

Abstract
The fluorescence emission of the single tryptophan (W233) of the mutant protein DD-carboxypeptidase from streptomyces is characterized by a red-edge excitation shift (REES), i.e., the phenomenon that the wavelength of maximum emission depends on the excitation wavelength. This phenomenon is an indication for a strongly reduced dynamic environment of the single tryptophan, which has a very low accessibility to the solvent. The REES shows, however, an unusual temperature and time dependence. This, together with the fluorescence lifetime analysis, showing three resolvable lifetimes, can be explained by the presence of three rotameric states that can be identified using the Dead-End Elimination method. The three individual lifetimes increase with increasing emission wavelength, indicating the presence of restricted protein dynamics within the rotameric states. This is confirmed by time-resolved anisotropy measurements that show dynamics within the rotamers but not among the rotamers. The global picture is that of a protein with a single buried tryptophan showing strongly restricted dynamics within three distinct rotameric states with different emission spectra and an anisotropic environment.
Keywords: tryptophan, fluorescence, lifetime, red-edge excitation shift, DD peptidase
Organic fluorophores in viscous or rigid environments and low temperatures often show particular spectroscopic behavior: Their wavelength of maximum emission increases with an increase in the wavelength of excitation. This phenomenon was first observed some 30 yr ago in low temperature glasses and has since been called the red-edge excitation shift (REES) and variants of it (Gallay and Purkey 1970; Weber and Shinitzky 1970; Itoh and Azumi 1973, 1975; Azumi et al. 1976; Demchenko 1981; Lakowicz and Keating-Nakamoto 1984; Chattopadhyay and Mukherjee 1999; Chattopadhyay et al. 2003). It is now well established that REES in organized assemblies, where it is most common, arises primarily because of the heterogeneity of solvation sites around the assemblies, which also contribute to inhomogeneous broadening of the absorption spectra (Klymchenko and Demchenko 2002; Mandal et al. 2004). An accurate self-explaining theoretical treatment of the red-edge effect was given by Demchenko and Sytnik (1991b), describing the fluorescence properties of 9,9′-Bianthryl with a two-state model. However, such a model is not applicable to tryptophan fluorescence. In a rigid environment, such as in a vitrified polar solvent at low temperature, the red-edge relaxation can be followed as a function of time. For example, time-resolved fluorescence spectroscopy has been used to probe the molecular motions at the aqueous interfaces of biological macromolecules and membranes. From such time-dependent Stokes shift (FSS) experiments, it has been inferred that water motions in the hydration layer are slowed down by one to three orders of magnitude (Nilsson and Halle 2005). Recently, the discovery that certain ionic liquids display REES (Mandal et al. 2004) and FSS (Mandal and Samanta 2005) at room temperature attracted much attention and inspired a promising theoretical model (Hu and Margulis 2006).
In proteins, REES was first observed for human serum albumin and, today, the same theoretical model used for organic fluorophores is also used to explain the red-edge effect in proteins (Demchenko 2002). A comprehensive study on single tryptophan proteins (Demchenko 1988) has shown that only tryptophan residues in class I and II of the Burstein classification (Reshetnyak et al. 2001) (λmax between 320 and 340 nm) show REES. Concerning highly buried tryptophan residues (λmax < 320nm), the tryptophan environment is generally considered to be too hydrophobic to show solvent dipolar relaxation (Demchenko 2002). For peptides and denaturated proteins, REES is not observed (Demchenko 2002) due to the high flexibility of these residues and the rapidly relaxing solvent in the denatured state. A notable exception is denatured spectrin (Chattopadhyay et al. 2003), which seems to have residual structure in the denatured state. For multi-tryptophan proteins, it should be realized that REES could be the trivial result of a different environment for each Trp, leading to a heterogeneity of individual spectra and a coupling of red excitation/red emission spectra.
In this study, we investigate the spectroscopic characteristics of the single tryptophan protein DD-carboxypeptidase-transpeptidase from Streptomyces R61 (R61). This exo-cellular peptidase has been used as a model for the membrane-bound dd-peptidases involved in bacterial cell wall biosynthesis (Frere and Joris 1985). R61 consist of 347 amino acids, two of which are tryptophan (Kelly et al. 1985). W233 is essential for the correct folding and activity of the enzyme, while W271 could be exchanged with lysine with no particular modification of the protein's biophysical and kinetic characteristics (Bourguignonbellefroid et al. 1992). We therefore produced the single tryptophan version of R61 by substituting tryptophan 271 with a lysine by site-specific mutagenesis as described elsewhere (Bourguignonbellefroid et al. 1992). Throughout this study we will refer to R61 as the single tryptophan version of the enzyme (W271L), if not otherwise specified. R61 has a quantum yield (Φ) of 0.09 and a fluorescence emission maximum (λmax) at 317 nm upon excitation at 295 nm at room temperature and pH 8 (Bourguignonbellefroid et al. 1992). In this protein we could observe a pronounced red-edge excitation shift. The REES shows, however, an unusual temperature and time dependence, which we can explain by the existence of different rotational conformers (rotamers) of the tryptophyl side chain, probing different environments. We also observe a shift in the fluorescence lifetimes with emission wavelength, which is attributed to nanosecond dynamics within the rotameric states.
Results
Steady-state fluorescence of R61: The red-edge effect
In good agreement with previously published data (Bourguignonbellefroid et al. 1992), R61 showed a λmax of 317 nm at room temperature (RT) and pH 8 upon excitation at 295 nm. According to the classification of tryptophan residues in proteins made by Reshetnyak and coworkers (Reshetnyak and Burstein 2001), W233 from R61 belongs to the class S of tryptophans, which includes buried tryptophan residues that can form exciplexes (hydrogen-bond complexes in the excited state) with polar residues in the surroundings in the stoichiometry of 1:1. The Stokes shift of R61 depends on the excitation wavelength (Fig. 1). At 8°C, the λmax increased from 315 to 340 nm as the λex was changed from 275 to 310 nm, producing a remarkable large red shift of 25 nm (Fig. 1A). Note that the majority of the REES occurs at excitation wavelengths higher than 290 nm, such that Tyrosine fluorescence does not contribute. This effect, which was also observed at room temperature (data not shown), has never been described for a tryptophan of class S in the Burstein classification (Demchenko and Lodokhin 1988; Demchenko 2002). Interestingly, the shape of the emission spectrum also changed as a function of the excitation wavelength as shown in Figure 1B for the normalized fluorescence spectra of the protein at 275 nm (spectrum 1), 295 nm (spectrum 2), 300 nm (spectrum 3), and 305 nm (spectrum 4). It is clear that the spectrum nr 1 (excitation at 275 nm) contains a large contribution of Tyr fluorescence, since there are 14 Tyr residues in the protein, but the other spectra with excitations at 295 nm and higher must be due to tryptophan fluorescence. In particular, at excitation wavelengths higher than 300 nm, the spectra of R61 fluorescence were wider than at lower λex, and two extra shoulders appeared at ∼340 and ∼350 nm (Fig. 1B). The extra shoulders in spectras 2 and 3 eventually corresponded to the maximum Stokes shift of the spectrum 4, after red-edge excitation.
Figure 1.

(A) Emission maxima of the steady-state fluorescence of 10 μM of R61 at 8°C in relation to the excitation wavelength. All spectra were corrected for the instrumental response and the Raman scattering of the solution. (B) Spectra of the steady-state fluorescence of 10 μM of R61 upon excitation at 275 nm (1), 295 nm (2), 300 nm (3), and 305 nm (4). The conditions were as in A.
The fluorescence properties of R61 were also investigated as a function of temperature. The thermal stability of R61 was calculated by measuring the temperature dependence of the steady-state fluorescence of the protein. The unfolding of a protein containing a buried tryptophan is accompanied by an increase in fluorescence at 350 nm due to the exposure of the tryptophan to the solvent. However, since the processes that quench the fluorescence of a chromophore are temperature dependent (Yu et al. 1995), the quantum yield of tryptophan also decreases with increasing temperature. To compensate for the latter effect, we plotted the ratio of the fluorescence value at 350 nm over the value at 317 nm (F350/317) as a function of temperature (Fig. 2A). The F350/317 of R61 was constant from 5°C to 48°C, and then it rapidly increased to its maximum value for temperatures higher than 65°C. This graph most likely describes the temperature unfolding of the protein. The midpoint of the transition was 55.9 ± 0.2°C (after fitting to a transition curve using Sigmaplot 8.0). Crucial to our study, we noticed that the λmax of R61 spectra also slightly changed with temperature (Fig. 2B). Interestingly, the λmax of R61 fluorescence decreased with increasing the temperature and not vice versa, as usually expected in similar systems (see below). At 48°C, which is close to the unfolding temperature of the enzyme (Fig. 2), the λmax was 316.4 nm, .3 nm lower than the λmax at 7.5°C. This behavior was observed at 295 nm (Fig. 2B) and 280 nm excitation wavelengths (data not shown). Of course, at higher temperatures a normal red shift was observed due to the denaturation of the protein.
Figure 2.

(A) Temperature unfolding of 5 μM of R61 in TRIS-EDTA buffer at pH 8. Every data point was calculated by dividing the value of fluorescence at 318 nm by the fluorescence value at 350 nm upon excitation at 295 nm. Each point is the average of four repetitions. All spectra were corrected for the instrumental response and the Raman scattering of the solution. (B) Temperature dependence of the maximum emission of the steady-state spectra of R61 (conditions were as in Fig. 1A).
Time-resolved fluorescence properties of W233
The time-resolved fluorescence analysis of W233 gave good fits to three exponential decays with χ2 values of ∼1.1. Three exponential fits were an improvement over double-exponential fits, which gave χ2 values of ∼1.9 and nonrandom weighted autocorrelation and residual functions. The fluorescence lifetimes were calculated from the exponential decays at several wavelengths using emission filters every 10(±5) nm from 320 to 380 nm (Fig. 3; Table 1). In this wavelength range, the average lifetime (<τ>) of R61 fluorescence linearly increased from 2.70 ± 0.2 ns at 320 nm to 3.83 ± 0.07 ns at 380 nm (Fig. 3, inset).
Figure 3.

Time-resolved fluorescence decays of R61 (5 μM) at different emission wavelength in Tris-EDTA buffer at pH 8 and 22°C. The traces were normalized to 10,000 counts. The instrument response function is 1 and the decays 2, 3, 4, and 5 are obtained using emission filters at 320, 340, 360, and 380 nm, respectively. For clarity, the decays at 330, 350, and 370 nm are omitted. The inset shows the average lifetime of the time-resolved fluorescence decays of R61 as a function of the emission wavelength. Excitation was done at 295 nm.
Table 1.
Time-resolved fluorescence data for W233 of R61 (5 μM) at 25°C in 50 mM TRIS-EDTA buffer at pH 8

The increase of <τ> over the accessible wavelength range was mainly due to two reasons: the increase of the relative contribution of the longest lifetime to the overall fluorescence from 320 to 380 nm and the increase of the values of the individual lifetimes in the same wavelength range (Table 1). gτ3, for example, increased from 7.4 ± 0.1 ns at 320 nm to 10.3 ± 0.3 ns at 380 nm, and its amplitude increased from 3.4 ± 0.1% to 12.0 ± 1.0% (Table 1).
The measured lifetimes were then grouped in three classes, namely τ1 (shortest lifetime), τ2 (medium lifetime), and τ3 (longest lifetime), and the decay-class associated spectra (DCAS) were calculated from the contribution of each lifetime to the steady-state fluorescence using Equation 1. The DCAS revealed that the three classes of lifetimes have different fluorescence emission maxima (Fig. 4A). τ2 emission was centered at wavelengths lower than 320 nm, probably coinciding with the steady-state λmax, and corresponded to a tryptophan in the Class S of the Burstein classification, while τ1 and τ3 showed emission maxima analogous to more solvent-exposed tryptophans. The λmax of τ1 was at ∼340 nm and the λmax of τ3 at ∼350 nm, corresponding to tryptophans in the class II and class III of the Burstein classification, respectively (Fig. 4A). The time-resolved decay-associated spectra (TRES) for the fluorescence of R61 were constructed from the time-dependent fluorescence decays and the steady-state fluorescence of the protein. This allowed the plot of the time-dependent Stokes shift for the frequency of the emission maximum [vmax(t)] as a function of time (Equation 2). The entire Stokes shift, from time zero to infinite time should correspond to the observable fraction of solvation of the relaxation process. To monitor the relaxation process we used the correlation function (Ct) that corresponded to the normalized time-dependent Stokes shift (Fig. 4). The relaxation process was completed after 25 ns, and the relaxation time, calculated from the area below the curve described by the C(t), was 14.0 ns (Table 2). The time dependence of the maximum Stokes shift [and C(t)] of the relaxation process of R61 showed a complex behavior (Fig. 4B). The maximum Stokes shift initially increased from 31,328 cm−1 (319.2 nm) at t0 [vmax(0)] to a maximum value of 31,546 cm−1 (317 nm) after 3.0 ns and then asymptotically decreased to 28,332 cm−1 (353 nm) at the maximum relaxation frequency [vmax(∞)] (Table 2).
Figure 4.

(A) Decay-class associated spectra of τ1 (●, right scale), τ2 (▲, left scale), and τ3 (○, right scale) components of R61 fluorescence. The excitation wavelength was 295 nm and the conditions are as in Figure 1. (B) Correlation function of R61 fluorescence in H2O (●) and D2O (○) as calculated from the steady-state and time-resolved fluorescence data (Equation 2).
Table 2.
Relaxation parameters of W233 of R61

Despite the low accessibility of W233, the fluorescence properties of R61 depended on the solvent used. The λmax of R61 was not affected by deuteriumoxide, and the shape of the fluorescence spectrum only slightly changed (data not shown). Contrary to the steady-state fluorescence, the time-resolved fluorescence of R61 in D2O, however, suggested that water is important in the relaxation process of the enzyme. At 320 nm, the three lifetimes were not very different from the lifetimes measured in water. The solvent-induced isotope effect (SIE = τD2O/τH2O) of τ1 was 0.91, of τ2 was 1.14, and of τ3 1.0 (Table 3). Upon increasing the emission wavelength to 380 nm, however, the SIE of τ1 gradually decreased to a value of 0.79, the SIE of τ2 remained roughly constant, while the SIE of τ3 increased up to 1.7 (Table 3). In D2O, the time correlation of the maximum Stokes shift of R61 fluorescence was similar to the solvent relaxation measured in water (Fig. 4B), but shifted to longer time. The C(t) value also reached a maximum at around 3 ns (3.2 ns, Fig. 4B), while the vmax(∞) value was 10 nm red shifted (363 nm, Table 2). In deuterated solvent the relaxation process was slightly slower than in water (20.1 ns, Table 2).
Table 3.
Time-resolved fluorescence data for W233 in D2O

Identification of the W233 rotamers
The multiexponential time-resolved fluorescence emission from single-tryptophan protein is often linked in terms of heterogeneity associated with rotamers of the tryptophan side chain (see below). Using the Dead-End Eimination method (DEE) implemented in the Brugel software, Hellings et al. (2003) were able to measure the relative energies of several tryptophan residues in different rotameric states. Intriguingly, the investigators found that virtually all tryptophan residues considered were stable in more than one rotameric state. In most cases, the number of clusters corresponded to the number of lifetimes measured experimentally, and the amplitude of fluorescence lifetimes corresponded well with the distance of the Cε3 atom of tryptophan to the carbonyl peptide bond of the tryptophan residue itself. For certain proteins, the existence of tryptophan rotamers could also be confirmed by X-ray crystallography (Hellings et al. 2004; Moors et al. 2006).
Using the DEE approach, we calculated the possible rotamers of W233 every 10° of the χ1 and χ2 angles (Fig. 5; Table 4). Within the 1296 structures calculated, only 99 (7.6%) displayed a potential energy lower than zero. The tryptophan conformations with negative nonbonded potential energy were concentrated around three clusters (Fig. 5). The most negative point of each cluster was considered as a possible rotamer for W233 within the R61 structure. The rotamer with the most negative potential energy (−28.6 kJ mol−1), R1, was the rotamer found in the crystal structure (χ1 = 60 and χ2 = −90). The second rotamer, R2 (χ1 = 60 and χ2 = 90), which, in comparison to R1, displayed the indole ring flipped 180° over the χ2 angle, had slightly lower potential energy (−25.1 kJ mol−1). Finally, R3 (χ1 = −140 and χ2 = −120) was the least stable rotamer (−14.9 kJ mol−1). The indole ring of this rotamer was perpendicular to the position of tryptophan in R1 and R2. (See Table 4 for the relative orientation of the tryptophan in the three rotamers of R61.) In the absence of other quenchers, multiexponential decays in proteins have been explained by different tryptophan rotamers that have different electron transfer donor–acceptor distances between the excited state of tryptophan and the π* antibonding orbital of the closest carbonyl bond of the peptide backbones. From the structures calculated for the three rotamer clusters, we were able to measure the distance between any phenyl carbon of the indole group of W233 and the closest carbonyl in the surroundings of the tryptophan (Table 4). These distances were taken as the donor–acceptor distances for the electron transfer reaction and were 3.4 Å, 3.8 Å, and 4.7 Å for R1, R2, and R3, respectively (Table 4). Fitting these distances to the empirical equation calculated for the electron transfer rate from the excited state of tryptophan to the nearest peptide (Adams et al. 2002) gave the theoretical lifetimes of 1.0, 1.9, and 8.9 ns for R1, R2, and R3 respectively (Table 4).
Figure 5.

Distribution of rotamer clusters (R1, R2, and R3) for W233 in R61. The torsion angles χ1 and χ2 were plotted against the nonbonded energy of the tryptophan residue in relation to the complete protein. The low-energy rotameric conformations are orientated around three distinctive clusters.
Table 4.
Predicted thermodynamic and fluorescence information of the three R61 tryptophan rotamers as calculated from the DEE method

Time-resolved fluorescence anisotropy
In order to estimate the mobility of both the tryptophan residue and the global protein, we measured the time-resolved anisotropy decay of R61. The best fit was obtained using a double-exponential decay, resulting in a fast and slow correlation time (φ1 = 0.73 ± 0.20 ns [25.5%], φ2 = 16.04 ± 1.9 ns [74.5%]) and a r0 value of 0.284. For globular proteins, the rotational correlation time is approximately related to the molecular weight (M) of the protein by
 |
where v is the partial specific volume of the protein, and h is the hydration, T is the temperature in Kelvin, R is the gas constant, and η is the viscosity. The approximated correlation time for R61 at 293°K is 14.6 ns, which is comparable to the slow correlation time resulting from the fitted anisotropy decay data. Comparison of the slow correlation time contribution with the calculated value for the total protein points to the fast correlation time being the result of segmental motions of the W233 residue. The fast contribution to the overall anisotropy decay can therefore be used as a measure of the local mobility of the trytophan residue. Using the additivity of anisotropy contributions and their relation to the angular displacement of the fluorophore:
 |
with γ the angle of displacement reached after the fast phase of the anisotropy decay and g2 the fractional contribution of the slow rotational correlation time. The displacement angle (γ) can be considered as a measure of the mobility of the tryptophan residue. For W233, this resulted in a γ value of 24°. (This is an average angle for all tryptophan rotamers. Determination of a rotation angle for each rotamer is not possible due to the high number of parameters involved in the model and the limited accuracy of the anisotropy data.) The movement of W233 in R61 can therefore be regarded as sufficiently restricted, such that fast interconversions between the rotameric states can be ruled out, since a far greater angular displacement would be needed to reach one rotameric position from any other (Fig. 5). The second component of anisotropy decay is of the same magnitude as the overall rotational correlation time calculated for the protein. Therefore, slow local mobility could be hidden by the rotation of the molecule as a whole.
Discussion
The single tryptophan in R61 shows very interesting spectroscopic characteristics. This protein not only is the most blue-shifted protein to display a REE and the only tryptophan in the Burstein Class S (Demchenko and Lakokhin 1988; Demchencko 2002), but also displays the largest red-edge effect measured in proteins to date (23.7 nm).
The time-resolved fluorescence of R61 is composed of three lifetimes. Surprisingly, these lifetimes increase with increasing emission wavelength (Table 1), suggesting that the dynamics of the surrounding of the tryptophan residues are in the same timescale as the fluorescence decay. Most of the fluorescence of R61 comes from the second lifetime (τ2), which counts for ∼80% of the total fluorescence at 320 nm and ∼50% at 380 nm. The DCAS (Fig. 4A) of R61 fluorescence show that τ2 is a relatively blue-shifted emission corresponding to the λmax of the steady-state fluorescence (317 mn). The DCAS of τ1 and τ3, on the other hand, are red shifted (λmax ∼350 and ∼340 nm, respectively), and their contribution increases with increasing emission wavelength (Table 1). This induces the fluorescence of R61 to shift in time to longer wavelengths (Fig. 4B). Such a red shift is often interpreted as a “solvent effect.” In certain constrained environments, the solvent dipolar relaxation time (τR) is similar to or longer than the fluorescence emission time (τF) and only long-lived excited molecules have sufficient time to experience a full solvent dipolar reorganization. The data presented here, therefore, suggest that the relaxation process of R61 fluorescence is relatively slow and in a similar timescale as the fluorescence emission. The correlation function of the relaxation process of R61 fluorescence, however, did not display the usual kinetic function (Fig. 4B). In fact, the correlation function is expected to decay exponentially from a maximum value at the blue edge of the spectrum to a minimum value at the red edge of the spectrum, since the time-dependent Stokes shift is usually considered the spectroscopic observation of the dielectric relaxation of the solvent dipoles upon fluorophore excitation (Kinoshita and Nishi 1988; Simon 1988; Studenov et al. 1991; Reynolds et al. 1996; Bardeen et al. 1999). Such exponential decay is observed in many other systems (see, for example, Chapman et al. 1990; Mandal et al. 2004; Humpolickova et al. 2005). In this system, however, the correlation function of R61 fluorescence first showed a blue shift from 319.2 to 317 nm in the first 3 ns of the relaxation process due to the rapid decay of a red-emitting contribution (τ1). And, only then did the expected exponential decay to the red edge of the spectrum due to the slow dynamics of the protein environment appear. The whole relaxation time of the process was 14.0 ns.
A time-dependent Stokes shift is usually observed in semi-rigid environments such as glasses, polymers, and other organized assemblies like micelles, at low temperature. As several investigators explained, this phenomenon is due to heterogeneity of the solvent distribution in space and to the slow reorientation rate of the solvent molecules around the changed excited state dipole of the solute. In other words, different excitation energies select different subsets of solute molecules with a specific solvation (heterogeneity in space). The so-formed excited states do not loose relaxation energy because of the rigidity of the system and emit photons at relatively high energies (blue shift). In these systems, the emission wavelength is related to the excitation wavelength: red-shifted λex produce red-shifted λem. Accordingly, increasing the solvent relaxation rate (e.g., by increasing the temperature or decreasing the solvent viscosity) results in more solvent molecules having the time to relax around the excited-state dipole of the fluorophore; thus, the fluorescence photons should increasingly be emitted from lower energetic states, and the emission should increasingly be red shifted (Demchenko and Ladokhin 1988; Demchenko and Sytnik 1991a; Demchenko 2002). The temperature dependence of R61 fluorescence is, for this reason, very surprising. The λmax of this protein decreased from low to high temperature, reaching its minimum value of 316.4 nm at 48°C. This unexpected result seems to suggest that the protein matrix around the tryptophan becomes more rigid with increasing temperature, reaching its maximum value just before the unfolding of the protein. A second unexpected finding was the shape of the emission spectra at red-edge excitation. If multiple solvation sites are responsible for the REES we observed here, then we also expect a homogeneous Gaussian distribution around the λmax. The presence of shoulders in the spectra at λex higher than 300 nm suggests otherwise, e.g., that this system is dynamically heterogeneous and that the spectra arise from more than one distinct state with different reorganization energies. Finally, since the REES and the TRES are a static and dynamic representation of the same solvent relaxation process, the υmax at red edge emission should be the same as the vmax(∞), but this is not the case.
So, what is the meaning of the TRES and the REES observed in the fluorescence of R61? We believe that the only possible explanation of the data collected in this study is the existence of multiple tryptophan rotamers within the structure of the protein.
Both in solution and in proteins, tryptophan can exist in several low-energy rotameric states: the four possible combinations of χ1 = ±60 and χ2 = ±90, and the two rotamers with χ1 = 0 and χ2 ± 90, where χ1 and χ2 are the torsion angles formed by Cα–Cβ and Cβ–Cγ of the tryptophan. Many studies have linked the multifluorescence exponential decays of model compounds to the presence of multiple conformational states of tryptophan, which do not interconvert in the fluorescence timescale (Donzel et al. 1974; Fleming et al. 1978; Szabo and Rayner 1980; Chang et al. 1983; Petrich et al. 1983; Colucci et al. 1990; Hellings et al. 2003, 2004). In the calculations we performed, we found that W233 is thermodynamically stable in three different conformational states, which are exposed to different environments given by the reorganization of the protein side chains. Each rotamer could be linked to a fluorescence lifetime, suggesting that the fluorescence of R61 is, in fact, the result of separate fluorescence decays of three different pools of proteins.
As a consequence, the time-dependent Stokes shift observed for R61 is most likely not the result of the decay of a single ensemble of molecules with different solvation states (heterogeneity in space or heterogeneity in the excited state), but the result of the relaxation of the same tryptophan residue in three different conformations with independent time-resolved fluorescence decays (heterogeneity in the ground state). Similarly, the REES is probably due to an increasingly selective photo-excitation of the tryptophan rotamer with λmax at ∼340 nm. The spectrum obtained exciting at 305 nm could then correspond to the spectrum of the rotamer described by τ3 (λmax = 340 nm), while the shoulder at 350 nm could correspond to the λmax of the rotamer described by τ1 (λmax = 350 nm). In addition, the existence of tryptophan rotamers could also explain the counterintuitive temperature dependence of R61 fluorescence. Assuming that the shortest lifetime class has the highest nonradiate rate constant, and that this contribution will be reduced most prominently by increasing temperature, a red-emitting component disappears most rapidly and a blue shift should result.
Inspection of the structure of the tryptophan rotamers of R61 revealed that in all three conformations, the tryptophan resides in a hydrophobic pocket in a rather rigid environment. No solvent is accessible to the tryptophan (Table 3). Therefore, the relatively high emission maxima of τ1 and τ3 must be related to the reorganization of two conserved crystal water molecules or other side chains in the vicinity of the tryptophan. The effect of the deuterated solvent on the time-resolved fluorescence of R61 might suggest the predominant role of water on the relaxation process. Molecular dynamic simulations are currently in process to assess the role of solvent in the relaxation dynamic of R61 fluorescence.
Conclusions
This work analyzed the complex fluorescence spectroscopy of R61. We found strong evidence that supports the existence of multiple tryptophan rotamers in this protein, and at the same time, we observe a REES and time-dependent Stokes shift for each rotamer in proteins.
We think that the unusual temperature dependence of the λmax and the multiexponential decay of the R61 fluorescence are due to the fact that the tryptophan in R61 exists is more than one conformation or rotameric state, which does not interconvert during the fluorescence lifetime of the protein. We believe that each rotamer possesses a specific fluorescence decay, Stokes shift and SIE; the amplitudes of which also suggest a relatively large difference in the dipole interaction of the tryptophan rotamers with the surroundings. The observed REES is, therefore, most likely due to the specific photo-selection of different rotameric populations of tryptophan with a different λmax and a different lifetime. In this view, the red shift of the TRES measured for R61 is due to the uncorrelated fluorescence decays of the rotameric populations, where the shortest lifetime arises from a rotamer that is more red shifted than the rotamers with the longer lifetimes.
In conclusion, we suggest that the REES and the time dependence of the Stokes shift of R61 is not due to the heterogeneity in the solvation of the excited state as commonly believed, but in the heterogeneity of the protein environment. The increase of each lifetime with increasing emission wavelength can be explained by slow local dynamics within the rotameric state. This behavior is in contrast to the protein GB1, where the whole fluorescence behavior has been explained in terms of protein dynamics (Toptygin et al. 2006).
Materials and Methods
All chemicals were purchased from Sigma except for D-glycogen, which was obtained from Janssen Chimica. R61 was expressed and purified as described elsewhere (Bourguignonbellefroid et al. 1992). Purity was checked with SDS-chromatography.
Steady-state fluorescence
Steady-state spectra with correction for the emission and adsorption spectra were recorded using a PTI fluorimeter (Quanta Master) in a 10-mm quartz cuvette (Hellma) closed with a Teflon lid. The temperature was controlled by a water bath connected to the cuvette holder and measured in the cuvette using a thermocouple (Fluke). The signal measured was corrected for the Raman scattering of the solution. The maximum Stokes shift was calculated from the zero value of the first derivative of the steady-state fluorescence spectrum.
Time-resolved fluorescence
Time-correlated single photon counting was used to measure the time-resolved fluorescence of R61. The laser source was a Millenia Pro, diode-pumped CW visible laser system from Spectra-Physics. The picoseconds laser pulses were created using a Tsunamimode-locked Ti:sapphire laser system from Spectra-Physics (model 3950C/D), and the frequency was selected at 1.6 MHz by a Spectra-Physics pulse selector (Model 3980). The so-formed laser pulses were frequency tripled to 295 nm by the Spectra-Physics prism harmonic separator (model GWU23PL). The fluorescence traces were collected at the magic angle (54.7°C) using a PMA 182 photomultiplier from Picoquant with a resolution of 40 ps. The instrument response function (IRF) was measured by recording the signal of a diluted solution of D-glycogen using an emission filter at 295 nm. The fluorescence decays were measured at different emission wavelengths using several wavelength filters spanning the emission spectra. The filters had a bandwidth of ±5 mm and were used every 10 nm from 320 to 380 nm. Measurements on N-acetyl-tryptophan-amide, showing one and the same lifetime at all wavelengths of emission, showed that the color effect in the photomultiplier is negligible (data not shown).
The true fluorescence decay data were obtained by deconvolution of the recorded data with the IRF and fitting to multiexponential function using the Marquardt-Levenberg nonlinear least-squares algorithm. The software used was FluoFit v3.3 from Picoquant. The quality of the fitting was calculated by χ2 values approaching unity and nonrandom-weighted autocorrelation and residual functions.
The decay-class associated spectra (DCAS) of the time-resolved fluorescence of R61 were calculated using the following equation:
where I(i, λ) is the steady-state fluorescence associated to each lifetime at a wavelength λ, I(λ) the overall steady-state fluorescence at wavelength λ, and aλ,I and τλ,i are the fluorescence amplitudes and lifetimes at λ.
The wavelength-dependent decays and the steady-state fluorescence were used to calculate the time-resolved emission spectra at different times. From the maximum Stokes shift so measured, the normalized correlation function C(t) was calculated using the following equation:
where [vmax(t)], [vmax(0)], and [vmax(∞)] are the Stokes shifts in wave numbers at time t, time zero, and infinite time, respectively.
Time-resolved anisotropy
Time-resolved fluorescence anisotropy measurements were performed using the same experimental setup as described for time-resolved fluorescence. The anisotropy decay was obtained by measuring the intensity of the parallel en perpendicular components of the fluorescence emission at 320 and 340 nm, after excitation at 295 nm, using wavelength filters with a bandwidth of ±5 mm. The resulting data was fitted to the multiexponential form of the time-resolved anisotropy decay law (Weber 1977).
 |
where r0 is the anisotropy at time zero, and gi and φi are the amplitude fraction and rotational correlation time for component i, respectively (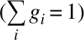 )
)
Computational determination of the tryptophyl side chain rotamers
The possible tryptophan rotameric conformations of W233 were predicted using of a method based on the Dead-End Elimination algorithm (DEE) (Desmet et al. 1992; De Maeyer et al. 2000) developed in our group (Hellings et al. 2003). In order to determine the possible rotameric clusters of W233, the tryptophan residue was iteratively rotated by 10° over the χ1 and χ2 angles, resulting in 1296 starting structures. The backbone and the tryptophan residue were kept fixed, while the protein side chains within a sphere of 8 Å around W233 were allowed to evolve by molecular dynamics. For each structure, the Global Minimum Energy Conformation (GMEC), the total nonbonded energy (Etotnb), and the nonbonded energy of W233 in relation to the complete protein (EWnb) were determined. These values were used to determine the rotameric clusters. Only the conformations with a negative Etotnb and EWnb value were considered and plotted as a function of the considered tryptophan χ1 and χ2 angles. For these allowed conformations, the distance between the indole Cε3 atom and the carbonyl carbon of the peptide bond was calculated. All calculations were performed using the Brugel software package (Delhaise et al. 1984) on an Apple G5 cluster (2 Ghz). The solvent-accessible surfaces for tryptophan in the different rotameric states were calculated with the “Survol option,” also in the Brugel software package, using a probe sphere with a standard radius of 0.14 nm (Alard and Wodak 1991).
Acknowledgments
Financial support was received in the pursuance of the contract P5/33 “Protein structure and function in the post-genomic era” in the framework of the Belgian federal program “Interuniversity Attraction Poles–Phase V.” Financial support from the Impuls financing of the K.U.Leuven BioMacS was appreciated. A.J. is supported by the Fund for Scientific Research (Flanders) grant G.0252.05
Footnotes
Reprint requests to: Yves Engelborghs, Laboratory of Biomolecular Dynamics, Department of Chemistry, University of Leuven, Celestijnenlaan 200 G, B-3001 Leuven, Belgium; e-mail: Yves.Engelborghs@fys.kuleuven.be; fax: 32-16-32-79-74.
Abbreviations: DCAS, decay class associated spectra; REES, red-edge excitation shift; SIE, solvent-induced isotope effect; FSS, fluorescence Stokes shift
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073147608.
References
- Adams, P.D., Chen, Y., Ma, K., Zagorski, M.G., Sonnichsen, F.D., McLaughlin, M.L., Barkley, M.D. Intramolecular quenching of tryptophan fluorescence by the peptide bond in cyclic hexapeptides. J. Am. Chem. Soc. 2002;124:9278–9286. doi: 10.1021/ja0167710. [DOI] [PubMed] [Google Scholar]
- Alard, P., Wodak, S.J. Detection of cavities in a set of interpenetrating spheres. J. Comput. Chem. 1991;12:918–922. [Google Scholar]
- Azumi, T., Itoh, K., Shiraishi, H. Shift of emission band upon excitation at long wavelength absorption-edge.3. Temperature-dependence of shift and correlation with time-dependent spectral shift. J. Chem. Phys. 1976;65:2550–2555. [Google Scholar]
- Bardeen, C.J., Rosenthal, S.J., Shank, C.V. Ultrafast solvation processes in polar liquids probed with large organic molecules. J. Phys. Chem. A. 1999;103:10506–10516. [Google Scholar]
- Bourguignonbellefroid, C., Wilkin, J.M., Joris, B., Aplin, R.T., Houssier, C., Prendergast, F.G., Vanbeeumen, J., Ghuysen, J.M., Frere, J.M. Importance of the 2 tryptophan residues in the Streptomyces R61 exocellular DD-peptidase. Biochem. J. 1992;282:361–367. doi: 10.1042/bj2820361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M.C., Petrich, J.W., Mcdonald, D.B., Fleming, G.R. Non-exponential fluorescence decay of tryptophan, tryptophylglycine, and glycyltryptophan. J. Am. Chem. Soc. 1983;105:3819–3824. [Google Scholar]
- Chapman, C.F., Fee, R.S., Maroncelli, M. Solvation dynamics in N-methylamides. J. Phys. Chem. 1990;94:4929–4935. [Google Scholar]
- Chattopadhyay, A., Mukherjee, S. Red-edge excitation shift of a deeply embedded membrane probe: Implications in water penetration in the bilayer. J. Phys. Chem. B. 1999;103:8180–8185. [Google Scholar]
- Chattopadhyay, A., Rawat, S.S., Kelkar, D.A., Ray, S., Chakrabarti, A. Organization and dynamics of tryptophan residues in erythroid spectrin: Novel structural features of denatured spectrin revealed by the wavelength-selective fluorescence approach. Protein Sci. 2003;12:2389–2403. doi: 10.1110/ps.03302003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci, W.J., Tilstra, L., Sattler, M.C., Fronczek, F.R., Barkley, M.D. Conformational studies of a constrained tryptophan derivative—implications for the fluorescence quenching mechanism. J. Am. Chem. Soc. 1990;112:9182–9190. [Google Scholar]
- Delhaise, P., Bardiaux, M., Wodak, S. Interactive computer animation of macromolecules. J. Mol. Graph. 1984;2:103–106. [Google Scholar]
- De Maeyer, M., Desmet, J., Lasters, I. The dead-end elimination theorem: Mathematical aspects, implementation, optimizations, evaluation, and performance. Methods Mol. Biol. 2000;143:265–304. doi: 10.1385/1-59259-368-2:265. [DOI] [PubMed] [Google Scholar]
- Demchenko, A.P. Dependence of human-serum albumin fluorescence-spectrum on the excitation wavelength. Ukr. Biokhim. Zh. 1981;53:22–27. [PubMed] [Google Scholar]
- Demchenko, A.P. Red-edge-excitation fluorescence spectroscopy of single-tryptophan proteins. Eu. Biophys. J.(with Biophys. Let.) 1988;16:121–129. doi: 10.1007/BF00255522. [DOI] [PubMed] [Google Scholar]
- Demchenko, A.P. The red-edge effects: 30 years of exploration. Luminescence. 2002;17:19–42. doi: 10.1002/bio.671. [DOI] [PubMed] [Google Scholar]
- Demchenko, A.P., Ladokhin, A.S. Red-edge-excitation fluorescence spectroscopy of indole and tryptophan. Luminescence. 1988;15:369–379. doi: 10.1007/BF00254724. [DOI] [PubMed] [Google Scholar]
- Demchenko, A.P., Sytnik, A.I. Site selectivity in excited-state reactions in solutions. J. Phys. Chem. 1991a;95:10518–10524. [Google Scholar]
- Demchenko, A.P., Sytnik, A.I. Solvent reorganizational red-edge effect in intramolecular electron-transfer. Proc. Natl. Acad. Sci. 1991b;88:9311–9314. doi: 10.1073/pnas.88.20.9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet, J., De Maeyer, M., Hazes, B., Ignace, L. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- Donzel, B., Gauducho, P., Wahl, P. Study of conformation in excited-state of 2 tryptophanyl diketopiperazines. J. Am. Chem. Soc. 1974;96:801–808. [Google Scholar]
- Fleming, G.R., Morris, J.M., Robbins, R.J., Woolfe, G.J., Thistlethwaite, P.J., Robinson, G.W. Non-exponential fluorescence decay of aqueous tryptophan and 2 related peptides by picosecond spectroscopy. Proc. Natl. Acad. Sci. 1978;75:4652–4656. doi: 10.1073/pnas.75.10.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere, J.M., Joris, B. Penicillin-sensitive enzymes in peptidoglycan biosynthesis. CRC Crit. Rev. Microbiol. 1985;11:299–396. doi: 10.3109/10408418409105906. [DOI] [PubMed] [Google Scholar]
- Gallay, W.C., Purkey, R.M. Role of heterogeneity of the solvation site in electronic spectra in solution. Proc. Natl. Acad. Sci. 1970;67:1116–1121. doi: 10.1073/pnas.67.3.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellings, M., De Maeyer, M., Verheyden, S., Hao, Q., Van Damme, E.J.M., Peumans, W.J., Engelborghs, Y. The dead-end elimination method, tryptophan rotamers, and fluorescence lifetimes. Biophys. J. 2003;85:1894–1902. doi: 10.1016/s0006-3495(03)74617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellings, M., Engelborghs, Y., Deckmyn, H., Vanhoorelbeke, K., Schiphorst, M.E., Akkerman, J.W.N., De Maeyer, M. Experimental indication for the existence of multiple Trp rotamers in von Willebrand factor A3 domain. Proteins. 2004;57:596–601. doi: 10.1002/prot.20227. [DOI] [PubMed] [Google Scholar]
- Hu, Z., Margulis, C.J. Heterogeneity in a room-temperature ionic liquid: Persistent local environments and the red-edge effect. Proc. Natl. Acad. Sci. 2006;103:831–836. doi: 10.1073/pnas.0507364103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpolickova, J., Stepanek, M., Prochazka, K., Hof, M. Solvent relaxation study of pH-dependent hydration of poly(oxyethylene) shells in polystyrene-block-poly(2-vinylpyridine)-block-poly(oxyethylene) micelles in aqueous solutions. J. Phys. Chem. A. 2005;109:10803–10812. doi: 10.1021/jp053348v. [DOI] [PubMed] [Google Scholar]
- Itoh, K., Azumi, T. Shift of emission band upon excitation at long wavelength absorption-edge.1. Preliminary survey for quinine and related compounds. Chem. Phys. Lett. 1973;22:395–399. [Google Scholar]
- Itoh, K.I., Azumi, T. Shift of emission band upon excitation at long wavelength absorption-edge.2. Importance of solute–solvent interaction and solvent reorientation relaxation process. J. Chem. Phys. 1975;62:3431–3438. [Google Scholar]
- Kelly, J.A., Knox, J.R., Moews, P.C., Hite, G.J., Bartolone, J.B., Zhao, H., Joris, B., Frere, J.M., Ghuysen, J.M. 2.8 Å structure of penicillin-sensitive D-alanyl carboxypeptidase-transpeptidase from Streptomyces-R61 and complexes with β-lactams. J. Biol. Chem. 1985;260:6449–6458. [PubMed] [Google Scholar]
- Kinoshita, S., Nishi, N. Dynamics of fluorescence of a dye molecule in solution. J. Chem. Phys. 1988;89:6612–6622. [Google Scholar]
- Klymchenko, A.S., Demchenko, A.P. Electrochromic modulation of excited-state intramolecular proton transfer: The new principle in design of fluorescence sensors. J. Am. Chem. Soc. 2002;124:12372–12379. doi: 10.1021/ja027669l. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R., Keating-Nakamoto, S. Red-edge excitation of fluorescence and dynamic properties of proteins and membranes. Biochemistry. 1984;23:3013–3021. doi: 10.1021/bi00308a026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, P.K., Samanta, A. Fluorescence studies in a pyrrolidinium ionic liquid: Polarity of the medium and solvation dynamics. J. Phys. Chem. B. 2005;109:15172–15177. doi: 10.1021/jp051844d. [DOI] [PubMed] [Google Scholar]
- Mandal, P.K., Sarkar, M., Samanta, A. Excitation-wavelength-dependent fluorescence behavior of some dipolar molecules in room-temperature ionic liquids. J. Phys. Chem. A. 2004;108:9048–9053. doi: 10.1021/jp060441q. [DOI] [PubMed] [Google Scholar]
- Moors, S.L., Hellings, M., De Maeyer, M., Engelborghs, Y., Ceulemans, A. Tryptophan rotamers as evidenced by X-ray, fluorescence lifetimes, and molecular dynamics modeling. Biophys. J. 2006;91:816–823. doi: 10.1529/biophysj.106.085100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson, L., Halle, B. Molecular origin of time-dependent fluorescence shifts in proteins. Proc. Natl. Acad. Sci. 2005;102:13867–13872. doi: 10.1073/pnas.0504181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrich, J.W., Chang, M.C., Mcdonald, D.B., Fleming, G.R. On the origin of non-exponential fluorescence decay in tryptophan and its derivatives. J. Am. Chem. Soc. 1983;105:3824–3832. [Google Scholar]
- Reshetnyak, Y.K., Burstein, E.A. Decomposition of protein tryptophan fluorescence spectra into log-normal components. II. The statistical proof of discreteness of tryptophan classes in proteins. Biophys. J. 2001;81:1710–1734. doi: 10.1016/S0006-3495(01)75824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshetnyak, Y.K., Koshevnik, Y., Burstein, E.A. Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 2001;81:1735–1758. doi: 10.1016/S0006-3495(01)75825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, L., Gardecki, J.A., Frankland, S.J.V., Horng, M.L., Maroncelli, M. Dipole solvation in nondipolar solvents: Experimental studies of reorganization energies and solvation dynamics. J. Phys. Chem. 1996;100:10337–10354. [Google Scholar]
- Simon, J.D. Time-resolved studies of solvation in polar media. Acc. Chem. Res. 1988;21:128–134. [Google Scholar]
- Studenov, V.I., Mazurenko, Y.T., Bakhshiev, N.G. Effect of dipole–dipole orientation interactions on polarized electronic-spectra of impurity molecules in nematic liquid-crystals. Optika I Spektroskopiya. 1991;71:60–65. [Google Scholar]
- Szabo, A.G., Rayner, D.M. Fluorescence decay of tryptophan conformers in aqueous-solution. J. Am. Chem. Soc. 1980;102:554–563. [Google Scholar]
- Toptygin, D., Gronenbron, A.M., Brand, L. Nanosecond relaxation dynamics of protein GB1 identified by the time-dependent red shift in the fluorescence of tryptophan and 5-fluorotryptophan. J. Phys. Chem. B. 2006;110:26292–26302. doi: 10.1021/jp064528n. [DOI] [PubMed] [Google Scholar]
- Weber, G. Theory of differential phase fluorometry—detection of anisotropic molecular rotations. J. Phys. Chem. 1977;66:4081–4091. [Google Scholar]
- Weber, G., Shinitzky, M. Failure of energy transfer between identical aromatic molecules on excitation at the long wave edge of the absorption spectrum. Proc. Natl. Acad. Sci. 1970;65:823–830. doi: 10.1073/pnas.65.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H.T., Vela, M.A., Fronczek, F.R., McLaughlin, M.L., Barkley, M.D. Microenvironmental effects on the solvent quenching rate in constrained tryptophan derivatives. J. Am. Chem. Soc. 1995;117:348–357. [Google Scholar]