the loss of definitive nucleoli by phase contrast microscopy. This finding indicates that the sensitivity to YB1-C-mediated nucleolar disassembly was restored by re-supply of B23.
DISCUSSION
This study identified B23 as a key mediator of nucleolar disassembly induced by FRGY2a and YB1. An emerging hypothesis is that FRGY2a and YB1 sequester B23 and associated molecules from nucleoli, triggering the disassembly of the nucleoli. Depletion of B23 alone by siRNA did not disassemble nucleoli, which may seem contradictory to the hypothesis; however, it should be emphasized that selective prevention of B23 production by siRNA and removal of existing B23 that is embedded within the nucleoli, interacting with many proteins and RNA, can cause different consequences. It is possible that indirect displacement of pre-ribosomal components and other B23-interacting proteins/RNA is the real driving factor for nucleolar disassembly, as suggested by co-pull-down of pre-ribosomal proteins with B23 (Fig. 4B). This possibility is supported by a report showing that B23 is a component of large pre-rRNA ribonucleoprotein (RNP) complexes that contain pre-RNA, ribosomal proteins, fibrillarin, and another nucleolar protein, nucleolin (22). Although FRGY2a and YB1 may also interact with nucleolar RNA by charge interactions through basic/aromatic domains, such a nonspecific interaction alone cannot account for the requirement of B23 for nucleolar disassembly. EGFP-FRGY2a remained bound to the rDNA region of the nucleolar remnants after B23 was completely dispersed (Fig. 2A, pattern D), implying additional interactions between FRGY2a and unidentified molecules. The functional significance of this observation is unknown.
B23 was originally identified as a heavily phosphorylated protein in the granular component of nucleoli (18, 23). It has numerous functions including ribosome biosynthesis (18, 24), nuclear import (25), centrosome duplication (26), insulation of enhancers (27), and cell growth regulation by interaction with p53 (28) and the ARF tumor suppressor (29). In addition, B23 contains intrinsic RNase activity, which may be important for pre-rRNA processing (24, 30). Consistent with roles in these essential functions, B23 knock-out mice exhibit aberrant organogenesis and die around embryonic day 12, primarily due to severe anemia (21).
Nucleoli can undergo dynamic structural reorganization in many circumstances, including mitotic disassembly/reassembly and segregation by chemicals such as the cancer drug actinomycin D and the adenosine analogue 5,6-dichloro-β-D-ribofuranosyl-benzimidazole (9, 31). In addition to these global reorganizations, many nucleolar proteins are constantly and rapidly shuttling between nucleoli and nucleoplasm without disrupting the overall nucleolar integrity (32). Despite extensive efforts in nucleolar research, little is known about the molecules that induce such dynamic nucleolar reorganization through direct interaction with nucleolar components. This work and our previous study (3) represent the first identification of a direct mediator of nucleolar disassembly and its interacting protein within nucleoli. At this stage, the only known in vivo nucleolar dynamics in which FRGY2a is involved is nucleolar disassembly in the context of somatic cell nuclear cloning. It is possible that FRGY2a is important for the induction of the disassembly of endogenous nucleoli in Xenopus eggs and subsequent maintenance of the disassembled nucleoli until the midblastula transition. Targeted disruption of Msy2, the mouse ortholog of Frgy2a, impairs spermatogenesis and oogenesis, but the abnormality is confined to germ cell development, reflecting the germ cell-specific expression of the gene (33). Nucleolar disassembly and reassembly are not as obvious in mouse embryos as in Xenopus embryos, and potential abnormalities in nucleolar morphology have not been reported in these mice. Since these knock-out mice survive beyond the newborn stage, the embryonic nucleoli are, presumably, almost intact. YB1 knock-out mice face embryonic lethality around embryonic day 13 (34), implying that these cells can process mitotic nucleolar disassembly without major abnormality, in the absence of YB1. This can be explained by at least two possibilities; either YB1 is irrelevant to mitotic nucleolar disassembly, or a compensation mechanism exists for the loss of YB1 in the nuclei. Close examination of mitotic nucleolar disassembly, as well as chemically induced nucleolar segregation in these cells, will be necessary to clarify potential involvement of YB1 in these nucleolar dynamics.
Recent studies have shown that a number of non-ribosomal proteins are localized in the nucleolus, largely for unknown reasons. They include proteins involved in cell cycle regulation (35), tumor suppression (36–38), aging (39), human hereditary diseases (38, 40), and telomere length regulation (41). This ever expanding list suggests the importance of the nucleolus not only for regulating many normal cellular functions but also in understanding the pathology of numerous human diseases. Therefore, if we wish to ultimately treat certain diseases, it will be essential to understand how nucleolar structure is organized and how its dynamics are regulated. Molecular analysis of nucleolar disassembly by FRGY2a will provide one promising approach to answer these questions.
Acknowledgments
We are grateful to J. Fowler and J. Haroldson for technical support and M. O’Conner and E. Coucouvanis for critical reading of the manuscript. We thank V. Evdokimova, B. McStay, and M. Schmidt-Zachmann for the generous contributions of reagents. We appreciate the electron microscopy work of G. Ahlstrand and the mass spectrometry work of L. Higgins and T. Krick. Large scale culture of HeLa cells was purchased from the National Cell Culture Center.
Footnotes
This work was supported in part by grants from the National Institutes of Health (Grant R01 GM068027), the University of Minnesota Graduate School, and the Minnesota Medical Foundation (to N. K.)
The abbreviations used are: EGFP, enhanced green fluorescent protein; siRNA, short interfering RNA; RNP, ribonucleoprotein.
References
- 1.Verheggen C, Le Panse S, Almouzni G, Hernandez-Verdun D. J. Cell Biol. 1998;142:1167–1180. doi: 10.1083/jcb.142.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurdon JB. J. Mol. Biol. 1965;12:27–35. doi: 10.1016/s0022-2836(65)80279-0. [DOI] [PubMed] [Google Scholar]
- 3.Gonda K, Fowler J, Katoku-Kikyo N, Haroldson J, Wudel J, Kikyo N. Nat. Cell Biol. 2003;5:205–210. doi: 10.1038/ncb939. [DOI] [PubMed] [Google Scholar]
- 4.Sommerville J, Ladomery M. FASEB J. 1996;10:435–443. doi: 10.1096/fasebj.10.4.8647342. [DOI] [PubMed] [Google Scholar]
- 5.Tafuri SR, Wolffe AP. Proc. Natl. Acad. Sci. U. S. A. 1990;87:9028–9032. doi: 10.1073/pnas.87.22.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolffe AP. BioEssays. 1994;16:245–251. doi: 10.1002/bies.950160407. [DOI] [PubMed] [Google Scholar]
- 7.Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M. BioEssays. 2003;25:691–698. doi: 10.1002/bies.10300. [DOI] [PubMed] [Google Scholar]
- 8.Simard R. Int. Rev. Cytol. 1970;28:169–211. doi: 10.1016/s0074-7696(08)62543-7. [DOI] [PubMed] [Google Scholar]
- 9.Hernandez-Verdun D, Roussel P, Gebrane-Younes J. J. Cell Sci. 2002;115:2265–2270. doi: 10.1242/jcs.115.11.2265. [DOI] [PubMed] [Google Scholar]
- 10.Dignam JD, Martin PL, Shastry BS, Roeder RG. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- 11.Evdokimova V, Ruzanov P, Imataka H, Raught B, Svitkin Y, Ovchinnikov LP, Sonenberg N. EMBO J. 2001;20:5491–5502. doi: 10.1093/emboj/20.19.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kikyo N, Wade PA, Guschin D, Ge H, Wolffe AP. Science. 2000;289:2360–2362. doi: 10.1126/science.289.5488.2360. [DOI] [PubMed] [Google Scholar]
- 13.Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI. Curr. Biol. 2002;12:1–11. doi: 10.1016/s0960-9822(01)00650-9. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt-Zachmann MS, Hugle B, Scheer U, Franke WW. Exp. Cell Res. 1984;153:327–346. doi: 10.1016/0014-4827(84)90604-9. [DOI] [PubMed] [Google Scholar]
- 15.Cairns C, McStay B. Nucleic Acids Res. 1995;23:4583–4590. doi: 10.1093/nar/23.22.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang YF, Homer C, Edwards SJ, Hananeia L, Lasham A, Royds J, Sheard P, Braithwaite AW. Oncogene. 2003;22:2782–2794. doi: 10.1038/sj.onc.1206357. [DOI] [PubMed] [Google Scholar]
- 17.Davydova EK, Evdokimova VM, Ovchinnikov LP, Hershey JW. Nucleic Acids Res. 1997;25:2911–2916. doi: 10.1093/nar/25.14.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt-Zachmann MS, Hugle-Dorr B, Franke WW. EMBO J. 1987;6:1881–1890. doi: 10.1002/j.1460-2075.1987.tb02447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zirwes RF, Schmidt-Zachmann MS, Franke WW. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11387–11392. doi: 10.1073/pnas.94.21.11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brady SN, Yu Y, Maggi LB, Jr, Weber JD. Mol. Cell. Biol. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 22.Pinol-Roma S. Mol. Biol. Cell. 1999;10:77–90. doi: 10.1091/mbc.10.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feuerstein N, Mond JJ. J. Immunol. 1987;139:1818–1822. [PubMed] [Google Scholar]
- 24.Hingorani K, Szebeni A, Olson MO. J. Biol. Chem. 2000;275:24451–24457. doi: 10.1074/jbc.M003278200. [DOI] [PubMed] [Google Scholar]
- 25.Szebeni A, Herrera JE, Olson MO. Biochemistry. 1995;34:8037–8042. doi: 10.1021/bi00025a009. [DOI] [PubMed] [Google Scholar]
- 26.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 27.Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G. Mol. Cell. 2004;13:291–298. doi: 10.1016/s1097-2765(04)00029-2. [DOI] [PubMed] [Google Scholar]
- 28.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nat. Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 29.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y. Mol. Cell. 2003;12:1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 30.Savkur RS, Olson MO. Nucleic Acids Res. 1998;26:4508–4515. doi: 10.1093/nar/26.19.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung AK, Lamond AI. Crit. Rev. Eukaryotic Gene Expression. 2003;13:39–54. doi: 10.1615/critreveukaryotgeneexpr.v13.i1.40. [DOI] [PubMed] [Google Scholar]
- 32.Chen D, Huang S. J. Cell Biol. 2001;153:169–176. doi: 10.1083/jcb.153.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Medvedev S, Yu J, Tang LC, Agno JE, Matzuk MM, Schultz RM, Hecht NB. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5755–5760. doi: 10.1073/pnas.0408718102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu ZH, Books JT, Ley TJ. Mol. Cell. Biol. 2005;25:4625–4637. doi: 10.1128/MCB.25.11.4625-4637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visintin R, Hwang ES, Amon A. Nature. 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- 36.Tao W, Levine AJ. Proc. Natl. Acad. Sci. U. S. A. 1999;96:6937–6941. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nat. Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 38.Mekhail K, Gunaratnam L, Bonicalzi M-E, Lee S. Nat. Cell Biol. 2004;6:642–647. doi: 10.1038/ncb1144. [DOI] [PubMed] [Google Scholar]
- 39.Marciniak RA, Lombard DB, Johnson FB, Guarente L. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winokur ST, Shiang R. Hum. Mol. Genet. 1998;7:1947–1952. doi: 10.1093/hmg/7.12.1947. [DOI] [PubMed] [Google Scholar]
- 41.Wong JM, Kusdra L, Collins K. Nat. Cell Biol. 2002;4:731–736. doi: 10.1038/ncb846. [DOI] [PubMed] [Google Scholar]