

Abstract
The mechanism by which ATP-sensitive K+ (KATP) channels open in the presence of inhibitory concentrations of ATP remains unknown. Herein, using a four-state kinetic model, we found that the nucleotide diphosphate UDP directed cardiac KATP channels to operate within intraburst transitions. These transitions are not targeted by ATP, nor the structurally unrelated sulfonylurea glyburide, which inhibit channel opening by acting on interburst transitions. Therefore, the channel remained insensitive to ATP and glyburide in the presence of UDP. “Rundown” of channel activity decreased the efficacy with which UDP could direct and maintain the channel to operate within intraburst transitions. Under this condition, the channel was sensitive to inhibition by ATP and glyburide despite the presence of UDP. This behavior of the KATP channel could be accounted for by an allosteric model of ligand-channel interaction. Thus, the response of cardiac KATP channels towards inhibitory ligands is determined by the relative lifetime the channel spends in a ligand-sensitive versus -insensitive state. Interconversion between these two conformational states represents a novel basis for KATP channel opening in the presence of inhibitory concentrations of ATP in a cardiac cell.
Keywords: KATP channel, nucleotide diphosphate, kinetic model, allosteric model, sulfonylurea
introduction
ATP-sensitive K+ (KATP) channels transduce cellular metabolic events into membrane potential changes (Ashcroft and Ashcroft, 1990; Lazdunski, 1994; Seino et al., 1996; Bryan and Aguilar-Bryan, 1997), which in heart muscle leads to shortening of action potential duration during ischemia (Nichols and Lederer, 1991; Findlay, 1994; Terzic et al., 1995). The defining property of KATP channels is their inhibition by intracellular ATP (Noma, 1983). In cardiomyocytes, however, the ATP concentration (∼5–10 mM) exceeds by >100-fold the IC50 value for KATP channel closure. Thus, a change of two orders of magnitude in the ATP concentration would be required for channels to open, which does not occur even under extreme cellular hypoxia (Weiss and Hiltbrand, 1985; Decking et al., 1995, 1997), suggesting that additional modulators of KATP channel opening are important.
In this regard, intracellular nucleotide diphosphates are of particular importance since they favor opening of KATP channels even within a cytosolic environment of high ATP concentration (Ashcroft and Ashcroft, 1990; Nichols and Lederer, 1991; Weiss and Venkatesh, 1993; Findlay, 1994; Terzic et al., 1994d ; Elvir-Mairena et al., 1996). However, the mechanism of this action of nucleotide diphosphates remains controversial. A conventional assumption has been that nucleotide diphosphates competitively antagonize ATP at an inhibitory binding site on the channel protein (Dunne and Petersen, 1986; Kakei et al., 1986; Misler et al., 1986; Findlay, 1987; Bokvist et al., 1991; Nichols and Lederer, 1991; Ueda et al., 1997). However, this mechanism cannot fully explain KATP channel opening since altered concentrations of cytosolic ATP and/or nucleotide diphosphates are not readily detectable, nor do they correlate with changes in KATP channel function. Moreover, nucleotide diphosphates, such as ADP or UDP, induce channel opening in the absence of ATP (Findlay, 1988; Lederer and Nichols, 1989; Tung and Kurachi, 1991; Allard and Lazdunski, 1992; Forestier and Vivaudou, 1993; Terzic et al., 1994a ) and can lose their ability to antagonize ATP-dependent channel inhibition under certain operative conditions of the channel (Deutsch and Weiss, 1993; Terzic et al., 1994a ). Such nonuniform regulation of KATP channel opening by nucleotide diphosphates has also been observed with other inhibitory ligands including sulfonylurea drugs (Venkatesh et al., 1991; Brady et al., 1996b , 1998) and diadenosine polyphosphates (Jovanovic et al., 1996, 1997). These findings suggest that an operative condition-dependent response of KATP channels is a fundamental property of the channel, which may be the basis for channel opening in the presence of inhibitory ligands.
To determine whether nucleotide diphosphates induce an alteration in channel behavior that could account for the observed response of KATP channels towards inhibitory ligands, we investigated the action of UDP on transitional states of the cardiac KATP channel. Based on a kinetic model of channel behavior, we demonstrate that UDP drives the channel into a state that is insensitive towards inhibitory ligands. Interconversion between ligand-sensitive and -insensitive states could be interpreted using an allosteric model that predicted the outcome of the interaction between an inhibitory ligand and the KATP channel in the presence of a nucleotide diphosphate. Transition of the channel into a ligand-insensitive channel state provides a means for KATP channel opening even in the presence of high concentrations of inhibitory ligands within a cardiomyocyte.
materials and methods
Isolated Cardiomyocytes
Ventricular myocytes were isolated by enzymatic dissociation (Alekseev et al., 1996a ). Solutions were prepared based on a “low Ca2+ medium” containing (mM): 100 NaCl, 10 KCl, 1.2 KH2PO4, 5 MgSO4, 20 glucose, 50 taurine, 10 HEPES, pH 7.2–7.3. Guinea pigs were anesthetized with pentobarbital (1 ml/100 mg body weight i.p.). After cardiotomy, the heart was retrogradely perfused (at 37°C) with: medium 199 (Sigma Chemical Co., St. Louis, MO) for 2–3 min, followed by Ca2+ EGTA-buffered low Ca2+ medium (pCa 7) for 80 s, and finally low Ca2+ medium containing pronase E (8 mg/100 ml; Serva Biochemicals, Heidelberg, Germany), proteinase K (1.7 mg/100 ml; Boehringer Mannheim Biochemicals, Indianapolis, IN), bovine serum albumin (0.1 g/100 ml, fraction V; Sigma Chemical Co.), and 200 μM CaCl2. Ventricles were separated from atria and cut into small fragments (6–10 mm3) in the low Ca2+ medium enriched with 200 μM CaCl2. Single cells were then isolated by stirring the tissue (at 37°C) in a solution containing pronase E and proteinase K supplemented with collagenase (5 mg/10 ml; Worthington Biochemical Corp., Freehold, NJ). After 10 min, the first aliquot was removed, filtered through a nylon sieve, centrifuged (at 300– 400 rpm, 1 min), and washed twice. Remaining tissue fragments were reexposed to collagenase, and isolation continued for two to three such cycles. Isolated cardiomyocytes were stored in low Ca2+ medium with 200 μM CaCl2. Rod-shaped cardiomyocytes with clear striations and a smooth surface were used for electrophysiological recordings. Experiments were performed with the approval of the Institutional Animal Care and Use Committee (Mayo Clinic).
Single-Channel Recording
Fire-polished pipettes, coated with Sylgard (resistance ∼5 MΩ), were filled with “pipette solution” containing (mM): 140 KCl, 1 CaCl2, 1 MgCl2, 5 HEPES-KOH, pH 7.3. Cardiac cells were superfused with “internal solution” containing (mM): 140 KCl, 1 MgCl2, 5 EGTA, 5 HEPES-KOH, pH 7.3, in the absence or presence of nucleotides (UDP or ATP) and/or glyburide (Sigma Chemical Co.), and recordings made at room temperature (20– 22°C) as described (Terzic et al., 1994c ; Terzic and Kurachi, 1996). Glyburide was dissolved in dimethylsulfoxide as concentrated stock solution, and the final concentration of dimethylsulfoxide was <0.1%, which did not affect KATP channels. UDP (Boehringer Mannheim Biochemicals) and ATP (potassium salt; Sigma Chemical Co.) were dissolved in internal solution before use. Single-channel recordings in the inside-out configuration were monitored online on a high-gain digital storage oscilloscope (VC-6025; Hitachi Ltd., Tokyo, Japan) and stored on tape using a PCM converter system (VR-10; Instrutech Corp., Great Neck, NY). Data were reproduced, low-pass filtered at 4 kHz (−3 dB) by a Bessel filter (902; Frequency Devices Inc., Haverhill, MA), sampled at 80-μs rate, and further analyzed using “BioQuest” software (Alekseev et al., 1997a , 1997b ).
Analysis of Channel Activity
The threshold for judging the open state of KATP channels was set at half single channel amplitude. The degree of channel activity was assessed by digitizing segments of current records, expressed as nP o, where n represents the number of channels in the patch and P o the probability of each channel to be open.
Kinetic schemes for cardiac KATP channels are commonly represented by one open and two closed states (Kakei and Noma, 1984). Herein, in prolonged records of single channel activity (under symmetrical K+ concentration and negative membrane potential), distribution of total dwell time could be best fit by three exponents, thus requiring consideration of an additional closed state as proposed for more detailed schemes of KATP channel behavior (Gillis et al., 1989; Furukawa et al., 1993). Accordingly, channel kinetic analysis was performed based upon a four-state kinetic scheme with three forward and three backward rate constants:
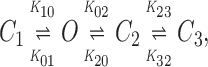 |
1 |
where transitions between the open (O) and the first closed (C1) state represent transitions within a burst (intraburst kinetics), whereas transitions between the open (O) and the second (C2) and third (C3) closed states define interburst kinetics.
Rate constants (see Scheme 1) were calculated based on parameters obtained from the fit of separated distributions of intraburst and interburst closed times, since relative areas under exponents that correspond to interburst transitions were negligible compared with intraburst events. A critical time (t cutoff) was used to define the maximal duration of an event that could still be interpreted as closure within a burst of channel activity and was used to construct distributions of open and closed events within and between bursts. The value for t cutoff ≥ 3.5 ms was determined based upon the relationship (Gillis et al., 1989):
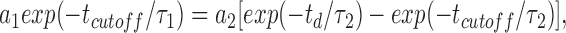 |
where τ1 is the time constant of closed intervals within a burst; τ2 is the time constant of closed intervals between bursts; a 1 and a 2 are areas of exponential fits corresponding to τ1 and τ2, respectively; and t d is the “dead time;” i.e., time of underestimated events that equals the double sampling rate (∼100 μs). Fitting closed and open time distributions by the sum of exponents was carried out using minimization of the χ2 criterion with the Nelder-Meed method of deformed polyhedron (Alekseev et al., 1996b ).
Characteristic open time (τo) was interpreted as:
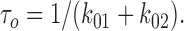 |
2 |
Characteristic intraburst closed time (τc,1), corresponding to gaps within burst, was interpreted as the lifetime in the C1 state:
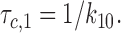 |
3 |
Similarly, the first characteristic interburst closed time (τc,2) was interpreted as the lifetime in the C2 state:
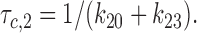 |
4 |
Finally, the second characteristic interburst closed time (τc,3) can be approximated as (see Sakmann and Trube, 1984; Gillis et al., 1989):
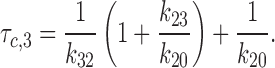 |
5 |
In addition, the number of intraburst closures per burst was expressed as:
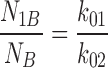 |
6 |
where N IB is the number of events within a burst, and N B is the number of bursts or ofgaps between bursts, distribution of which was defined by τc,2 and τc,3, with representative relative areas under each exponent, a 2 and a 3 (a 2 + a 3 = 1), respectively. Therefore:
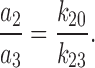 |
7 |
Eqs. 2–7 were then solved for each of the three forward and three backward rates of transition (see Scheme 1):
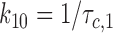 |
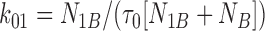 |
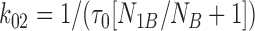 |
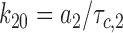 |
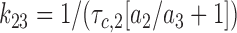 |
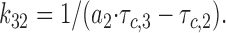 |
8 |
Calculated rates of transition were used as a quantitative tool to describe the effect of nucleotides and sulfonylurea drugs on KATP channel kinetics.
Results are expressed as mean ± SEM; n refers to the number of myocytes used in each analysis.
results
UDP Directs Cardiac KATP Channel Activity Towards Intraburst Transitions
The single channel behavior of cardiac KATP channels is characterized by clustering of channel openings in groups (bursts) separated by prolonged closures (gaps between bursts; Fig. 1 A). Within a burst, distributions of closed and open times were fitted by corresponding single exponents (characteristic times τc,1 = 0.48 ± 0.03 ms and τo = 2.21 ± 0.18 ms, respectively; n = 5; Fig. 1 B, left). Between bursts, distribution of gaps required at least two exponents (Fig. 1 C, left). Thus, in addition to the open and closed channel states that define intraburst (C1 ↔ O) transitions, two closed states were required to describe interburst (↔ C2 ↔ C3) KATP channel behavior (Fig. 1 D, left).
Figure 1.

UDP induces loss of interburst transition. (A) Single KATP channel records (80 s in duration) in the absence (top) and presence (bottom) of 1 mM UDP. Records under both conditions are presented on compressed and extended time scales. Zero-current level is indicated by 0 pA in the compressed, and by a dotted line in the extended time record. (B, left) In the absence of UDP, open and closed (inset) time distributions of intraburst events were fitted by single exponents (solid lines) with characteristic times τo andτc,1. (right) Distribution of openings within a burst in the presence of 1 mM of UDP. (C, left) In the absence of UDP, distribution of gaps between bursts was fitted by the sum of two exponents (solid line) with characteristic times τc,2 andτc,3. Dashed lines correspond to individual exponents. (right) In the presence of UDP, there were no interburst events, and the distribution of closed times could be fitted by a single exponent with a characteristic time, τc, essentially identical to the mean closed time for intraburst events (τc,1) obtained in the absence of UDP. Kinetic scheme constructed based on calculated rates of transitions (in s−1) using Eq. 8 in the absence (left) and presence (right) of 1 mM UDP. Holding potential was −60 mV.
The nucleotide diphosphate UDP (1 mM) eliminated gaps between bursts, promoting the channel to operate within a sustained burst (Fig. 1 A; Table I). UDP did not change the distribution of open times (Table I) that could be fitted by a single exponent (characteristic time τo = 2.36 ± 0.22 ms; n = 4; Fig. 1 B, right), not statistically different from the value obtained in the absence of UDP (P < 0.05; see above). However, since UDP eliminated gaps between bursts, the total distribution of closed times became a single exponent (Fig. 1 C, right) with a characteristic time τc,1 = 0.48 ± 0.04 ms (n = 4). This value was identical to the parameter defining closures within bursts of channel activity in the absence of UDP (P < 0.05; see above). In terms of the four-state linear scheme of KATP channel activity, UDP reduced channel operation to one closed and one open state with a rate of transition (Fig. 1 D, right), identical to the rate of intraburst transition measured in the absence of UDP (Fig. 1 D, left). Thus, UDP directed the KATP channel to operate exclusively within intraburst transitions (Fig. 1 D, right).
Table I.
Parameters of Open and Closed Time Distributions of KATP Channel Activity Obtained under Different Experimental Conditions
| Sustained channel activity | Partial rundown | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −UDP | +UDP | −ATP | +ATP | −Glyburide | +Glyburide | −UDP | +UDP+ATP | −UDP | +UDP | |||||||||||
| Number of events within burst (NIB) | 27115 | 23094 | 18022 | 3825 | 13022 | 11255 | 9113 | 9471 | 38094 | 33183 | ||||||||||
| Number of events between burst (NB) | 27 | 2 | 23 | 255 | 14 | 31 | 14 | 4 | 224 | 125 | ||||||||||
| Gaps within a burst (τc,1), ms | 0.44 | 0.44 | 0.49 | 0.53 | 0.53 | 0.54 | 0.5 | 0.5 | 0.53 | 0.50 | ||||||||||
| Gaps between bursts, fast (τc,2), ms | 5.34 | — | 9.4 | 12.1 | 9.8 | 21 | 12 | — | 11.9 | 3.05 | ||||||||||
| Gaps between burst, slow (t𝔠,3), ms | 39.3 | — | 230 | 700 | 544 | 1500 | 383 | — | 842 | 35.4 | ||||||||||
| Relative area of τc,2 (a 2) | 0.6 | — | 0.66 | 0.27 | 0.77 | 0.58 | 0.74 | — | 0.83 | 0.52 | ||||||||||
| Relative area of τc,3 (a 3) | 0.4 | — | 0.34 | 0.73 | 0.23 | 0.42 | 0.26 | — | 0.17 | 0.48 | ||||||||||
| Open time (τo), ms | 2.83 | 2.9 | 2.3 | 2.2 | 2.1 | 2.1 | 2.3 | 2.4 | 1.9 | 1.9 | ||||||||||
ATP and Glyburide Affect KATP Channel Behavior Outside Intraburst Transitions
In the absence of ligands, KATP channel activity displayed intraburst and interburst transitions (Fig. 2, a1 and b1). ATP (100 μM; Fig. 2 A, a2) or glyburide (1 μM; Fig. 2 B, b2) blocked KATP channel activity without affecting single channel amplitude (n = 48). Neither ATP (n = 5) nor glyburide (n = 4) significantly changed mean closed (τc,1) and mean open (τo) times that define intraburst kinetics (Table I). However, both inhibitory ligands did prolong the fast (τc,2) and slow (τc,3) characteristic times that define the distribution of gaps between bursts (Table I) and increased the number of prolonged channel closures (Table I; relative areas a 1 and a 2). This was associated with a significant change in transition rates defining interburst, without a change in rates (k 10 and k 01) defining intraburst (C1 ↔ O; Fig. 2) transitions. Specifically, ATP (100 μM) promoted escape of the channel from intraburst transitions to the C2 closed state by increasing the k 02 rate from 0.55 to 28 s−1, and delayed initiation of a burst by decreasing the backward k 20 rate from 70 to 22 s−1 (Fig. 2 A). Similarly, but to a lesser extent than ATP, glyburide (1 μM) increased by threefold the k 02 rate, and decreased by over twofold the backward k 20 rate (Fig. 2 B). Consequently, the mean duration of a burst (2,210 ms, n burst = 23 and 2,380 ms, n burst = 14 in the absence of ATP and glyburide, respectively) was reduced by each of the inhibitory ligands (56 ms, n burst = 255 and 850 ms, n burst = 31 in the presence of ATP and glyburide, respectively).
Figure 2.

Inhibitory ligands act on KATP channels outside intraburst transition. Portions of original single channel records in the absence (a1 and b1) and presence (a2) of 100 μM ATP or 1 μM glyburide (b2). Corresponding kinetic schemes with calculated rates of transitions (in s−1, Eq. 8) are provided for each record in the absence and presence of ATP (A) and in the absence and presence of glyburide (B). Holding potential was −60 mV throughout.
In agreement with experimental data, calculated mean burst duration, using rates defining intraburst transitions and the rate leading away from these transitions (Sakmann and Trube, 1984):
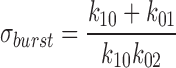 |
8 |
was 2,200 vs. 44 ms in the absence and presence of ATP, and 2,500 vs. 960 ms in the absence and presence of glyburide.
Since the lifetime the channel spends in a specific state is defined by the reciprocal of the sum of transition rates that lead away from this state:
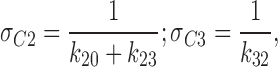 |
9 |
the lifetime the KATP channel spent in C2 or C3, in the presence of ATP (100 μM), was 12.2 and 175 ms, respectively. Although these values were within the range of values obtained in the absence of ATP (σC2 = 9.1 ± 1.4 ms, and σC3 = 207 ± 84 ms; n = 4), ATP by accelerating k 23 and by reducing k 20 rates (Fig. 2 A) promoted the channel to operate within C2 ↔ C3 closed states, away from intraburst transitions. Thereby, ATP significantly increased the combined lifetime the KATP channel spent within interburst transitions (Sakmann and Trube, 1984):
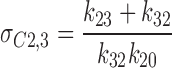 |
10 |
from 87.8 ms in the absence to 524 ms in the presence of ATP (Fig. 2 A).
Glyburide (1 μM) reduced the lifetime in either the C2 (from 9.7 to 20 ms) or the C3 (from 417 to 833 ms) closed states (Fig. 2 B). The mean lifetime (σC2,3) the channel spent within C2 ↔ C3 closed states increased from 132 ms in the absence to 589 ms in the presence of glyburide (Fig. 2 B). Thus, ATP and glyburide, despite apparent differences in the mechanism of channel inhibition, act outside the intraburst C1 ↔ O transition, shorten burst duration, and prolong the time the KATP channel remains within C2 and C3 closed states.
UDP Prevents Ligand Inhibition of KATP Channels by Favoring Intraburst Activity
In the absence of UDP, KATP channel activity exhibited intraburst and interburst transitions (Fig. 3, A and B, Table I) and was sensitive to the inhibitory action of ATP (200 μM). Washout of ATP restored channel activity, which was directed towards intraburst transition by 1 mM UDP (Fig. 3, A and C). Intraburst forward and backward rates of transition were similar before (2,000 and 434 s−1), and after (2,000 and 420 s−1) addition of UDP and ATP. However, UDP eliminated gaps between bursts in all patches so tested (n = 4), which induced an apparent insensitivity of the KATP channel towards ATP (Fig. 3 A).
Figure 3.

Loss of interburst events induced by UDP associated with loss of ATP or glyburide sensitivity of KATP channels. (A) Single-channel record of KATP channel activity inhibited by ATP (200 μM) in the absence but not in the presence of UDP (1 mM). (B) Single-channel record of segment 1 (in A) presented on an extended time scale with distributions for gaps between burst (left), gaps within burst (middle), and open gaps (right). Corresponding kinetic scheme with rates of transitions (s−1, Eq. 8) is provided. (C) Single-channel record of segment 2 (in A), obtained in the presence of UDP (1 mM) and ATP (200 μM) and presented on an extended time scale with distributions for gaps between burst (left), gaps within burst (middle), and open gaps (right). In the presence of both UDP and ATP, essentially no gaps between bursts were visible, while no effect on intraburst kinetics was observed. A corresponding kinetic scheme, without interburst transitions and with calculated rates of intraburst transitions (s−1), is provided. (D) Single-channel record of KATP channel activity depicting UDP-induced antagonism of glyburide-mediated channel inhibition. Holding potential was −60 mV throughout.
The effect of UDP was also tested in the presence of glyburide, a nonnucleotide inhibitory ligand of the channel. As in the case of ATP (Fig. 3 A), maintenance of channel activity within intraburst transitions by UDP also prevented glyburide (3 μM) to block KATP channel activity (Fig. 3 D; n = 6). Thus, regardless of the structure of the inhibitory ligand, UDP could apparently shield the KATP channel from ATP or glyburide by “trapping” the channel within ligand-insensitive transitions.
UDP Is Not the Sole Determinant of Sensitivity to Inhibitory Ligands
The efficacy with which UDP antagonizes ATP- and glyburide-inhibitory gating has been reported to vary with the operative condition of the KATP channel (Terzic et al., 1994a ; Brady at al., 1998). As shown in Fig. 4 A, sustained spontaneous KATP channel activity was only partially sensitive to ATP (300 μM) in the presence of UDP (2 mM), yet fully sensitive in the absence of UDP (n = 10). With channel “rundown,” UDP restored channel activity but could no longer antagonize ATP (Fig. 4 A, see also Terzic et al., 1994a ). Similarly, after rundown, UDP induced channel opening that was inhibited by glyburide (1 μM; Fig. 4 B; n = 5). However, restoration of spontaneous channel activity, by pretreatment with MgATP (5 mM), was associated with return of UDP- induced antagonism of glyburide inhibition (Fig. 4 B, see also Brady et al., 1998). Thus, channel rundown appears to decrease the efficacy with which UDP antagonizes the action of inhibitory ligands.
Figure 4.

Dual responsive behavior of KATP channels, in the presence of UDP, towards ATP and glyburide. (A) After patch excision, KATP channel activity was vigorous and sustained at maximal level by UDP (2 mM), under which condition ATP (300 μM) produced only partial channel inhibition. Removal of UDP was associated with channel inhibition by ATP. Such effect was reproducible. With time after patch excision, KATP channel activity was observed to rundown. Under partial rundown, UDP enhanced channel activity, but only partially antagonized ATP- induced channel inhibition. The dotted line corresponds to the zero-current level. (B) Conversion of rundown to spontaneous KATP channel activity by Mg-ATP switches on the UDP-induced antagonism of glyburide-dependent channel block. A 10-min long pretreatment of rundown KATP channels with 5 mM Mg-ATP restored spontaneous channel activity and with it the UDP-induced antagonism of glyburide-dependent channel block lost in rundown channels. In the absence of UDP, channel activity was readily inhibited by glyburide. The dotted line with original trace corresponds to the zero-current level. nP o values, corresponding to the trace record, were calculated over 1.02-s-long intervals. Holding potential was −60 mV throughout.
Rundown Prevents UDP from Holding KATP Channels within Intraburst Transitions
Progression of rundown of KATP channel activity was associated with an increased number of prolonged closures (Fig. 5 A, top). Intraburst channel properties (τc,1 and τ0, Fig. 5 B, Table I) were unchanged with rundown, but both fast and slow time components defining gaps between burst (τc,2 and τc,3) were prolonged (Fig. 5 C, Table I). As the process of rundown accelerated, the k 02 rate responsible for exit of the channel from a burst (from 0.52 ± 0.07 s−1, n = 4, under sustained activity to 3.1 s−1 during rundown; Fig. 5), mean burst duration of partial rundown channels was shorter when compared with sustained channel activity (τburst = 440 ms; n burst = 224 vs. τburst = 2,456 ± 315 ms; n = 4). Furthermore, KATP channels under partial rundown spent a significantly longer time in the C3 state (Eq. 10) when compared with channels under sustained channel activity (670 ms vs. 207 ± 84 ms; n = 4), although the apparent lifetime in the C2 state (12 ms vs. 9.1 ± 1.4 ms, n = 4; Eq. 10) was similar under both conditions. Thus, channel rundown directed KATP channels away from intraburst and towards interburst transitions.
Figure 5.

Rundown prevents UDP from locking KATP channels within intraburst transitions. (A) Single KATP channel records during partial rundown in the absence (top) and presence (bottom) of 1 mM UDP. Zero-current level indicated by 0 pA. (B) Intraburst kinetic properties. UDP (right) did not affect distributions of closed (top) and open (bottom) times within bursts of channel activity. Both distributions were well-fitted by single exponents, with τc,1 and τo representing characteristic closed and open times, respectively. (C) Distribution of gaps between bursts in the absence (left) and presence (right) of UDP. Under both conditions, distributions needed to be fitted by the sum of two exponents with characteristic times τc,2 andτc,3. Solid lines correspond to the sum of both exponents drawn by fitting, whereas dashed lines correspond to individual exponents. Holding potential was −60 mV. (D) Kinetic scheme constructed based on calculated rates of transitions (in s−1, Eq. 8) in the absence (left) and presence (right) of 1 mM UDP.
To determine the efficacy with which UDP acts on KATP channels driven towards the C3 state as rundown progresses, UDP (1 mM) was applied to partially rundown channels (Fig. 5 A, bottom). UDP increased channel activity and reduced the number of channel closures (Fig. 5 A, bottom). Similar to its effect on sustained KATP channel activity, UDP did not affect intraburst kinetics; i.e., UDP did not significantly change rates defining the C1 ↔ O transition (k 10 and k 01) of partially rundown channels (Fig. 5, A and B). However, in contrast to sustained KATP channel activity (Fig. 1), UDP did not eliminate interburst events in rundown channels, the distribution of which remained biexponential (Fig. 5 C). Also, in contrast to its effect on sustained channel activity where UDP eliminated the O ↔ C2 transition (from 0.35 to ∼0 s−1; Fig. 1, Table I), UDP only decreased the transition rate (k 02) associated with burst closure of partially rundown channels (i.e., the O ↔ C2 transition changed from 3.1 to 2.0 s−1; Fig. 5 D). Under this condition, the KATP channel could still transit between intraburst and interburst states despite the presence of UDP (Fig. 5, A and C). Mean burst duration (Eq. 9) of partially rundown channels increased in the presence of UDP from τburst = 440 ms (n burst = 224) to τburst = 684 ms (n burst = 125), similar to calculated values of τburst (412 and 631 ms in the absence and presence of UDP, respectively; Eq. 9). Acting on partially rundown channels, UDP apparently reduced the lifetime the channel spent within interburst transitions (3 and 15 ms for C2 and C3, respectively; Fig. 5 A; Eq. 10). Thus, under such conditions, although UDP still directed partially rundown channels towards intraburst ligand-insensitive transitions, the nucleotide diphosphate could not maintain the channel within a ligand-insensitive state.
Allosteric Model of Ligand/Channel Interaction
It has been reported that UDP shifts to the right the concentration–response curve to ATP (Terzic et al., 1994a ), whereas it completely eliminates the sensitivity of the KATP channel to glyburide (Brady et al., 1998). Based on the kinetic model, it was not possible to quantitatively predict the different response of KATP channels to inhibitory ligands. Therefore, an allosteric model of ligand/protein interaction (Monod et al., 1965), previously applied to analyze ligand regulation of ion channels (Karlin, 1967; Hosoya et al., 1997; Tibbs et al., 1997), was used. The features of such allosteric models are that (a) the allosteric protein (i.e., the channel complex) interconverts within two distinct conformational states: ligand-sensitive (S) and -insensitive (I); (b) the channel complex possesses two sets of binding sites (n and m), one for the inhibitor (A), and the second for the activator (B); (c) each set of binding sites is equivalent within a state, but exhibits different microscopic dissociation constants (KA,S or KB,S in S and KA,I or KB,I in I) between states; and (d) binding of an inhibitor or an activator shifts, in opposite directions, the equilibrium between the two conformational states and thus increases the fraction of total protein (channel complex) in the state with higher affinity for a particular ligand and decreases the apparent (macroscopic) affinity towards the other ligand.
The equilibrium constant (L) in the absence of a ligand was expressed by the ratio between the channel lifetime in ligand-insensitive (i.e., mean burst duration; Eq. 9) and -sensitive (i.e., lifetime spent in C2 plus C3; Eq. 11) states:
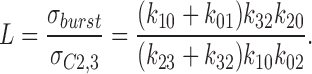 |
11 |
From our experimental data (i.e., Figs. 1 and 2), calculation of L revealed a value in the range of 150–200 under spontaneous channel activity. This indicates that under sustained spontaneous channel activity the equilibrium between ligand-sensitive and -insensitive states is significantly shifted towards ligand-insensitive states, which corresponds to a prolonged burst of channel activity.
Channel inhibition is a function of the fraction of the protein in the S state:
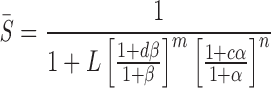 |
12 |
where d = KB,S /KB,I, β = [B]/KB,S, c = KA,S/KA,I, and α = [A]/KA,S. For A, the allosteric inhibitor (e.g., glyburide or ATP), c < 1. For B, the allosteric activator (i.e., UDP), d > 1.
In the absence of UDP, the concentration dependence of KATP channel inhibition by glyburide (A; Fig. 6
A, •) was well fitted by the 1-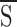 function (Eq. 13) using the following parameters: L = 200; KA,S = 1 nM, KA,I = 1 mM, n = 1 (Fig. 6
A, solid line). In the presence of 1 mM UDP, the observed loss of sensitivity of the KATP channel towards the sulfonylurea (Fig. 6
A, ○) was well described by the allosteric model (L = 200; KA,S = 1 nM, KA,I = 1 mM, n = 1; KB,S = 3.5 mM, KB,I = 0.1 mM, m = 4). The model predicts that at millimolar concentrations of the activator (UDP) the effect of glyburide on KATP channel activity will be fully antagonized even at tens of micromoles of the inhibitor (Fig. 6
A, solid line).
function (Eq. 13) using the following parameters: L = 200; KA,S = 1 nM, KA,I = 1 mM, n = 1 (Fig. 6
A, solid line). In the presence of 1 mM UDP, the observed loss of sensitivity of the KATP channel towards the sulfonylurea (Fig. 6
A, ○) was well described by the allosteric model (L = 200; KA,S = 1 nM, KA,I = 1 mM, n = 1; KB,S = 3.5 mM, KB,I = 0.1 mM, m = 4). The model predicts that at millimolar concentrations of the activator (UDP) the effect of glyburide on KATP channel activity will be fully antagonized even at tens of micromoles of the inhibitor (Fig. 6
A, solid line).
Figure 6.

Use of an allosteric model to predict the effect of glyburide or ATP on KATP channel opening in the absence and presence of UDP. (A) Concentration dependence of glyburide-induced KATP channel inhibition in the absence (•) and presence (○) of 1 mM UDP. Data points are from five to nine patches. Relative effect of glyburide was calculated in each patch as a ratio between slopes of cumulative nP o measured in the presence over the value obtained in the absence of glyburide (see Brady et al., 1998). Solid curves were constructed using Eq. 13 at various concentrations of UDP (see text for values of parameters). (B) Concentration dependence of ATP-induced KATP channel inhibition in the absence (▪) and presence (▾) of 5 mM UDP. Data points are from 4 to 10 patches. Relative effect of ATP was calculated in each patch as a ratio between nP o values measured in the presence over the value obtained in the absence of ATP (see Terzic et al., 1994a ). Curves 1 and 1′ (at 0 mM UDP), 2 and 2′ (at 5 mM UDP) were constructed using Eqs. 13 and 14, respectively. See text for values of parameters.
In the absence of UDP, the concentration dependence of KATP channel inhibition by ATP (A; Fig. 6
B, ▪) was also well fitted by the 1-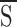 function (Eq. 13) using the following parameters: L = 200; KA,S = 8.5 μM, KA,I = 70 mM, n = 3.6 (Fig. 6
B, curve 1). However, in the presence of UDP (5 mM; KB,S = 3.5 mM, KB,I = 0.1 mM, m = 4), the allosteric model predicted a rightward shift but failed to precisely fit (Fig. 6
B, curve 2) the experimentally obtained data defining the concentration response of ATP-induced KATP channel inhibition under this condition (Fig. 6
B, ▾). Since varying the number of binding sites, cooperativity, and/or dissociation constants for ATP did not improve the fit, we further developed the model taking into account the existence of an additional presumed binding site for ATP not affected by nucleotide diphosphate regulation (Tucker et al., 1997; Ueda et al., 1997). Therefore, we added to the 1-
function (Eq. 13) using the following parameters: L = 200; KA,S = 8.5 μM, KA,I = 70 mM, n = 3.6 (Fig. 6
B, curve 1). However, in the presence of UDP (5 mM; KB,S = 3.5 mM, KB,I = 0.1 mM, m = 4), the allosteric model predicted a rightward shift but failed to precisely fit (Fig. 6
B, curve 2) the experimentally obtained data defining the concentration response of ATP-induced KATP channel inhibition under this condition (Fig. 6
B, ▾). Since varying the number of binding sites, cooperativity, and/or dissociation constants for ATP did not improve the fit, we further developed the model taking into account the existence of an additional presumed binding site for ATP not affected by nucleotide diphosphate regulation (Tucker et al., 1997; Ueda et al., 1997). Therefore, we added to the 1-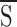 function (Eq. 13) an additional allosteric regulation-independent inhibitory process to account for both mechanisms of ATP inhibitory action:
function (Eq. 13) an additional allosteric regulation-independent inhibitory process to account for both mechanisms of ATP inhibitory action:
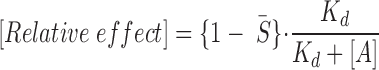 |
13 |
where K d is the dissociation constant to this additional ATP binding site. In this adjusted model, K d = 610 μM could fully account for the ATP-induced channel inhibition in the absence (Fig. 6 B, curve 1′) and presence (Fig. 6 B, curve 2′) of UDP. The prediction based on the developed model can be interpreted to mean that UDP antagonized ATP-induced KATP channel inhibition through one set of ATP-binding sites, thus reducing channel inhibition to the other set of ATP-binding sites.
Despite the lack of reliable means to quantify the rundown process, the allosteric model could, in principle, also be used to describe the effects of ligands under rundown of KATP channel activity. Based on our experimental data (Fig. 5), rundown shifted the equilibrium constant (L) from 150–200 under spontaneous channel activity to 10–20 under partial rundown, and to even lower values with further progression of rundown. This was associated with a prolongation of the lifetime the channel spent in C2 and C3 closed states. In contrast to spontaneous channel activity, where UDP increased L → ∞•(by k 02 → 0), under partial rundown UDP elevated L only up to 70–90 (i.e., Fig. 5). However, such a change in L was not sufficient to restore the experimentally obtained channel sensitivity towards ATP and glyburide (Terzic et al., 1994a ; Brady et al., 1998). This could indicate that besides the effect on L, rundown could also alter other parameters of channel/ ligand interaction. For instance, rundown could decrease the affinity of ligands for the I state, which would promote the effect of inhibitors (Thuringer and Escande, 1989; Deutsch and Weiss, 1993). Thus, the use of the allosteric model could explain the nonuniform responsive behavior of KATP channels to inhibitory ligands in the presence of UDP depending on the operative condition of the channel.
discussion
The present study demonstrates that cardiac KATP channels can be directed to operate within ligand-insensitive conformational states. The switch into ligand-insensitive behavior was induced by the nucleotide diphosphate, UDP. Interconversion between ligand-sensitive and -insensitive states represents a novel mechanism of KATP channel regulation. The property of a nucleotide diphosphate to direct cardiac KATP channels towards a state that is insensitive towards inhibitory ligands could provide a mechanistic basis for channel opening in the presence of inhibitory concentrations of ATP within an intact cell.
Ligand–Channel Interaction and Kinetic Model
To distinguish between conformational transitions that define KATP channel activity, we applied a linear kinetic model used previously (Sakmann and Trube, 1984; Gillis et al., 1989; Nichols et al., 1991; Furukawa et al., 1993; Takano and Noma, 1993). This entropic model does not describe all conformations through which a channel transits, but it does allow description of end points of sequential conformational transitions accessible to direct measurement. Although this simplified model was developed for inward KATP channel current as recorded under present experimental conditions (with symmetrical K+ solutions and at a holding potential of −60 mV), intraburst and interburst transitions can also be distinguished for outward KATP channel currents despite more complex intraburst kinetics (Zilberter et al., 1988; Larsson et al., 1993; Alekseev et al., 1997b ). Herein, we found that the inhibitory ligands, ATP and glyburide, inhibited KATP channel activity by acting upon conformational states that define interburst behavior without affecting intraburst channel transitions. This is in agreement with previous studies that have also shown that inhibitory ligands target specific rates of channel kinetics (Gillis et al., 1989; Qin et al., 1989; Nichols et al., 1991; Takano and Noma, 1993; Benz and Kohlhardt, 1994; Smith et al., 1994). We further found that UDP could keep the channel within a burst, preventing interburst transitions. Therefore, the observed effect of UDP to antagonize channel inhibition by ATP and glyburide could be attributed to the limitation of channel behavior within ligand-insensitive intraburst conformational transitions. Such a mechanism could explain the altered responsiveness of cardiac KATP channels towards ATP and sulfonylureas observed in the presence of nucleotide diphosphates (Nichols and Lederer, 1991; Venkatesh et al., 1991; Virag et al., 1993; Findlay, 1994; Terzic et al., 1994a ; Brady et al., 1998). Based on the kinetic model used, the present study provides evidence that the response of the cardiac KATP channel depends not only on the concentration of an inhibitor, but also on the lifetime the channel spends within ligand-sensitive states. This concept may not be limited to UDP. Indeed, it has been shown that other agents, such as potassium channel openers (Fan et al., 1990; Terzic et al., 1994b ), intracellular protons (Vivaudou and Forestier, 1995; Alekseev et al., 1997a ), cytoskeleton disrupters (Brady et al., 1996a ), or channel trypsinization (Deutsch and Weiss, 1994), which also promote the KATP channel to operate within a burst, decrease the sensitivity of the channel towards inhibitory ligands. Furthermore, combined application of ADP and the opener diazoxide potentiated the ability of these agents to antagonize ATP inhibition of KATP channels by prolonging the lifetime the channel spends within a burst (Larsson et al., 1993). Conversely, ATP and related nucleotides that direct the KATP channel to operate within interburst transitions were shown to enhance the sensitivity of KATP channels towards sulfonylureas (Virag et al., 1993; Schwanstecher et al., 1994).
Application of an Allosteric Model to the Regulation of KATP Channels
The dual nature of KATP channel behavior, in terms of ligand-sensitive and -insensitive states, drew parallelism with interconversion of an allosteric protein between two significant conformational states with different affinities to ligands (Monod et al., 1965; Karlin, 1967). This allosteric model predicted the observed change in the ATP- and glyburide-dependent inhibitory gating of the channel induced by a UDP-mediated shift in the equilibrium towards the ligand-insensitive state of the KATP channel. The difference in the microscopic affinities for the two conformational states (106 for glyburide and ∼104 for ATP) predicted by the present allosteric model is consistent with the existence of ligand-insensitive and -sensitive states of the channel. In fact, the allosteric model predicted that at millimolar concentrations of UDP the cardiac KATP channel loses its sensitivity towards glyburide. This is in accord with previous studies that have established that under spontaneous KATP channel activity, nucleotide diphosphates, such as UDP or ADP, antagonize sulfonylurea-induced channel inhibition (Venkatesh et al., 1991; Virag et al., 1993; Brady et al., 1998). Although it is difficult to compare microscopic with apparent dissociation constants, high and low affinities for sulfonylurea binding have been previously reported (Fosset et al., 1988; Aguilar-Bryan et al., 1992). Thus, due to the negligible affinity of glyburide towards the ligand-insensitive (I) state, UDP by shifting the equilibrium of the KATP channel towards this particular state could effectively antagonize the effect of the sulfonylurea. In the case of ATP, for which the model predicts an additional, nucleotide diphosphate-independent, ATP-inhibitory channel gating, UDP could produce only a rightward shift in the concentration response curve of ATP-induced channel inhibition, as previously experimentally observed with UDP (Terzic et al., 1994a ) or other nucleotide diphosphates such as ADP (Findlay, 1988; Lederer and Nichols, 1989). In contrast to UDP, channel rundown shifted the equilibrium towards the ligand-sensitive state of the KATP channel. This increased sensitivity of the channel towards inhibitory ligands is in accord with experimental findings that have shown that rundown enhances the inhibitory action of ATP on cardiac KATP channel activity (Thuringer and Escande, 1989; Deutsch and Weiss, 1993). Since rundown is believed to be associated with changes in the phosphorylation status of the KATP channel or associated proteins (Trube and Hescheler, 1984; Findlay and Dunne, 1986; Findlay, 1987; Ohno-Shosaku et al., 1987; Takano et al., 1990; Furukawa et al., 1996; Hilgemann and Ball, 1996), the equilibrium between the S and I channel states may be dependent upon a phosphorylation process. Treatment of rundown membrane patches with Mg-ATP (but not with ATP alone or with nonhydrolyzable ATP analogs), through presumed “rephosphorylation” of channel proteins, restored spontaneous cardiac KATP channel activity, and with it the efficacy of UDP to antagonize ATP- and glyburide-induced channel inhibition (see also Terzic et al., 1994a ; Brady et al., 1998).
Relevance to the Molecular Structure of KATP Channels
Results predicted by the allosteric model applied herein to the native cardiac KATP channel are in agreement with the reported structure and stoichiometry of the recombinant KATP channel complex (Inagaki et al., 1995, 1996; Isomoto et al., 1996; Clement et al., 1997; Tucker et al., 1997). This channel is a heteromultimer that combines four Kir6.2 and four SUR subunits into an octamer (Clement et al., 1997; Inagaki et al., 1997). It has been suggested that ATP binds to both the pore-forming Kir6.2 (Tucker et al., 1997) and the regulatory SUR (Bernardi et al., 1992; Ueda et al., 1997) subunits. In view of this, the requirement of two sets of binding sites for ATP could be interpreted to indicate two separate ATP-binding sites on each subunit of the channel complex. The allosteric model further predicts four binding sites for UDP on the KATP channel complex. This apparently correlates with the previously observed binding of a nucleotide diphosphate to only one of the channel subunits, the SUR subunit (Bernardi et al., 1992; Nichols et al., 1996; Gribble et al., 1997; Trapp et al., 1997; Tucker et al., 1997; Ueda et al., 1997). The binding of sulfonylureas to the KATP channel is also presumed to occur on the SUR subunit (Aguilar-Bryan et al., 1995; Inagaki et al., 1995, 1996; Clement et al., 1997). However, in terms of the allosteric model and in contrast to nucleotides, KATP channel regulation by glyburide was characterized by lack of cooperativity (see also Venkatesh et al., 1991).
Intraburst kinetics that define ligand-insensitive transitions are apparently associated with conformational fluctuation of Kir6.2 itself (Alekseev et al., 1997b ; Tucker et al., 1997), whereas interburst kinetics are modulated by association of Kir6.2 with the SUR subunit (Inagaki et al., 1996). In view of the proposed structure of the KATP channel complex (Inagaki et al., 1995, 1996, 1997; Clement et al., 1997; Tucker et al., 1997), the kinetic and allosteric properties of channel behavior may provide the basis for a mechanistic model of the UDP- induced changes in the ATP- and glyburide-dependent regulation of KATP channel gating (Fig. 7). Such a model implies the existence of two inhibitory gating pathways. The first, mediated through binding of inhibitory ligands to the SUR subunit, appears to be sensitive to UDP regulation. The second, mediated through binding of ATP to Kir6.2, appears to be insensitive to UDP regulation. Disruption of the first inhibitory gating pathway by UDP switches the channel into sulfonylurea- insensitive behavior and decreases the channel sensitivity towards ATP (Fig. 7). Phosphorylation(s) of the channel protein can apparently restore the ability of UDP to disrupt the first inhibitory pathway lost after rundown of channel activity.
Figure 7.

Scheme of UDP-induced change in ATP- and glyburide-dependent inhibitory channel gating. This mechanistic model, which takes into account the proposed structure of the KATP channel (Inagaki et al., 1995, 1996, 1997; Clement et al., 1997; Tucker et al., 1997), as well as the kinetic and allosteric properties of channel behavior, suggests the existence of two inhibitory gating mechanisms of KATP channels labeled 1 and 2. The gating mechanism number 1 transduces inhibitory signals from glyburide (Glyb) and ATP-binding sites on the SUR channel subunit. This inhibitory gating can be intercepted after binding of a nucleotide-diphosphate (NDP) to the SUR subunit. Presumed dephosphorylation of the channel affects nucleotide diphosphate–dependent regulation of channel gating. The gating mechanism number 2 transduces inhibitory signals from the ATP-binding site on the Kir6.2 channel subunit, which appears to be insensitive to nucleotide-diphosphate regulation.
Concluding Remarks
Although the present study used UDP as a nucleotide diphosphate, the observed effect on KATP channel behavior may also be attributable to other nucleotide diphosphates, such as ADP. In contrast to UDP, the presence of the adenine moiety could make the interpretation of the effect of ADP more complex due to possible competitive interaction of ADP with an ATP-binding site (Ueda et al., 1997). Despite this, similar effects of ADP (MgADP) in modulating the KATP channel inhibitory gating and postrundown channel behavior have previously been demonstrated (Dunne and Petersen, 1986; Tung and Kurachi, 1991; Venkatesh et al., 1991; Weiss and Venkatesh, 1993; Findlay, 1988, 1994; Elvir-Mairena et al., 1996), as well as the ability of MgADP to prolong burst duration (Larsson et al., 1993).
The property of cardiac KATP channels to interconvert between ligand-sensitive and -insensitive states, described herein, resembles other ion channels that also show differential sensitivity towards ligands depending on their operative state, including “use-dependent” blockade of Na+ and Ca2+ channels (Lee and Tsien, 1983; Hill et al., 1989; Ragsdale at al., 1994; Nuss et al., 1995). Entry of the KATP channel into a ligand-insensitive state by a nucleotide diphosphate could provide a basis for cardiac KATP channel opening during hypoxia or ischemia despite rather constant levels of ATP within the cell.
Acknowledgments
This work was supported by grants from the Bruce and Ruth Rappaport Program in Vascular Biology and Gene Delivery, the Miami Heart Research Institute, the American Heart Association, the George M. Einseberg Cardiovascular Research Fund, and the Harrington Professorship Fund (to A. Terzic). P.A. Brady is supported by a Clinician-Investigator Fellowship from General Mills.
references
- Aguilar-Bryan L, Nichols CG, Rajan AS, Parker C, Bryan J. Co-expression of sulfonylurea receptors and KATPchannels in hamster insulinoma tumor (HIT) cells. Evidence for direct association of the receptor with the channel. J Biol Chem. 1992;267:14934–14940. [PubMed] [Google Scholar]
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Alekseev AE, Gomez LA, Aleksandrova LA, Brady PA, Terzic A. Opening of cardiac sarcolemmal KATPchannels by dinitrophenol separate from metabolic inhibition. J Membr Biol. 1997a;157:203–214. doi: 10.1007/s002329900229. [DOI] [PubMed] [Google Scholar]
- Alekseev AE, Kennedy ME, Navarro B, Terzic A. Burst kinetics of co-expressed Kir6.2/SUR1 clones: comparison of recombinant with native ATP-sensitive K+channel behavior. J Membr Biol. 1997b;159:161–168. doi: 10.1007/s002329900279. [DOI] [PubMed] [Google Scholar]
- Alekseev AE, Jovanovic A, Lopez JR, Terzic A. Adenosine slows the rate of K+-induced membrane depolarization in ventricular cardiomyocytes: possible implication in hyperkalemic cardioplegia. J Mol Cell Cardiol. 1996a;28:1193–1202. doi: 10.1006/jmcc.1996.0110. [DOI] [PubMed] [Google Scholar]
- Alekseev AE, Markevich NI, Korystova AF, Terzic A, Kokoz YM. Comparative analysis of the kinetic characteristics of L-type calcium channels in cardiac cells of hibernators. Biophys J. 1996b;70:786–797. doi: 10.1016/S0006-3495(96)79618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard B, Lazdunski M. Nucleotide diphosphates activate the ATP-sensitive potassium channel in mouse skeletal muscle. Pflügers Arch Eur J Physiol. 1992;422:185–192. doi: 10.1007/BF00370419. [DOI] [PubMed] [Google Scholar]
- Aschroft FM, Ashcroft SJH. Properties and functions of ATP-sensitive K+channels. Cell Signal. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- Benz I, Kohlhardt M. Distinct modes of blockade in cardiac ATP-sensitive K+channels suggest multiple targets for inhibitory drug molecules. J Membr Biol. 1994;142:309–322. doi: 10.1007/BF00233438. [DOI] [PubMed] [Google Scholar]
- Bernardi H, Fosset M, Lazdunski M. ATP/ADP binding sites are present in the sulfonylurea binding protein associated with brain ATP-sensitive K+channels. Biochemistry. 1992;31:6328–6332. doi: 10.1021/bi00142a023. [DOI] [PubMed] [Google Scholar]
- Bokvist K, Ämmälä C, Ashcroft FM, Berggren P-O, Larsson O, Rorsman P. Separate processes mediate nucleotide- induced inhibition and stimulation of the ATP-regulated K+-channels in mouse pancreatic β-cells. Proc R Soc Lond Ser B Biol Sci. 1991;243:139–144. doi: 10.1098/rspb.1991.0022. [DOI] [PubMed] [Google Scholar]
- Brady PA, Alekseev AE, Aleksandrova LA, Gomez LA, Terzic A. A disrupter of actin microfilaments impairs sulfonylurea-inhibitory gating of cardiac KATPchannels. Am J Physiol. 1996a;271:H2710–H2716. doi: 10.1152/ajpheart.1996.271.6.H2710. [DOI] [PubMed] [Google Scholar]
- Brady, P.A., A.E. Alekseev, and A. Terzic. 1998. Operative condition-dependent response of cardiac ATP-sensitive K+ channels toward sulfonylureas. Circ. Res. In press. [DOI] [PubMed]
- Brady PA, Zhang S, Lopez JR, Jovanovic A, Alekseev AE, Terzic A. Dual effect of glyburide, an antagonist of KATP channel, on metabolic inhibition-induced Ca2+loading in cardiomyocytes. Eur J Pharmacol. 1996b;308:343–349. doi: 10.1016/0014-2999(96)00327-5. [DOI] [PubMed] [Google Scholar]
- Bryan J, Aguilar-Bryan L. The ABCs of ATP-sensitive potassium channels—more pieces of the puzzle. Curr Opin Cell Biol. 1997;9:553–559. doi: 10.1016/s0955-0674(97)80033-6. [DOI] [PubMed] [Google Scholar]
- Clement JP, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of K-ATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Decking UKM, Reffelmann T, Schrader J, Kammermeier H. Hypoxia-induced activation of K-ATP channels limits energy depletion in the guinea pig heart. Am J Physiol. 1995;269:H734–H742. doi: 10.1152/ajpheart.1995.269.2.H734. [DOI] [PubMed] [Google Scholar]
- Decking UKM, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- Deutsch N, Weiss JN. ATP-sensitive K+channel modification by metabolic inhibition in isolated guinea-pig ventricular myocytes. J Physiol. 1993;465:163–179. doi: 10.1113/jphysiol.1993.sp019671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch N, Weiss JN. Effects of trypsin on cardiac ATP-sensitive K+channels. Am J Physiol. 1994;266:H613–H622. doi: 10.1152/ajpheart.1994.266.2.H613. [DOI] [PubMed] [Google Scholar]
- Dunne MJ, Petersen OH. Intracellular ADP activates K+channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett. 1986;208:58–62. doi: 10.1016/0014-5793(86)81532-0. [DOI] [PubMed] [Google Scholar]
- Elvir-Mairena JR, Jovanovic A, Gomez LA, Alekseev AE, Terzic A. Reversal of the ATP-liganded state of ATP-sensitive K+channels by adenylate kinase. J Biol Chem. 1996;271:31903–31908. doi: 10.1074/jbc.271.50.31903. [DOI] [PubMed] [Google Scholar]
- Fan Z, Nakayama K, Hiraoka M. Multiple actions of pinacidil on adenosine triphosphate-sensitive potassium channels in guinea-pig ventricular myocytes. J Physiol. 1990;430:273–295. doi: 10.1113/jphysiol.1990.sp018291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay I. The effects of magnesium upon adenosine trisphosphate-sensitive potassium channels in a rat insulin-secreting cell line. J Physiol. 1987;391:611–629. doi: 10.1113/jphysiol.1987.sp016759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay I. Effects of ADP upon the ATP-sensitive K+channel in rat ventricular myocytes. J Membr Biol. 1988;101:83–92. doi: 10.1007/BF01872823. [DOI] [PubMed] [Google Scholar]
- Findlay I. Interactive regulation of the ATP-sensitive potassium channel of cardiac muscle. J Cardiovasc Pharmacol. 1994;24:S6–S11. [PubMed] [Google Scholar]
- Findlay I, Dunne MJ. ATP maintains ATP-inhibited K+channels in an operational state. Pflügers Arch Eur J Physiol. 1986;407:238–240. doi: 10.1007/BF00580683. [DOI] [PubMed] [Google Scholar]
- Forestier C, Vivaudou M. Modulation by Mg2+and ADP of ATP-sensitive potassium channels in frog skeletal muscle. J Membr Biol. 1993;132:87–94. doi: 10.1007/BF00233054. [DOI] [PubMed] [Google Scholar]
- Fosset M, De Weille JR, Green RD, Schmid-Antomarchi H, Lazdunski M. Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+channels. J Biol Chem. 1988;263:7933–7936. [PubMed] [Google Scholar]
- Furukawa T, Virag L, Sawanobori T, Hiraoka M. Stilbene disulfonates block ATP-sensitive K+channels in guinea pig ventricular myocytes. J Membr Biol. 1993;136:289–302. doi: 10.1007/BF00233668. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Yamane T, Terai T, Katayama Y, Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+channel to the actin cytoskeleton. Pflugers Archiv Eur J Physiol. 1996;431:504–512. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Gee WM, Hammoud A, McDaniel ML, Falke LC, Misler S. Effects of sulfonamides on a metabolite-regulated ATPi-sensitive K+channel in rat pancreatic β-cells. Am J Physiol. 1989;257:C1119–C1127. doi: 10.1152/ajpcell.1989.257.6.C1119. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the walker a motifs of SUR1 in KATPchannel activation by Mg-ADP and diazoxide. EMBO (Eur Mol Biol Organ) J. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RJ, Duff HJ, Sheldon RS. Class I antiarrhythmic drug receptor: biochemical evidence for state-dependent interaction with quinidine and lidocaine. Mol Pharmacol. 1989;36:150–159. [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Yamada M, Matsumoto S, Higashiguchi O, Horio Y, Matsuzawa Y, Kurachi Y. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+channel. J Biol Chem. 1996;271:24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- Jovanovic A, Zhang S, Alekseev AE, Terzic A. Diadenosine polyphosphate-induced inhibition of cardiac KATPchannels: operative state-dependent regulation by a nucleoside diphosphate. Pflügers Arch Eur J Physiol. 1996;431:800–802. doi: 10.1007/BF02253848. [DOI] [PubMed] [Google Scholar]
- Jovanovic A, Alekseev AE, Terzic A. Intracellular diadenosine polyphosphates: a novel family of inhibitory ligands of the ATP-sensitive K+channel. Biochem Pharmacol. 1997;54:219–225. doi: 10.1016/s0006-2952(97)00262-1. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Ball R. Regulation of cardiac Na+, Ca2+ exchange and KATP potassium channels by PIP2 . Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- Hosoya Y, Yamada M, Ito H, Kurachi Y. A functional model for G protein activation of the muscarinic K+channel in guinea pig atrial myocytes. Spectral analysis of the effect of GTP on single-channel kinetics. J Gen Physiol. 1996;108:485–495. doi: 10.1085/jgp.108.6.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakei M, Noma A. Adenosine-5′-triphosphate-sensitive single potassium channel in the atrioventricular node cell of the rabbit heart. J Physiol. 1984;352:265–284. doi: 10.1113/jphysiol.1984.sp015290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakei M, Kelly RP, Ashcroft SJH, Ashcroft FM. The ATP-sensitivity of K+channels in pancreatic β-cells is modulated by ADP. FEBS Lett. 1986;208:63–66. doi: 10.1016/0014-5793(86)81533-2. [DOI] [PubMed] [Google Scholar]
- Karlin A. On the application of “a plausible model” of allosteric proteins to the receptor for acetylcholine. J Theor Biol. 1967;16:306–320. doi: 10.1016/0022-5193(67)90011-2. [DOI] [PubMed] [Google Scholar]
- Larsson O, Ammala C, Bokvist K, Fredholm B, Rorsman P. Stimulation of the KATPchannel by ADP and diazoxide requires nucleotide hydrolysis in mouse pancreatic β-cells. J Physiol. 1993;463:349–365. doi: 10.1113/jphysiol.1993.sp019598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazdunski M. ATP-sensitive potassium channels: an overview. J Cardiovasc Pharmacol. 1994;24:S1–S5. [PubMed] [Google Scholar]
- Lederer WJ, Nichols CJ. Nucleotide modulation of the activity of rat heart ATP-sensitive K+channels in isolated membrane patches. J Physiol. 1989;419:193–211. doi: 10.1113/jphysiol.1989.sp017869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialyzed heart cells. Nature. 1983;302:790–794. doi: 10.1038/302790a0. [DOI] [PubMed] [Google Scholar]
- Misler S, Falke LC, Gillis K, McDaniel ML. A metabolite-regulated potassium channel in rat pancreatic β cells. Proc Natl Acad Sci USA. 1986;83:7119–7123. doi: 10.1073/pnas.83.18.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ, Cannel MB. ATP dependence of KATPchannel kinetics in isolated membrane patches from rat ventricle. Biophys J. 1991;60:1164–1177. doi: 10.1016/S0006-3495(91)82152-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Tomaselli GF, Marban E. Cardiac sodium channels (hH1) are intrinsically more sensitive to block by lidocaine than are skeletal muscle (μ1) channels. J Gen Physiol. 1995;106:1193–1209. doi: 10.1085/jgp.106.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Zünkler BJ, Trube G. Dual effects of ATP on K+currents of mouse pancreatic β-cells. Pflügers Arch Eur J Physiol. 1987;408:133–138. doi: 10.1007/BF00581342. [DOI] [PubMed] [Google Scholar]
- Qin D, Takano M, Noma A. Kinetics of the ATP-sensitive K+channel revealed with oil-gate concentration jump method. Am J Physiol. 1989;257:H1624–H1633. doi: 10.1152/ajpheart.1989.257.5.H1624. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Trube G. Voltage-dependent inactivation of inward-rectifying single-channel currents in the guinea-pig heart cell membrane. J Physiol (Camb) 1984;347:659–683. doi: 10.1113/jphysiol.1984.sp015089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanstecher C, Dickel C, Panten U. ATP-sensitive K+channel in mouse beta-cells. Br J Pharmacol. 1994;111:302–310. doi: 10.1111/j.1476-5381.1994.tb14060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Inagaki N, Namba N, Gonoi T. Molecular biology of the β-cell ATP-sensitive K+channel. Diabetes Rev. 1996;4:177–190. [Google Scholar]
- Smith PA, Williams BA, Ashcroft FM. Block of ATP-sensitive K+channels in isolated mouse pancreatic beta-cells by 2,3-butanedione monoxime. Br J Pharmacol. 1994;112:143–149. doi: 10.1111/j.1476-5381.1994.tb13044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano M, Noma A. The ATP-sensitive K+channel. Prog Neurobiol. 1993;41:21–30. doi: 10.1016/0301-0082(93)90039-u. [DOI] [PubMed] [Google Scholar]
- Takano M, Qin D, Noma A. ATP-dependent decay and recovery of K+channels in guinea-pig cardiac myocytes. Am J Physiol. 1990;258:H45–H50. doi: 10.1152/ajpheart.1990.258.1.H45. [DOI] [PubMed] [Google Scholar]
- Terzic A, Findlay I, Hosoya Y, Kurachi Y. Dualistic behavior of ATP-dependent K+channel towards intracellular nucleotide diphosphates. Neuron. 1994a;12:1049–1058. doi: 10.1016/0896-6273(94)90313-1. [DOI] [PubMed] [Google Scholar]
- Terzic A, Jahangir A, Kurachi Y. HOE-234, a second generation K+ channel opener, antagonizes the ATP-dependent gating of cardiac ATP-sensitive K+channels. J Pharmacol Exper Ther. 1994b;68:818–825. [PubMed] [Google Scholar]
- Terzic A, Jahangir A, Kurachi Y. Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+channel-opening drugs. Am J Physiol. 1995;269:C525–C545. doi: 10.1152/ajpcell.1995.269.3.C525. [DOI] [PubMed] [Google Scholar]
- Terzic A, Kurachi Y. Actin microfilament disrupters enhance KATPchannel opening in patches from guinea-pig cardiomyocytes. J Physiol (Camb) 1996;492:395–404. doi: 10.1113/jphysiol.1996.sp021316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzic A, Tung R, Inanobe A, Katada T, Kurachi Y. G proteins activate ATP-sensitive K+channels by antagonizing ATP-dependent gating. Neuron. 1994c;12:885–893. doi: 10.1016/0896-6273(94)90340-9. [DOI] [PubMed] [Google Scholar]
- Terzic A, Tung R, Kurachi Y. Nucleotide regulation of ATP-sensitive K+channels. Cardiovasc Res. 1994d;28:746–753. doi: 10.1093/cvr/28.6.746. [DOI] [PubMed] [Google Scholar]
- Tibbs GR, Goulding EH, Siegelbaum SA. Allosteric activation and tuning of ligand efficacy in cyclic-nucleotide-gated channels. Nature. 1997;386:612–615. doi: 10.1038/386612a0. [DOI] [PubMed] [Google Scholar]
- Thuringer D, Escande D. Apparent competition between ATP and the potassium channel opener RP-49356 on ATP-sensitive K+channels of cardiac myocytes. Mol Pharmacol. 1989;36:897–902. [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Activation and inhibition of K-ATP currents by guanine nucleotides is mediated by different channel subunits. Proc Natl Acad Sci USA. 1997;94:8872–8877. doi: 10.1073/pnas.94.16.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trube G, Hescheler J. Inward-rectifying channels in isolated patches of the heart cell membrane: ATP-dependence and comparison with cell-attached patches. Pflügers Arch Eur J Physiol. 1984;401:178–184. doi: 10.1007/BF00583879. [DOI] [PubMed] [Google Scholar]
- Tucker S, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Tung RT, Kurachi Y. On the mechanism of nucleotide diphosphate activation of the ATP-sensitive K+channel in ventricular cell of guinea-pig. J Physiol (Camb) 1991;437:239–256. doi: 10.1113/jphysiol.1991.sp018593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Inagaki N, Seino S. MgADP antagonism to Mg2+-independent ATP binding of the sulfonylurea receptor SUR1. J Biol Chem. 1997;272:22983–22986. doi: 10.1074/jbc.272.37.22983. [DOI] [PubMed] [Google Scholar]
- Venkatesh N, Lamp ST, Weiss JN. Sulfonylureas, ATP-sensitive K+ channels, and cellular K+loss during hypoxia, ischemia and metabolic inhibition in mammalian ventricle. Circ Res. 1991;69:623–637. doi: 10.1161/01.res.69.3.623. [DOI] [PubMed] [Google Scholar]
- Virag L, Furukawa T, Hiraoka M. Modulation of the effect of glibenclamide on KATPchannels by ATP and ADP. Mol Cell Biochem. 1993;119:209–215. doi: 10.1007/978-1-4615-3078-7_28. [DOI] [PubMed] [Google Scholar]
- Vivaudou M, Forestier C. Modification by protons of frog skeletal muscle KATPchannels: effect on ion conduction and nucleotide inhibition. J Physiol (Camb) 1995;486:629–645. doi: 10.1113/jphysiol.1995.sp020840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Hiltbrand B. Functional compartmentation of glycolytic versus oxidative metabolism in isolated rabbit heart. J Clin Invest. 1985;75:436–447. doi: 10.1172/JCI111718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Venkatesh N. Metabolic regulation of cardiac ATP-sensitive K+channels. Cardiovasc Drugs Ther. 1993;7:499–505. doi: 10.1007/BF00877614. [DOI] [PubMed] [Google Scholar]
- Zilberter Y, Burnashev N, Papin A, Portnov V, Hodorov B. Gating kinetics of ATP-sensitive single potassium channels in myocardial cells depends on electromotive force. Pflügers Arch Eur J Physiol. 1988;411:584–589. doi: 10.1007/BF00582382. [DOI] [PubMed] [Google Scholar]