

Abstract
Two high-resolution structures of a double mutant of bacterial cholesterol oxidase in the presence or absence of a ligand, glycerol, are presented, showing the trajectory of glycerol as it binds in a Michaelis complex-like position in the active site. A group of three aromatic residues forces the oxidized isoalloxazine moiety to bend along the N5-N10 axis as a response to the binding of glycerol in the active site. Movement of these aromatic residues is only observed in the glycerol-bound structure, indicating that some tuning of the FAD redox potential is caused by the formation of the Michaelis complex during regular catalysis. This structural study suggests a possible mechanism of substrate-assisted flavin activation, improves our understanding of the interplay between the enzyme, its flavin cofactor and its substrate, and is of use to the future design of effective cholesterol oxidase inhibitors.
Keywords: cholesterol oxidase, Michaelis complex, flavin, flavin activation, redox potential
Bacterial cholesterol oxidase is a bifunctional flavoenzyme that catalyzes the oxidation and isomerization of 3β-hydroxysteroids (Sampson and Vrielink 2003). Rhodococcus equi, an opportunistic pathogen, that infects immunocompromised individuals (Hondalus 1997), secretes high levels of cholesterol oxidase. While cholesterol oxidase does not appear to be important in the early stages of infection (Pei et al. 2006) and its exact role is unknown, it has been proposed that the bacteria require cholesterol oxidase for maintenance of the infection in the host macrophage (Linder and Bernheimer 1997). The enzyme is unique to bacteria and therefore constitutes an attractive target for the design of novel antibiotics. However, the design of specific and efficacious inhibitors of the enzyme requires a full understanding of the molecular details of substrate binding and catalysis.
There are two structurally diverse types of cholesterol oxidases: type I containing a non-covalently bound flavin adenine dinucleotide (FAD) cofactor and found in Streptomyces and Rhodococcus bacteria, and type II containing a covalently bound FAD cofactor and found in Brevibacterium and Burkholderia. Both types of enzyme catalyze the same reaction involving oxidation and isomerization of 3β-hydroxysteroids (Scheme 1), but the kinetic mechanisms are different, as are the redox potentials of the FAD cofactors. These functional differences are reflected in the large structural divergence between the two enzyme forms (Vrielink et al. 1991; Yue et al. 1999; Coulombe et al. 2001). Clearly, the differences in cofactor reactivity are dictated by the protein microenvironment, although the mechanism of this modulation is poorly understood.
Scheme 1.

Catalytic mechanism of type I cholesterol oxidase.
The exact structural state of the Michaelis (E•S) complex of either type of cholesterol oxidase is not known. The binding geometry of the substrate has been inferred for the type I enzyme from atomic resolution structural work (Lario et al. 2003) and the structure of a steroid substrate bound to the reduced enzyme (Li et al. 1993). This model places the steroid C3 atom within a hydrogen bond distance (∼3.0 Å) of N5 of FAD and in a position similar to those of hydride donors in many other flavoenzyme oxidases (Fraaije and Mattevi 2000). In this orientation, the 3β-OH of the substrate displaces a structurally conserved active site water molecule (Wat541). To confirm the accuracy of the Michaelis complex model and to better understand the effect of the protein microenvironment on substrate binding and turnover, a structure of the E•S complex of oxidized cholesterol oxidase with a substrate analog was pursued. The Michaelis complex model predicted the movement of Tyr446 and Phe444 (Lario et al. 2003). Structural studies in the presence of a bound ligand would help to establish if ligand binding causes a conformational rearrangement of active site residues and how such changes would affect the microenvironment in the vicinity of the flavin cofactor.
Previously, a structure of cholesterol oxidase from R. equi in complex with dehydroepiandrosterone (DHEA) was obtained by soaking the crystals in a solution of DHEA under anaerobic conditions (Li et al. 1993). Under the conditions of the soak, the enzyme underwent a single turnover event. The lack of oxygen in the soak solution precluded reoxidation of the cofactor, resulting in retention of excess substrate in the active site of the reduced enzyme. However, the altered electrostatics of the reduced enzyme resulted in a binding geometry inconsistent with the previously determined substrate-bound structures of flavin-dependent oxidases (Fraaije and Mattevi 2000).
Successful trapping of the true Michaelis complex requires soaking the crystal with substrate or an analog under aerobic conditions, choosing a mutant form of the enzyme that exhibits reduced enzyme turnover (kcat) while minimally affecting the affinity for the substrate (Km), and choosing a substrate or an analog that has higher solubility in the crystal stabilizing medium than cholesterol or DHEA. Cholesterol oxidase from Streptomyces sp. (a close homolog of the R. equi cholesterol oxidase) was chosen because its crystals yield X-ray diffraction data to very high resolution and because much biochemical information exists concerning its key mutants. A double mutant, H447Q/E361Q, of this enzyme was selected for this study since it has a ∼600-fold lower kcat than wild type with minimal effect on Km. (Yin 2002) The substitution of His447 by glutamine was expected to preserve the hydrogen-bonding interactions with the substrate, slowing down catalysis without a serious disruption of substrate binding (Kass and Sampson 1998b; Yue et al. 1999). The Glu361 substitution by Gln was previously shown to affect oxidation in concert with the H447Q mutation (Kass and Sampson 1998a).
Here we present two high-resolution structures of the H447Q/E361Q double-mutant form of Streptomyces sp. CO, one in the presence and one in the absence of a ligand, glycerol. The high-quality electron density maps allow an investigation of the hydrogen-bond network in the mutant protein and the conformational rearrangements that take place in the active site upon substrate binding. This study sheds light on some intriguing new mechanisms by which a flavoprotein is able to influence the reactivity of its flavin cofactor and provides information important to future design of potent cholesterol oxidase inhibitors.
Results
Glycerol binds to the active site of cholesterol oxidase
Two crystal structures of the H447Q/E361Q double mutant of cholesterol oxidase were determined at very high resolution (1.2 Å and 0.98 Å). In both cases, crystals were treated with a cryoprotectant solution containing 20% glycerol and flash frozen. No glycerol electron density was detected in crystals that underwent short (less than 10 sec) soaks in cryoprotectant (Fig. 1A). When the crystals were soaked in the cryoprotectant for ∼3 min, four distinct populations of glycerol in the substrate-binding cavity were observed, three of which are mutually exclusive alternate conformations of one molecule modeled at partial occupancy (Figs. 1B, 2). In addition, a covalent adduct between oxidized and dehydrated glycerol and N5 of the flavin was observed.
Figure 1.
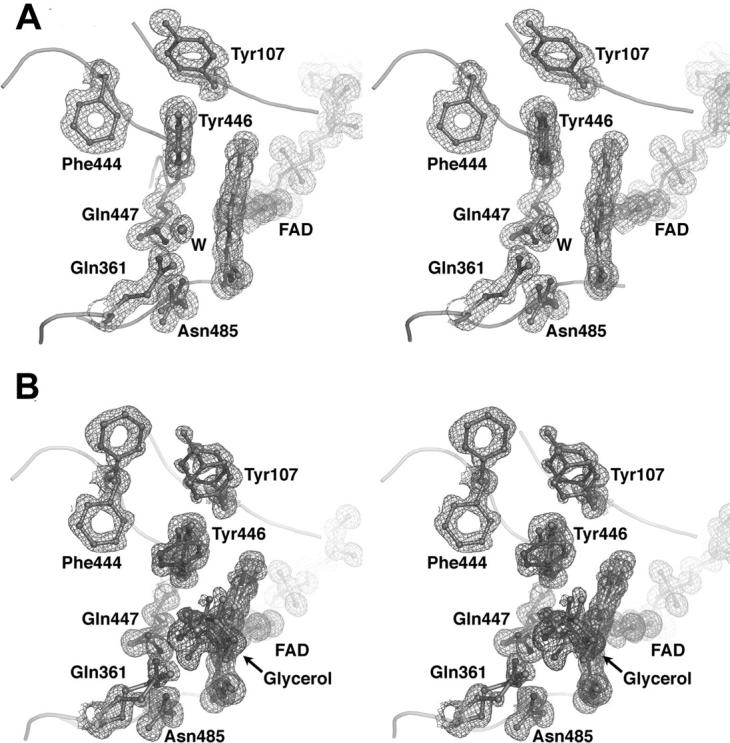
A stereographic representation of the cholesterol oxidase active site. Electron density of the active site in the absence (A) and presence (B) of glycerol. Electron density of the protein and FAD atoms and of the glycerol molecule bound in the active site was calculated with coefficients 2Fo−Fc and contoured at 1.0σ.
Figure 2.
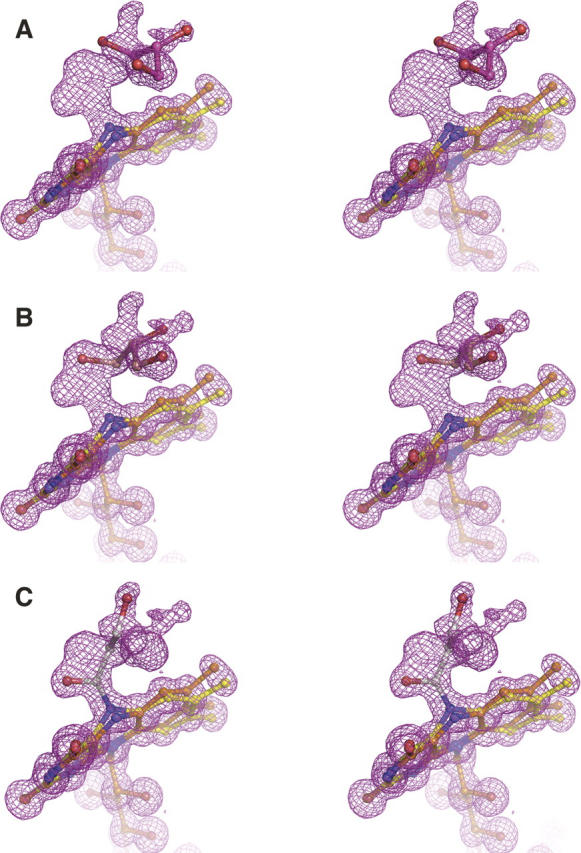
Glycerol populations in the cholesterol oxidase active site. Ball-and-stick representations of the three mutually exclusive ligand populations in the active site, fitted to 2Fo−Fc electron density (magenta mesh, contoured at 1.0σ). Glycerol molecules are shown separately and in different colors for clarity.
The first position occupied by glycerol is at the distal end of the substrate-binding cavity remote from the FAD cofactor (Fig. 3A). The second position of the ligand is near the flavin, and the glycerol is in hydrogen bond contact with NE2 of Gln361 (Fig. 3B). In the third position, the hydrogen bond with Gln361 is lost and a hydrogen bond with NE2 of Gln447 is formed, placing C3 of glycerol within 2.34 Å of N5 of FAD (Fig. 3C). The fourth population of ligand in the cholesterol oxidase active site is a covalent adduct with N5 of FAD (Fig. 3D). In this population, the distance between C3 and O3 of the ligand, which was refined without geometric restraints, is 1.23 Å. This distance corresponds to a C–O double bond. Furthermore, C3, O3, and C2 of glycerol and N5 of FAD all lay within the same plane, indicating that C3 is sp2 hybridized. Furthermore, in the adduct, the distance between C3 and N5 is 1.32 Å, consistent with partial double-bond character and typical of N–C=O linkages. The absence of electron density for O2 suggests that this hydroxyl group has been lost, although the mechanism of adduct formation has not been elucidated. Generation of hydrogen peroxide or consumption of oxygen was not observed with either wild-type or H447Q/E361Q cholesterol oxidase in aqueous solution containing glycerol (0.5–1 M). In solution, no reversible inhibition of wild-type cholesterol oxidase by glycerol was observed (up to 4 M). These data suggest that glycerol has a very weak affinity for the enzyme and will only bind in the absence of competitive ligands. In order to monitor binding directly by visible spectroscopy, an aerobic titration of 30 μM H447Q/E361Q cholesterol oxidase with glycerol was performed. The concentrated protein precipitated upon addition of glycerol. In a second experiment, an anaerobic solution of H447Q/E361Q cholesterol oxidase was electrochemically reduced using the method of Pellett and Stankovich (2002) and monitored by UV/visible spectroscopy. Glyceraldehyde was added to the reduced enzyme. Again, the concentrated protein precipitated before direct binding could be measured. Thus, in solution, direct binding could not be monitored. Moreover, time-dependent inactivation of cholesterol oxidase by glycerol (1 M) or glyceraldehyde (0.1 M) was not detected. These data suggest that a glycerol derivative undergoes covalent reaction with the FAD cofactor only upon irradiation in the X-ray beam, which generates free electrons.
Figure 3.
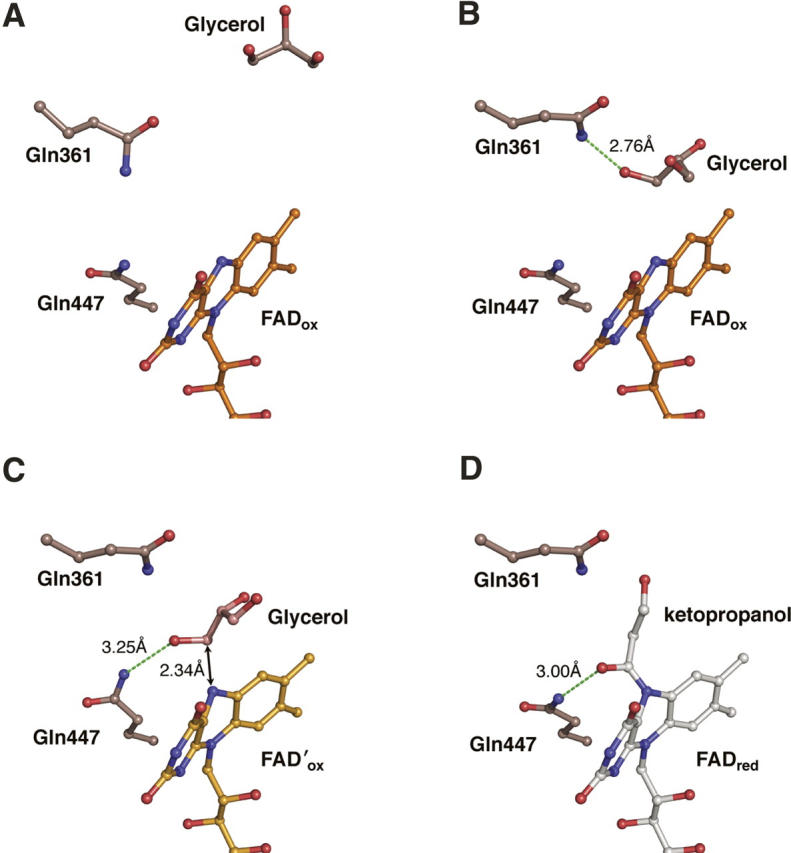
Glycerol binding and reaction with FAD. A time-resolved interpretation of multiple glycerol conformations found in the steroid-binding cavity and the active site. (A) Glycerol binds in the distal part of the steroid-binding cavity; (B) glycerol moves to the proximal part of the steroid-binding cavity and forms a hydrogen bond (green dashed line) with Gln361; (C) glycerol binds in the active site in a Michaelis complex-like orientation; and (D) the covalent FAD-ketopropanol adduct.
To test for glycerol binding to the wild-type enzyme in the crystal, a structure was determined from a wild-type crystal soaked for 10 min in 20% glycerol. Binding effects similar to those observed in the H447Q/E361Q structure were expected. However, electron density observed in the wild-type active site, while larger than a water molecule, could not be unambiguously interpreted as a glycerol molecule (data not shown). Moreover, a covalent bond between glycerol and FAD was not observed in the wild-type structure, though some perturbation of the active site could be seen.
Active site mobility
Comparison of the H447Q/E361Q and wild-type structures shows that OE1 of Gln447 superposes almost exactly with ND1 of His447 in the wild-type enzyme, whereas NE2 of Gln447 is found in two alternate positions within ∼1.5 Å of NE2 of His447 (Fig. 4A). In the glycerol-bound structure, each population of Gln447 makes a hydrogen bond with a population of the glycerol ligand. The first conformation (A) forms a hydrogen bond to the covalent adduct (the fourth glycerol population), and the second conformation (B) makes a long (3.25 Å) H-bond to O3 of glycerol in position three (Fig. 3C,D). The two conformations of Gln447 are also observed in H447Q/E361Q in the absence of glycerol, suggesting that the mobility of glutamine is not the result of ligand binding. Here, conformation B makes a H-bond contact with Wat541 (which occupies the same place as O3-glycerol in position three), while conformation A makes no contacts via NE2.
Figure 4.
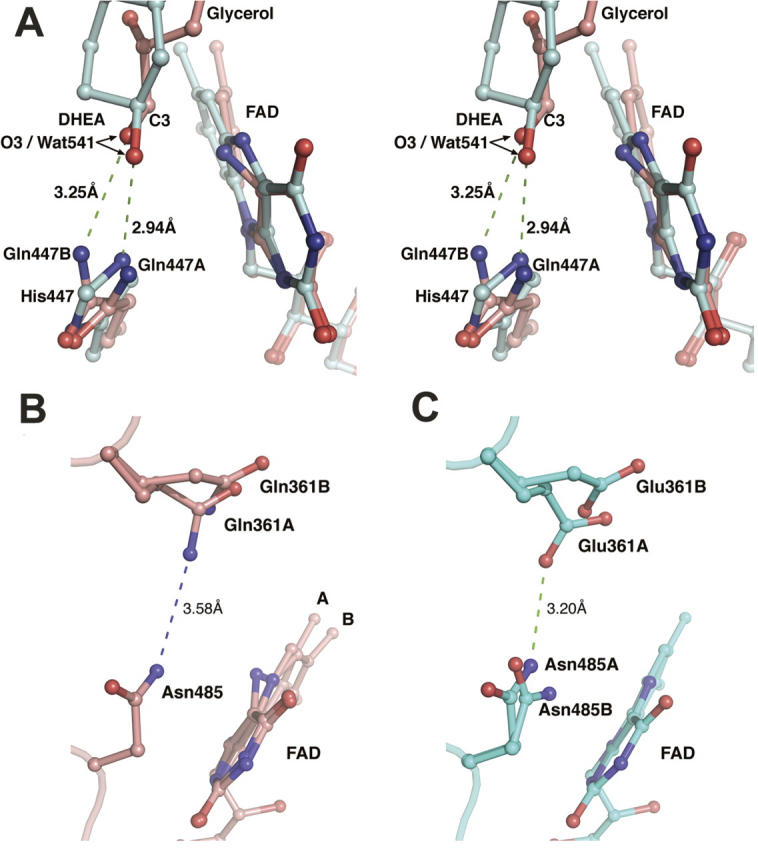
Effect of mutation on the cholesterol oxidase active site. (A) A stereographic representation of Gln447, glycerol, and FAD in the H447Q/E361Q double-mutant structure (pink) superposed with the proposed Michaelis complex model (cyan). For clarity, glycerol and FAD of the H447Q/E361Q structure are shown in conformations corresponding to the proposed Michaelis complex. Only ring A of the steroid substrate is shown. O3 of both DHEA and glycerol occupies the exact position of Wat541 found in corresponding structures in the absence of ligand; this position is labeled as O3/Wat541. (B, C) Comparison of alternate conformations of Gln361 and Asn485 in mutant (B) and wild-type (C) structures.
In the wild-type structure, Glu361 occupies two discrete conformations, correlated with the opening of a narrow hydrophobic channel that leads from the active site to the surface of the protein. This channel has been proposed to act as a possible access route for molecular oxygen during the oxidative half-reaction (Lario et al. 2003). The alternate mobility of Glu361 may also correlate with the roles of this residue in both oxidation and isomerization. In the glycerol-bound H447Q/E361Q structure, position 361 is now occupied by a glutamine residue, which also exhibits multiple conformations (Fig. 1B). In this case, conformation B is in a slightly different position than that of Glu361 in the wild-type structure (Fig. 4B,C) because NE2 of Gln361 repels ND2 of Asn485. Thus, the hydrogen bond between Glu361 and Asn485 in the wild-type structure (Fig. 4C) is eliminated by the E361Q mutation (Fig. 4B).
In the H447Q/E361Q glycerol-bound structure, Asn485 is found in only one conformation, which corresponds to conformation A in the wild-type structure. This conformation forms a hydrogen bond with the covalent FAD-ketopropanol adduct. In the H447Q/E361Q structure without glycerol, two conformations of Asn485 similar to those in the wild-type enzyme are observed. In the wild-type structure, conformation B was proposed to stabilize the reduced FAD cofactor via an NH ⋯ π interaction (Yin et al. 2001; Lario et al. 2003). In the H447Q/E361Q structure, the NH ⋯ π interaction is preserved in the absence of glycerol, but abolished by the steric clash with a covalently modified flavin.
Modification of FAD geometry by the “aromatic triad”
The conformation of the isoalloxazine moiety of the FAD cofactor is characterized by the angle ϕ between the pyrimidine and dimethylbenzene rings. Electron density for the FAD cofactor in the glycerol-bound structure is consistent with two conformations of the isoalloxazine moiety (Figs. 1B, 2), “planar” (ϕ = 166.54° ± 0.65) and “bent” (ϕ = 149.55° ± 0.59). In the absence of glycerol, only the planar conformation of isoalloxazine is observed, with ϕ = 167.64° (±0.58°) (Fig. 1A). The ϕ angle of ∼150° has been associated with reduced isoalloxazine in solution studies (Hasford et al. 1997; Reibenspies et al. 2000), as well as predicted from molecular geometry calculations (Hall et al. 1987; Walsh and Miller 2003), although it is not always found in crystal structures of reduced flavins bound to flavoproteins (Lennon et al. 1999). The distortion of FAD geometry in cholesterol oxidase is correlated with the mobility of a group of aromatic residues in the vicinity of the dimethylbenzene ring of FAD: Tyr107, Phe444, and Tyr446. The binding of glycerol in the active site of cholesterol oxidase causes a steric clash with Tyr446 in the oxidized position. As a result, the phenol group of Tyr446 is rotated ∼90° around the CB–CG bond, the side chain of Phe444 is rotated 180° around the CA-CB bond, and Tyr107 is shifted to maintain the hydrophobic interactions between the three residues (Fig. 5). The rotation of Tyr446 also creates a steric clash with the planar isoalloxazine, inducing the observed change in ring geometry. Thus, distortion of FAD occurs as a result of glycerol binding through rearrangement of the “aromatic triad” of Tyr446, Phe444, and Tyr107.
Figure 5.

Effect of glycerol binding on the “aromatic triad” and FAD ϕ-angle. (A, B) A comparison of Tyr107, Phe444, Tyr446, and Gln447 conformations before (A) and after (B) glycerol binding. (C, D) A comparison of the isoalloxazine “butterfly bend” before (C) and after (D) glycerol binding; the angle (ϕ) between the dimethylbenzene and pyrimidine rings was calculated using SHELX.
The role of glycerol binding in the movement of the aromatic triad and the bending of the isoalloxazine moiety is supported by the structure determined in the absence of glycerol in the active site. In this structure, the aromatic triad residues are clearly seen in conformation A only. Therefore, during regular catalysis, the rearrangements of the triad and the bending of the flavin will occur only when the substrate enters the active site.
Discussion
Glycerol binding reveals the Michaelis complex
The structure reveals that glycerol is a ligand of the enzyme in the crystal state from which inferences about enzyme–substrate interactions can be drawn. One glycerol position in the active site very closely corresponds to the Michaelis complex model proposed previously (Lario et al. 2003). In this position, C3-glycerol is within hydrogen-bond distance of N5-FAD. The location of O3-glycerol exactly overlaps with Wat541 in the H447Q/E361Q structure without glycerol and is very close to Wat541 in the wild-type structure (Vrielink et al. 1991; Lario et al. 2003). This observation fits the proposed model for the Michaelis complex in which Wat541 mimics the position of the substrate 3β-OH. Moreover, the hydrogen-bonding interaction between a proton donor in position 447 and the ligand hydroxyl is preserved in this mutant protein, which supports a recent interpretation regarding the role of His447 in substrate positioning (Lario et al. 2003).
Typically, the distance between the hydride donor and N5-FAD in oxidoreductases is ∼3.5 Å, and the N10-N5-CH angle (where CH is the hydride donor) lies between 96°–117°. In the glycerol H447Q/E361Q structure, the N5-CH distance is 2.34 Å and the corresponding N10-N5-C3 angle is 129.2°. Furthermore, the hydride donor atom is usually shifted with respect to the N5-N10 axis in the direction of the pyrimidine ring by 0.2–1.1 Å. In contrast, C3-glycerol in the H447Q/E361Q structure is shifted ∼1.2 Å in the direction of the dimethylbenzene ring. Thus, the position of C3-glycerol with respect to N5-FAD deviates slightly from what has typically been observed in flavin-dependent oxidoreductases (Fraaije and Mattevi 2000).
The mutation of histidine to glutamine at position 447 is the most likely reason for the differences between substrate-binding geometry typically observed in flavoenzyme oxidases and that observed in the H447Q/E361Q cholesterol oxidase structure. In the absence of glycerol, Wat541 is found in the same position as O3-glycerol in the glycerol-bound structure. Hydrogen bonded to NE2 of Gln447, Wat541 is shifted 0.68 Å toward the dimethylbenzene ring of FAD, in comparison to the Wat541 position in the wild-type enzyme. Thus, it is likely that during regular catalysis of a steroid substrate by the wild-type enzyme, the hydride donor atom would occupy the predicted position (Fig. 4A). Indeed, the predicted Michaelis complex geometry is much closer to that observed in other flavoprotein oxidases.
Exposure of crystals of the wild-type enzyme to glycerol results in weaker binding of the ligand than that observed in the H447Q/E361Q mutant protein. Comparison of the wild-type and mutant active site regions suggests that the difference in binding is due to conformational differences between wild-type Glu361 and the Gln361 of the mutant protein (Fig. 4B,C). Conformation A of glutamate at position 361 lies closer to the active site than that of the glutamine, interfering with glycerol binding. In the H447Q/E361Q mutant protein, Gln361 is pushed away from the active site due to repulsion from Asn485, thus creating a larger cavity that better accommodates the glycerol molecule.
Despite these differences of glycerol binding in the wild-type and mutant enzymes, these structures offer a glimpse of how substrate binding might occur. It is clear from the H447Q/E361Q structure with glycerol that an alcohol hydroxyl group occupies the same position as Wat541 in the absence of ligand. This supports the previous hypothesis that 3β-OH of cholesterol displaces Wat541 during catalysis (Lario et al. 2003). The existence of a mutant cholesterol oxidase that binds glycerol more efficiently is fortunate because it allows observations to be made regarding changes of active site geometry induced by ligand binding.
The observation of a covalently modified FAD, clearly a result of the reaction with glycerol, is in conflict with the lack of observed glycerol turnover in solution. The contradiction may be explained by considering the effect of high-energy X-rays upon the system during data collection. It is well known that exposure of crystals to X-rays at typical experimental wavelengths (1.0–1.5 Å, which corresponds to ∼12 keV) results in radiolysis of water, which produces regions of electron deficiency in the form of hydroxyradicals (O'Neill et al. 2002). These “electron holes” propagate through the system even at temperatures as low as 100 K, at which the diffraction experiments are performed. Given that glycerol is an electron hole-trapping agent, (Davydov et al. 1994) the formation of a covalent adduct with FAD could proceed via a radical mechanism initiated by the X-ray beam and involving hydroxyradicals formed from the water molecules present in the crystal. However, the exact mechanism of this process is unclear.
An aromatic triad influences FADox reactivity
From the analysis of the mutually exclusive populations of glycerol, FAD and the residues in the aromatic triad (Tyr446, Phe444, and Tyr107), we propose that a 15° relative decrease in the ϕ angle is imposed upon the oxidized species of the isoalloxazine by the bound ligand via Tyr446 prior to hydride transfer. This observation is in contrast to theoretical calculations, in which the oxidized flavin is predicted to have much less flexibility along the N5-N10 axis than the reduced flavin, which prefers ϕ ≈ 150° over planarity (Hall et al. 1987; Walsh and Miller 2003).
Molecular orbital calculations suggest that while an energy input of only ∼1–2 kcal/mol is necessary to effect a 10° bend, ∼10 kcal/mol would be required to force a 30° bend (Hall et al. 1987; Walsh and Miller 2003). Though in all structures of CO, isoalloxazine has a butterfly bend of ∼15° prior to ligand binding, the extra 15° bend at ligand binding would still require ∼7 kcal/mol (Hall et al. 1987; Walsh and Miller 2003). It is unclear, given the structural information, from where this energy would come. The binding of glycerol in the Michaelis complex position is relatively weak; however, it is possible that the aromatic triad amplifies the impetus for distortion of the isoalloxazine geometry. The rotation of Tyr446 alone may be insufficient to force a butterfly bend, and the buttressing effect of Tyr107 and Phe444 may be required to cause the necessary change of the isoalloxazine geometry.
The conclusion that the 15° increase in the ϕ angle is the result of ligand binding is strengthened by an atomic resolution structure of a photoreduced Streptomyces sp. CO, which demonstrates that mere reduction of CO-bound FAD is insufficient to effect a significant change of isoalloxazine geometry. In the course of data collection, the region of a crystal of wild-type cholesterol oxidase bathed by the X-ray beam lost its yellow coloring, indicating reduction of the FAD cofactor (P. Lario and A. Vrielink, pers. comm.). The reduction occurred in the absence of any substrate or ligand in the active site, and the structure (Protein Data Bank [PDB] identification no. 1N1P) shows that FAD geometry is unchanged with respect to that observed in the oxidized enzyme. These observations strongly support the inference that the bending of the isoalloxazine is the result of ligand binding and not X-ray exposure. Furthermore, the H447Q/E361Q crystals retained their yellow color throughout data collection in both the presence and absence of glycerol, indicating that in both cases the enzyme remained oxidized. This would imply that the covalent FAD-ketopropanol adduct is also oxidized, but it is not possible to ascertain that at this time.
Distortion of flavin geometry is a commonly observed feature of flavoproteins (Lennon et al. 1999). Imposition of a “butterfly bend” upon the oxidized isoalloxazine has been shown to increase the flavin's redox potential in isolated isoalloxazine derivatives in aqueous solution (Hasford et al. 1997; Reibenspies et al. 2000). Recently, a study of a type II cholesterol oxidase from Brevibacterium sterolicum revealed a correlation between the ϕ angle of isoalloxazine and redox potential (Lim et al. 2006). Specifically, the structure of the wild-type enzyme contained a flavin cofactor with a noticeable butterfly bend, covalently attached to an active-site histidine residue. When the covalent attachment between the protein and the cofactor was eliminated by mutation, the flavin assumed an exactly planar conformation (ϕ = 180°), and a drop in redox potential from −101 mV to −204 mV was observed. In the case of the type I cholesterol oxidase from Streptomyces sp., the change of the ϕ angle is transient rather than permanent and is achieved not by covalent linkage but via the steric interactions with the aromatic triad upon substrate binding. However, the effect of this geometric distortion is most likely the same: to modulate the enzyme's redox potential. It is therefore very probable that mutations of the aromatic triad, and particularly Tyr446, will adversely affect the redox potential and thus the catalytic ability of cholesterol oxidase.
In the structure of R. equi cholesterol oxidase in complex with DHEA, Tyr446 is displaced in exactly the same manner as in the H447Q/E361Q Streptomyces sp. structure. In contrast, Phe444 is shifted only slightly in the R. equi structure, while Tyr107 is not shifted at all, which differs from the large conformational changes of these residues found in the glycerol-bound H447Q/E361Q structure. This may be because of the species-specific differences in the environment of the aromatic triad. In the R. equi enzyme Lys105 is in a strong hydrogen-bond contact with Tyr107, which may prevent Tyr107 from moving; also the position of the Lys105 side chain would block Phe444 from rotating fully around its CA-CB bond. In the Streptomyces sp. enzyme, position 105 is occupied by a valine, which does not prevent the movement of either Tyr107 or Phe444. Thus, participation of Tyr107 and Phe444 in the bending of FAD may be specific to the Streptomyces sp. cholesterol oxidase. In contrast, the role of Tyr446 appears to be conserved in both species. The future design of ligands or inhibitors should take these dynamics into consideration.
Structure-based design of effective inhibitors is complicated by the dynamic nature of enzymes, which cycle rapidly through a number of states during catalysis. However, the vast majority of crystallographic work on enzymes yields static, averaged structures that carry little information about how the state of the enzymes change with time. The advantage of crystallographic analysis at resolutions at and above 1.2 Å lies in the greater amount of structural information available. In the case of the H447Q/E361Q double mutation of cholesterol oxidase, a large number of alternate conformations of amino acid residues, ligand, and cofactor were unambiguously modeled from the high-quality electron density maps. Importantly, information about the trajectory of substrate binding and the catalytic mechanism were gained from analysis of these alternate populations. Two alternate states of a flavin cofactor are seen in a single crystal structure, and geometric distortion of isoalloxazine, which is predicted to influence the flavin's redox potential, is linked to substrate binding. This structural study of active-site plasticity has expanded our understanding of the interplay among the enzyme, its flavin cofactor, and its substrate, and is of use to the future design of effective cholesterol oxidase inhibitors.
Materials and Methods
Materials
Cholesterol, DHEA, horseradish peroxidase (HRP), para-hydroxyphenylacetic acid (p-HPAA), 12,12′-azino-bis [13-ethylbenziazoline-16-sulfonic acid] (ABTS), bovine serum albumin (BSA), safranin O, and methylviologen were purchased from Sigma Chemical Co.; all other chemicals and solvents were supplied by Fisher Scientific, unless otherwise specified. The following buffers were used: buffer A, 50 mM sodium phosphate buffer (pH 7.0); buffer B, buffer A + 0.025% Triton X-100 (w/v) + 0.020% BSA; and buffer C, 10 mM sodium phosphate buffer, 137 mM NaCl, and 2.7 mM KCl (pH 7.0).
Expression, purification, and crystallization of H447Q/E361Q double-mutant protein
The plasmid carrying the H447Q/E361Q mutant gene was used to transform BL21 pLysS strain of E. coli. The mutant protein was expressed by an 8-h induction at 21°C with 0.400 mM IPTG and purified in a similar manner to that previously reported (Yue et al. 1999). Briefly, cell paste containing the overexpressed recombinant cholesterol oxidase was lysed by French press, and the soluble fraction was isolated by ultracentrifugation. The protein was then precipitated by ammonium sulfate and isolated by centrifugation, reconstituted, and purified by ion exchange chromatography using DEAE-cellulose (DE52, Whatman) resin. Fractions containing cholesterol oxidase were pooled and precipitated by ammonium sulfate, reconstituted, and further purified by hydrophobic interaction chromatography (Pharmacia) using butyl-Sepharose resin. The recrystallization step typically applied before the second round of chromatography (Yin et al. 2002) was omitted in this purification, which did not affect the crystallization of the enzyme or its activity. Pure cholesterol oxidase was dialyzed into a 20 mM HEPES storage buffer (pH 7.0). Crystals of the H447Q/E361Q mutant protein were grown by the vapor diffusion method from a 2-μL hanging-drop comprising a 1:1 mixture of protein at 4.8 mg/mL and the precipitant solution (14%–16% polyethylene glycol [PEG] 8000, 75 mM MnSO4, and cacodylate buffer at pH 5.2). The drop was streak seeded using wild-type crystals of the enzyme (Stura and Wilson 1990). Crystals suitable for X-ray diffraction studies grew from a heavy brown precipitate within approximately 1 mo.
Data collection and processing
Data for the native structure were collected using a rotating anode X-ray generator (CuK radiation) and a Raxis IV detector (Rigaku Inc.); a single crystal was used for the entire collection. The crystal was briefly (∼5–10 sec) immersed in the cryoprotecting solution, consisting of the original precipitant solution with 20% glycerol, flash-frozen in liquid nitrogen, and mounted on the goniostat in preparation for X-ray data collection. The detector was tilted at an angle 2θ = 25° to maximize the resolution of the collected reflections. The X-ray diffraction images were processed, merged, and scaled using the HKL2000 suite of software (Otwinowski and Minor 1997); a resolution of 1.20 Å with reasonable completeness in the last shell was achieved (Table 1).
Table 1.
Data collection and refinement statistics
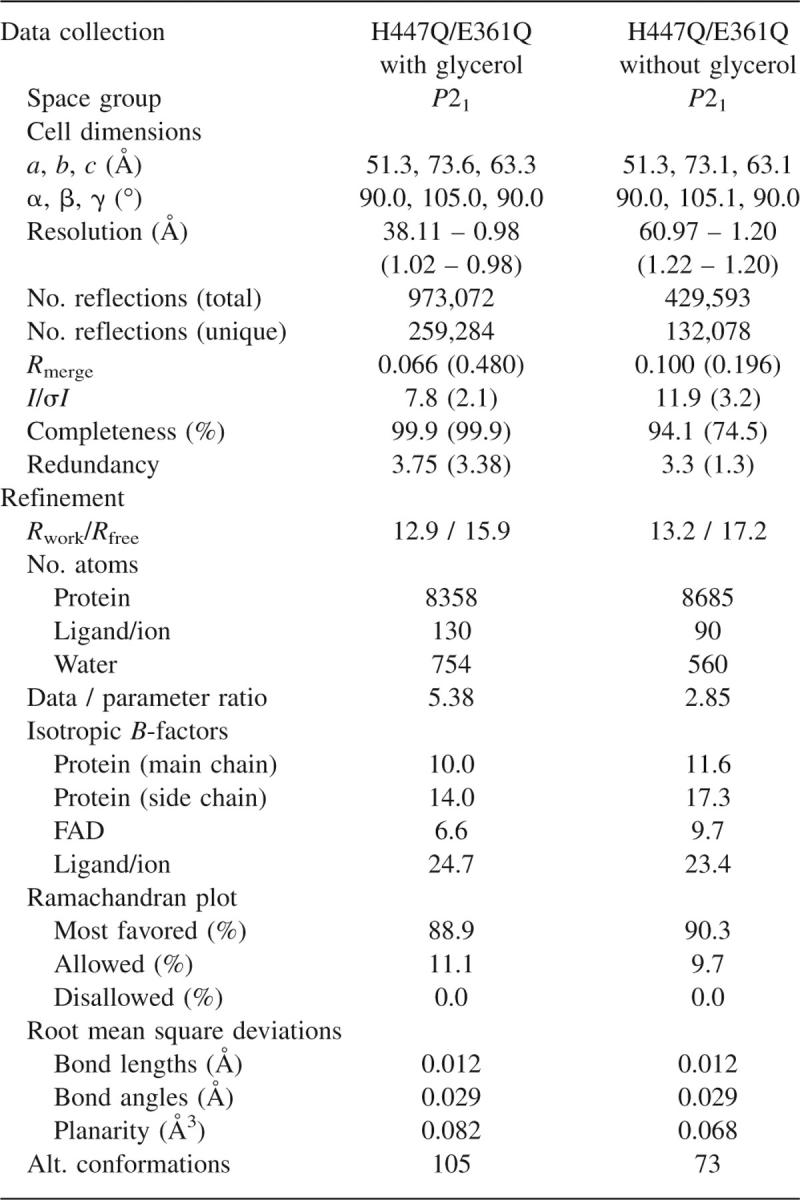
Data for the glycerol-bound structure were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 9-1 from a single crystal. The crystal was incubated in the cryoprotectant solution for ∼3 min and then flash-frozen in liquid nitrogen and mounted on the goniostat. Two sweeps of data collection were performed: one at low exposure to avoid overloaded reflections at low resolution and the other at high exposure to obtain reflections at highest possible resolution. Each data set was integrated individually using the program d*Trek (Pflugrath 1999). Subsequently, the two sets of integrated reflections were merged and scaled together to a final resolution of 0.98 Å (Table 1).
Structure refinement and analysis
The structures of the mutant protein were solved by difference Fourier techniques using the wild-type structure, previously determined to 0.95 Å resolution (Lario et al. 2003), modified to exclude all water molecules and the FAD cofactor. In addition, amino acid side chains that were mutated or were otherwise thought important in catalysis, namely, His447, Glu361, Met122, Val124, Val191, Phe359, Leu377, Asp459, and Asn485 were changed to alanines. Finally, all anisotropic temperature factor parameters were removed while the original isotropic temperature factor parameters were left unchanged. Initial refinement was carried out using Refmac5 (Murshudov et al. 1997). SigmaA-weighted electron density maps were calculated with coefficients 2Fo−Fc and Fo−Fc and used in model building. Very prominent positive difference electron density corresponding to the active site glycerol and the covalently modified FAD cofactor could be clearly seen in these first electron density maps. All of the truncated amino acid side chains (except Phe359, Gln361, and Leu377) were modeled at this point using the program Coot (Emsley and Cowtan 2004), and the models were subjected to another round of restrained least-squares refinement.
Subsequent refinement was carried out using the program SHELXH (Sheldrick and Schneider 1997). The free R-value was monitored during the course of refinement using a test data set composed of 5% of reflection selected randomly from the total set of collected data prior to structure determination and excluded from all rounds of refinement. A significant improvement in R-values and electron density map quality was observed when anisotropic temperature factors were refined for all non-hydrogen atoms in the structure. In the case of water molecules, anisotropic temperature factor parameters were restrained by using the ISOR and CONN commands. After completion of refinement, an additional 15 cycles were performed with the work and test sets combined. Final refinement statistics for both structures are reported in Table 1.
Water molecules were modeled only where strong peaks of difference density were observed and if at least one reasonable hydrogen bond was formed with the protein, the FAD cofactor, or other waters. Alternate positions for water molecules were included where indications were evident in the electron density. In addition, the occupancies of a few water molecules interacting with protein side chains in alternate conformations were refined by using the same free variable (FVAR) for the water molecule and the alternate conformation.
Alternate conformations were added where clearly indicated by the electron density. In the glycerol-bound structure, three contiguous regions of main chain (residues 48–50, 181–187, and 257–259) had to be modeled in alternate conformations. In the case of residues 257–259, the movement was also observed in the structure without glycerol and is consistent with previous observations at atomic resolution (Lario et al. 2003).
Hydrogen atoms were inserted in two steps nearing the final stages of refinement. First, hydrogen atoms bonded to all main-chain atoms and side-chain carbon atoms were placed using the “riding model,” where the position of the hydrogen is fixed with respect to the atom it “rides” on. Next, hydrogen atoms bonded to oxygen or nitrogen atoms on Asn, Arg, Gln, His, Ser, Thr, Trp, and Tyr were modeled, and His NH hydrogens were added only where difference density peaks were visible at a minimal level of 2.0. Modeling of hydrogen atoms improved the R-values by ∼1%, and the improved phases resulted in the detection of a few more water molecules and alternate conformations. In addition, two molecules of SO4 2− and three molecules of glycerol were modeled in the glycerol-bound structure. Of the three glycerol molecules, two were found in the substrate-binding cavity, one of which was modeled in three alternate conformations (Supplemental material Fig. S1). The relative occupancies of these conformations were 0.2 for conformation A (Fig. 4B), 0.4 for conformation B (Fig. 4C), and 0.4 for conformation C (Fig. 4D). Conformation C was modeled as covalently bound to FAD and missing O2. For this conformation, the quality of the electron density allowed unrestrained refinement of the distance between C1 and O1. Finally, the isoalloxazine moiety of FAD was modeled in two states with differing angles between the pyrimidine and dimethylbenzene rings (ϕ). The ϕ angle was quantified, using SHELX, as an angle between planes defined by N1, C2, N3, C4, C4A, C10A, N5, and N10 (pyrimidine ring) and C5A, C6, C7, C8, C9, C9A, N5, and N10 (dimethylbenzene ring). Standard deviations for all calculated geometric parameters were estimated using the full-matrix inversion method (Sheldrick and Schneider 1997). In contrast, no glycerol molecules and no alternate conformations of the cofactor were observed in the native structure. One molecule of SO4 2− was modeled with partial occupancy.
The final models were validated using the programs SFCHECK (Vaguine et al. 1999) and PROCHECK (Laskowski et al. 1993) from the CCP4 suite of software (Collaborative Computational Project and Number 4 1994). The refinement statistics are given in Table 1. The coordinates have been deposited in the Protein Data Bank (Berman et al. 2000) with the accession codes 3B6D (native) and 3B3R (glycerol bound).
Oxygen consumption measurements
Measurements were carried out at 37°C in buffer A + 1 M glycerol. The reaction was initialized by adding cholesterol oxidase to a final concentration of 5 μM. The dissolved oxygen level was monitored with a YSI model 53 oxygen meter as a function of time.
Hydrogen peroxide production measurements
Glycerol (1 M), HRP (2000 U/mL), p-HPAA (200 mM), and ABTS (100 mM) stock solutions were prepared in buffer A. The concentration of enzyme used in the assays were as follows: wild-type CO, 50 nM to 500 nM; H447Q/E361Q CO, 5 μM. The concentration of each reagent used in the assays were as follows: glycerol, 0.5–1 M; HRP, 40 U/mL; p-HPAA, 4 mM; or ABTS, 2 mM. To detect the formation of H2O2 with p-HPAA, the reaction was followed by excitation at 325 nm and monitoring the fluorescence emission at 415 nm; with ABTS, the absorbance was monitored at 600 nm as a function of time.
Assay for irreversible inhibition
Glycerol (up to 1 M) was incubated with 200 nM wild-type cholesterol oxidase or 10 μM H447Q/E361Q in buffer A at 37°C. At various time intervals, the amount of active cholesterol oxidase remaining was measured by removing an aliquot (100 μL), diluting to 1 mL with buffer B containing 50 μM cholesterol, and following the appearance of cholest-4-en-3-one at 240 nm. The inhibition assay for glyceraldehyde was carried out in an identical manner using a maximum concentration of 0.1 M glyceraldehyde due to its limited solubility in the buffer.
Assay for competitive inhibition
A stock solution of DHEA (3.7 mM) was prepared by dissolving DHEA in ethanol. This solution was filtered through a 0.45-μm nylon filter. Measurements of the activity of wild-type enzyme were carried out at 37°C in buffer C + 2 mM ABTS, 40 U/mL HRP, and 4% ethanol. Cholesterol oxidase was added at a final concentration of 40 nM to initiate the reaction. DHEA concentrations ranged from 6–150 μM, and glycerol concentrations were varied from 0–4 M.
Binding titration
H447Q/E361Q (30 μM) was electrochemically reduced as described by Pellett and Stankovich (2002) at pH 7.0, 25°C in buffer A, with 4 μM safranin O and 100 μM methylviologen under anaerobic conditions. One equivalent of glycerol or glyceraldehyde was added from a sidearm, and absorbance spectra were recorded from 330 to 825 nm.
Electronic supplemental material
Supplemental Figure S1 depicts three selected stages of refinement of the H447Q/E361Q glycerol-bound structure and represents the evolution of the original difference electron density corresponding to the active site glycerol molecule.
Acknowledgments
This work is supported by the National Institutes of Health Grants GM63262 to A.V. and HL53301 to N.S.S.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Alice Vrielink, School of Biomedical Biomolecular and Chemical Sciences, University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia; e-mail: alice.vrielink@uwa.edu.au; fax: 61-8-6488-1005.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073168207.
References
- Berman H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number4 1994. The CCP4 suite: Programs for protein crystallography. Acta Cryst. D 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Coulombe R., Yue, K.Q., Ghisla, S., and Vrielink, A. 2001. Oxygen access to the active site of cholesterol oxidase through a narrow channel is gated by an Arg-Glu pair. J. Biol. Chem. 276: 30435–30441. [DOI] [PubMed] [Google Scholar]
- Davydov R., Kuprin, S., Gräslund, A., and Ehrenberg, A. 1994. Electron paramagnetic resonance study of the mixed-valent diiron center in Escherichia coli ribonucleotide reductase produced by reduction of radical-free protein R2 at 77 K. J. Am. Chem. Soc. 116: 11120–11128. [Google Scholar]
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fraaije M.W. and Mattevi, A. 2000. Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem. Sci. 25: 126–132. [DOI] [PubMed] [Google Scholar]
- Hall L.H., Bowers, M.L., and Durfor, C.N. 1987. Further consideration of flavin coenzyme biochemistry afforded by geometry-optimized molecular orbital calculations. Biochemistry 26: 7401–7409. [DOI] [PubMed] [Google Scholar]
- Hasford J.K., Kemnitzer, W., and Rizzo, C.J. 1997. Conformational effects on flavin redox chemistry. J. Org. Chem. 62: 5244–5245. [Google Scholar]
- Hondalus M.K. 1997. Pathogenesis and virulence of Rhodococcus equi . Vet. Microbiol. 56: 257–268. [DOI] [PubMed] [Google Scholar]
- Kass I.J. and Sampson, N.S. 1998a. The importance of Glu361 position in the reaction catalyzed by cholesterol oxidase. Bioorg. Med. Chem. Lett. 8: 2663–2668. [DOI] [PubMed] [Google Scholar]
- Kass I.J. and Sampson, N.S. 1998b. Evaluation of the role of His447 in the reaction catalyzed by cholesterol oxidase. Biochemistry 37: 17990–18000. [DOI] [PubMed] [Google Scholar]
- Lario P.I., Sampson, N., and Vrielink, A. 2003. Sub-atomic resolution crystal structure of cholesterol oxidase: What atomic resolution crystallography reveals about enzyme mechanism and the role of the FAD cofactor in redox activity. J. Mol. Biol. 326: 1635–1650. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283–291. [Google Scholar]
- Lennon B.W., Williams Jr, C.H., and Ludwig, M.L. 1999. Crystal structure of reduced thioredoxin reductase from Escherichia coli: Structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 8: 2366–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Vrielink, A., Brick, P., and Blow, D.M. 1993. Crystal structure of cholesterol oxidase complexed with a steroid substrate: Implications for flavin adenine dinucleotide dependent alcohol oxidases. Biochemistry 32: 11507–11515. [PubMed] [Google Scholar]
- Lim L., Molla, G., Guinn, N., Ghisla, S., Pollegioni, L., and Vrielink, A. 2006. Structural and kinetic analyses of the H121A mutant of cholesterol oxidase. Biochem. J. 400: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder R. and Bernheimer, A.W. 1997. Oxidation of macrophage membrane cholesterol by intracellular Rhodococcus equi . Vet. Microbiol. 56: 269–276. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- O'Neill P., Stevens, D.L., and Garman, E.F. 2002. Physical and chemical considerations of damage induced in protein crystals by synchrotron radiation: A radiation chemical perspective. J. Synchrotron Radiat. 9: 329–332. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pei Y., Dupont, C., Sydor, T., Haas, A., and Prescott, J.F. 2006. Cholesterol oxidase (ChoE) is not important in the virulence of Rhodococcus equi . Vet. Microbiol. 118: 240–246. [DOI] [PubMed] [Google Scholar]
- Pellett J. and Stankovich, M. 2002. Potentiometric measurements of proteins. In Encyclopedia electrochemistry (eds. A.J. Bard and M. Stratmann), pp. 485–509. Wiley, VCH, Weinheim, Germany.
- Pflugrath J.W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55: 1718–1725. [DOI] [PubMed] [Google Scholar]
- Reibenspies J.H., Guo, F., and Rizzo, C.J. 2000. X-ray crystal structures of conformationally biased flavin models. Org. Lett. 2: 903–906. [DOI] [PubMed] [Google Scholar]
- Sampson N.S. and Vrielink, A. 2003. Cholesterol oxidases: A study of nature's approach to protein design. Acc. Chem. Res. 36: 713–722. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. and Schneider, T.R. 1997. SHELXL: High-resolution refinement. In Methods in enzymology (eds. C.W.J. Carter Jr and R.M. Sweet), pp. 319–343. Academic Press, Orlando, FL. [PubMed]
- Stura E. and Wilson, I.A. 1990. Analytical and production seeding techniques. Methods: A Companion to Methods in Enzymology 1: 38–49. [Google Scholar]
- Vaguine A.A., Richelle, J., and Wodak, S.J. 1999. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55: 191–205. [DOI] [PubMed] [Google Scholar]
- Vrielink A., Lloyd, L.F., and Blow, D.M. 1991. Crystal structure of cholesterol oxidase from Brevibacterium sterolicum refined at 1.8 Å resolution. J. Mol. Biol. 219: 533–554. [DOI] [PubMed] [Google Scholar]
- Walsh J.D. and Miller, A.-F. 2003. Flavin reduction potential tuning by substitution and bending. J. Mol. Struct. 623: 185–195. [Google Scholar]
- Yin Y. 2002. “Functional studies to probe the active site structure of cholesterol oxidase”. State University of New York, Stony Brook, NY. Ph.D thesis,
- Yin Y., Sampson, N.S., Vrielink, A., and Lario, P.I. 2001. The presence of a hydrogen bond between asparagine 485 and the pi system of FAD modulates the redox potential in the reaction catalyzed by cholesterol oxidase. Biochemistry 40: 13779–13787. [DOI] [PubMed] [Google Scholar]
- Yin Y., Liu, P., Anderson, R.G., and Sampson, N.S. 2002. Construction of a catalytically inactive cholesterol oxidase mutant: Investigation of the interplay between active site-residues glutamate 361 and histidine 447. Arch. Biochem. Biophys. 402: 235–242. [DOI] [PubMed] [Google Scholar]
- Yue Q.K., Kass, I.J., Sampson, N.S., and Vrielink, A. 1999. Crystal structure determination of cholesterol oxidase from Streptomyces and structural characterization of key active site mutants. Biochemistry 38: 4277–4286. [DOI] [PubMed] [Google Scholar]