

Abstract
N-Acetylglucosamine-1-phosphate uridyltransferase (GlmU) catalyzes the first step in peptidoglycan biosynthesis in both Gram-positive and Gram-negative bacteria. The products of the GlmU reaction are essential for bacterial survival, making this enzyme an attractive target for antibiotic drug discovery. A series of Haemophilus influenzae GlmU (hiGlmU) structures were determined by X-ray crystallography in order to provide structural and functional insights into GlmU activity and inhibition. The information derived from these structures was combined with biochemical characterization of the K25A, Q76A, D105A, Y103A, V223A, and E224A hiGlmU mutants in order to map these active-site residues to catalytic activity of the enzyme and refine the mechanistic model of the GlmU uridyltransferase reaction. These studies suggest that GlmU activity follows a sequential substrate-binding order that begins with UTP binding noncovalently to the GlmU enzyme. The uridyltransferase active site then remains in an open apo-like conformation until N-acetylglucosamine-1-phosphate (GlcNAc-1-P) binds and induces a conformational change at the GlcNAc-binding subsite. Following the binding of GlcNAc-1-P to the UTP-charged uridyltransferase active site, the non-esterified oxygen of GlcNAc-1-P performs a nucleophilic attack on the α-phosphate group of UTP. The new data strongly suggest that the mechanism of phosphotransfer in the uridyltransferase reaction in GlmU is primarily through an associative mechanism with a pentavalent phosphate intermediate and an inversion of stereochemistry. Finally, the structural and biochemical characterization of the uridyltransferase active site and catalytic mechanism described herein provides a basis for the structure-guided design of novel antibacterial agents targeting GlmU activity.
Keywords: conformational changes, active sites, structure/function studies, pyrophosphorylase, crystallography
According to the Center for Disease Control and Prevention, the last few decades have seen a remarkable increase in the occurrence of bacterial resistance to the major groups of antibacterial agents (Levy 2005). Of particular concern is that pathogenic organisms like Pseudomonas, Streptococcus, Staphylococcus, and Enterococcus have developed resistance to many or all of the important antibiotic classes, such as quinolones, penicillins, cephalosporins, aminoglycosides, tetracyclines, macrolides, and rifamycins. Thus, the increasing spread of drug-resistant microorganisms has a significant impact on current antibacterial therapy and provides a renewed impetus for the discovery of novel classes of antibacterial agents. The clinical efficacy of compounds that disrupt bacterial cell-wall biosynthesis, including the vancomycin, β-lactam, and cephalosporin classes of antibiotics, suggests that key enzymes within this pathway may be attractive targets for antibacterial discovery (Green 2002). N-acetylglucosamine-1-phosphate uridyltransferase (GlmU, also referred to as UDP-GlcNAc pyrophosphorylase) is an essential, bifunctional enzyme that catalyzes production of UDP-N-acetylglucosamine (UDP-GlcNAc), which is involved in the peptidoglycan and lipopolysaccharide biosynthetic pathways in both Gram-positive and Gram-negative bacteria (Anderson and Raetz 1987). In the first step of the reaction, GlmU catalyzes the transfer of the acetyl group from acetyl-CoA to glucosamine-1-phosphate to produce N-acetylglucosamine-1-phosphate (GlcNAc-1-P):
In the second reaction, uridyl monophosphate is transferred from UTP to GlcNAc-1-P to produce UDP-GlcNAc and pyrophosphate (PPi) (Mengin-Lecreulx and van Heijenoort 1993, 1994; Gehring et al. 1996; Pompeo et al. 1998):
The acetyltransferase and uridyltransferase activities of GlmU reside in two functionally autonomous catalytic sites (Mengin-Lecreulx and van Heijenoort 1993, 1994; Gehring et al. 1996; Pompeo et al. 1998). Kinetic studies demonstrate that the first step in the production of UDP-GlcNAc is the acetyltransferase reaction carried out within the C-terminal domain. The GlcNAc-1-P product is released from the acetyltransferase active site, where it then binds to the N-terminal uridyltransferase active site. There was no apparent substrate channeling between the active sites (Gehring et al. 1996).
The first crystal structure of GlmU was derived from a C-terminally truncated version from the Escherichia coli ortholog (Brown et al. 1999). This important work laid the groundwork for subsequent crystallographic studies of the full-length structures of GlmU from Escherichia coli (ecGlmU) (Olsen and Roderick 2001; Olsen et al. 2007) and from the Gram-positive bacteria Streptococcus pneumoniae (spGlmU) (Kostrewa et al. 2001; Sulzenbacher et al. 2001). Based on the similarity of protein folds and signature residues, the N-terminal uridyltransferase domain of GlmU belongs to the nucleoside triphosphate transferase (NTP-transferase) superfamily that includes the bacterial adenylyltransferases (Han et al. 2006), cytidylyltransferase (Kwak et al. 2002), and thymidylyltransferases (Blankenfeldt et al. 2000; Zuccotti et al. 2001; Sivaraman et al. 2002). These enzymes also require divalent cations for catalysis, specifically Mg2+, suggesting a common catalytic mechanism (Sivaraman et al. 2002). The C-terminal acetyltransferase domain of GlmU is highly unusual in that it is comprised of multiple hexapeptide repeats arranged in a left-handed parallel β-sheet helix (Kostrewa et al. 2001; Sulzenbacher et al. 2001; Olsen et al. 2007). The left-handed β-sheet helix (LßH) adopts the shape of a hollow equilateral triangular prism that is ∼20 Å wide and 50 Å long. Full-length GlmU exists in a homotrimeric arrangement with the acetyltransferase active site formed at the junction of the LßH domains of two neighboring GlmU molecules (Kostrewa et al. 2001; Sulzenbacher et al. 2001; Olsen et al. 2007).
Although ecGlmU and spGlmU crystal structures have been reported in apo form and in complex with the product UDP-GlcNAc, no substrate-bound crystal structures are available for the GlmU uridyltransferase active site, leading to some uncertainty concerning residues involved in interactions with UTP and GlcNAc-1-P and the structural rearrangement caused by binding of UDP-GlcNAc. Herein, we describe crystal structures of hiGlmU complexes with uridine, UMP, UDP, and the product UDP-GlcNAc. In addition, we report an X-ray structure of hiGlmU in apo form. The information derived from these structures was combined with biochemical characterization of the K25A, Q76A, D105A, Y103A, V223A, and E224A hiGlmU mutants in order to map these active-site residues to catalytic activity of the enzyme. A comparison of the hiGlmU crystal structures suggests that a large conformational change occurs in the uridyltransferase active site upon formation of the GlmU complex with substrates UTP and GlcNAc-1-P. Examination of the hiGlmU-binding conformations indicates that the substrate binding follows an ordered sequential mechanism in which UTP binding precedes GlcNAC-1-P binding. Finally, interactions between the substrate and product molecules and the enzyme active-site residues are compared with other GlmU orthologs, providing a basis for the structure-guided design of new antibacterial agents with the Gram-negative and Gram-positive profiles.
Results
Overall structure
The overall structures of hiGlmU (Fig. 1A) are similar to those previously reported for the full-length spGlmU and ecGlmU (Kostrewa et al. 2001; Sulzenbacher et al. 2001; Olsen et al. 2007). The uridyltransferase domain (residues 4–229) adopts a α/β fold structurally similar to Rossmann fold and binds uridine, UMP, UDP, and UDP-GlcNAc at the active site. The acetyltransferase active site is contained within the C-terminal domain and formed through the trimerization of GlmU molecules. While in the rhombohedral crystals (space group H32, a = b = 108 Å, c = 327 Å), hiGlmU forms exact trimers generated by a crystallographic threefold rotation axis, in the hexagonal crystals (space group P6322, a = b = 108 Å, c = 234 Å), the protein forms exact trimers generated by a crystallographic 63-screw axis. An average interface area calculated as an accessible surface area of the interfacing hiGlmU molecules (apo structure) using PISA (Krissinel and Henrick 2007) is 2212 Å2. Electron density is well-defined for all residues of the bifunctional enzyme, except residues K454–K456 at the C-terminal end that are disordered in all hiGlmU structures. The final structures have an excellent geometry as defined by PROCHECK (Laskowski et al. 1993). The Ramachandran plot generated using final coordinates of the UDP-bound crystal structure (resolution 2.3 Å) showed that 355 (90.6%) residues of the total 392 non-glycine and non-proline residues were in most favorable regions, while 35 (8.9%) residues occupied additionally allowed areas. Two residues, N146 and K352, occupied a generously allowed region, and there were no residues in the disallowed region. Structural analysis of the uridine, UMP, and UDP-bound structures indicate that the uridyltransferase active site of hiGlmU maintains in the same conformation as observed in the apo form (also referred to as an open conformation). However, the binding of the UDP-GlcNAc product induces a substantial structural rearrangement to a more closed conformation of the enzyme (Fig. 1B). The entire region of the upper N-terminal lobe, including residues L133-G140 and V150-K166 tilts ∼20° to anchor the GlcNAc moiety of UDP-GlcNAc. The conformational change is the most noticeable for residues E154 and K156 that are displaced 4 Å and 6 Å, respectively. A comparison of the open and closed conformations of the enzyme suggests that this structural change is required for completion of catalytic reaction.
Figure 1.
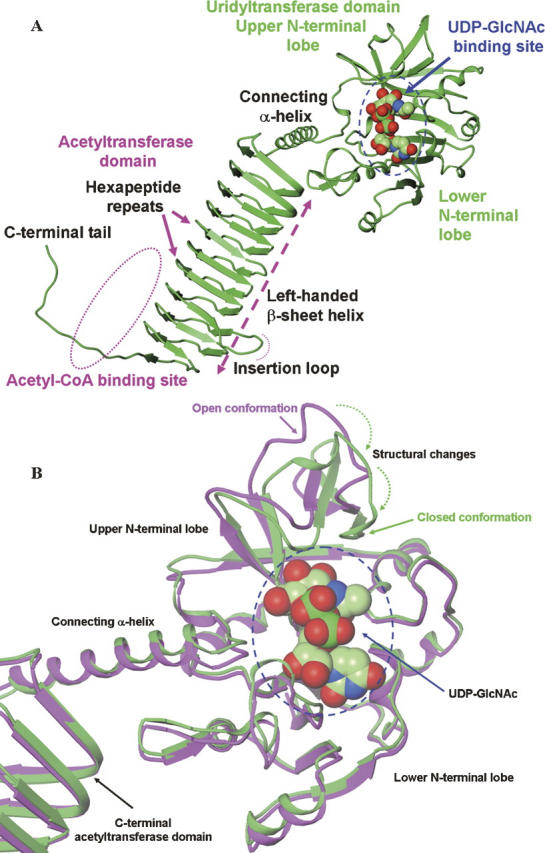
Structures of hiGlmU. (A) Ribbon representation of the bifunctional hiGlmU in a complex with the UDP-GlcNAc product bound at the uridyltransferase-binding site of the N-terminal urydiltransferase domain. (B) Structural changes at the uridyltransferase active site of hiGlmU upon the UDP-GlcNAc-binding event. Ribbon representation of the protein coordinates of hiGlmU in a complex with UDP is colored in magenta. In the UDP-bound complex, the uridyltrasnferase active site is in open conformation similar to the structure of hiGlmU in apo form. Ribbon representation of hiGlmU in a complex with UDP-GlcNAc is colored in green. Upon the substrate binding, the upper lobe of the uridyltransferase domain forms a closed conformation by tilting toward the lower N-terminal lobe and anchoring GlcNAc of UDP-GlcNAc (shown by two dotted arrows). The UDP-GlcNAc molecule is colored in the following atom colors: carbon, light green; nitrogen, blue; oxygen, red; phosphorus, dark green. UDP molecule is omitted for clarity.
Analysis of the GlmU uridyltransferase active site
The unambiguous electron density maps for UDP-GlcNAc, UDP, and uridine (Fig. 2A) describe a consistent binding mode of the ligands to the enzyme active-site pocket. Briefly, the uridyltransferase active site of hiGlmU is ∼22 Å long and 16 Å wide. The active site contains an array of conserved residues A12-G14, K25, Q76-T82, Y103-D105, T137-G140, E154, N169-G171, Q193-T199, and N227 that are arranged in a relatively open elongated surface depression with Y103 at its center (Table 1; Fig. 2B–D). The majority of the key active-site residues are highly conserved across the bacterial GlmU family with the exception of Y103, which is substituted to either alanine or cysteine in Gram-positive species.
Figure 2.

hiGlmU uridyltransferase active site. (A) Stereoview of the (Fo-Fc) OMIT electron density maps of UDP-GlcNAc, UDP, and uridine bound to the uridyltransferase active site. The maps are contoured at 3σ level. (B) Schematic diagram of the UDP-GlcNAc-bound uridyltransferase active site. Key residues of hiGlmU that form the uracil and catalytic subsite, GlcNAc-binding subsite and the liphophilic pocket are labeled. (C) Connolly binding-site surface of GlmU calculated using the UDP-GlcNAc bound crystal structures and colored by hydrophobicity (brown, hydrophobic; blue and red, hydrophilic). The UDP-GlcNAc substrate molecule is colored in the following atom colors: carbon, gray; nitrogen, blue, oxygen, red; phosphorus, green. (D) Schematic drawing of hydrogen bonds and salt bridges between UDP-GlcNAc and hiGlmU residues at the uridyltransferase-binding site.
Table 1.
Sequence alignment of the residues at the uridyltransferase active site

Analysis of the uridine-binding site
The uracil-binding and catalytic subsites consisting of the A12-G14, A18, K25, Q76-T82, and D105 structural segments forms a solvent-exposed pocket that contains Q76 at its bottom (Fig. 2B–D). The uracil moiety of the ligands utilizes characteristic hydrogen-bonded ionic interactions between the ring nitrogen atom N3 and the exocyclic oxygen O4 of the uracil base and the side-chain atoms of Q76, a key residue in the uracil recognition event (Table 2). To determine the necessity of key amino acid residues involved in substrate recognition and catalytic activity, six site-directed mutants were constructed (Supplemental material). The enzyme activities of these six mutant proteins (K25A, Q76A, Y103A, D105A, V223A, and E224A) were measured using the same assay format with 2.5 mM MgCl2 at different enzyme concentrations (Fig. 3). Table 3 compares the initial rates of these mutants to wild-type protein at the same concentration used in the standard assay protocol of 2.5 nM. As the kinetic studies indicated, the site-directed Q76A, K25A, and D105A mutant did not possess any measurable catalytic activity. This further supports our crystal structures and the role that these residues play in the overall kinetic activity of the enzyme, reinforcing their role in UTP recognition. In addition, the exocyclic oxygen atoms O2 and O4 of the uracil ring form a weak hydrogen bond to the peptide nitrogen atoms of residues A13 and G81, respectively, further stabilizing interactions at the uracil recognition site. Overall, the network of hydrogen bonds within the uridine-binding subsites is consistent among various GlmU orthologs (Table 2).
Table 2.
Conserved intermolecular interactions at the uridyltransferase active site of the GlmU/UDP-GlcNAc complexes

Figure 3.

Kinetic characterization of the selected hiGlmU mutants designed to target specific residues at the uridyltransferase active site. Comparison of the enzyme activities of the mutants as a function of enzyme concentration.
Table 3.
Comparison of initial rates of the mutants to hiGlmU wild type at 2.5 nM enzyme concentration under standard assay conditions

Adjacent to the uridine pocket lies the Y103 residue, which is substituted by either alanine or cysteine in the Gram-positive GlmU orthologs (Table 1). While our kinetic (Fig. 3) and crystallographic data indicate that these substitutions do not interfere with the production of UDP-GlcNAc by the enzyme, the central location of Y103 at the active site suggests that this residue can play a role in designing compound inhibitors with Gram-positive or Gram-negative selectivity profiles. The O2′ and O3′ of the ribose moiety of UDP-GlcNAc form a hydrogen bond with residues G14-N and D105-OD1, respectively, while the phosphodiester group, in the absence of a metal ion, is fully exposed to the solvent area and involved only in a water-mediated network of interactions with the protein. The catalytic subsite contains the signature sequence G-X-G-T-(R/S)-(X)4-P-K found in the majority of pyrophosphorylases shown by mutagenesis experiments to be essential for the pyrophosphorylase activity. With an exception of residue Q79 that swings almost 90 degrees around the χ2 torsion angle away from the nucleotide-recognition subsite, side chains of all residues at the uracil-binding and catalytic subsite are positioned optimally for binding of uridine, UMP, or UDP and remain in the same orientations upon binding. The mode of binding of UDP-GlcNAc in hiGlmU is similar to that observed in other GlmU crystal structures (Table 2). Due to the absence of a metal ion in our hiGlmU structures interacting with a nonbridge oxygen of each α- and β-phosphate groups of UDP-GlcNAc, a large portion of the upper N-terminal lobe of the uridyltransferase domain (residues K3-K111 and Q216-N227) of GlmU remains in an open conformation (structure superposition is guided by the Cα positions of the central β-sheet). This conformation is also observed in the metal-free ecGlmU structure complexed with UDP-GlcNAc. On the other hand, in the metal-bound ecGlmU and spGlmU structures (Kostrewa et al. 2001; Olsen and Roderick 2001; Sulzenbacher et al. 2001), this portion of the residues moves nearly 3 Å toward the bound UDP-GlcNAc molecule to add favorable interactions between the phosphodiester group of UDP-GlcNAc and residues K22/K25, D102/D105, and N227 (Table 2).
Because no metal ion was found at the active site in the present hiGlmU structures, we confirmed that this was a crystallization artifact, and the hiGlmU activity is enhanced in the presence of magnesium ions. To demonstrate the dependence on MgCl2 for hiGlmU activity, the assay was performed using MgCl2 concentrations bracketing the optimized assay concentration. The results of the assay indicated (data not shown) that the rate of the assay was influenced by MgCl2 concentration. No activity was observed in the absence of MgCl2, and there appears to be stoichiometric relationship of MgCl2 to enzyme concentration.
GlcNAc-binding site
The GlcNAc-binding subsite of hiGlmU is comprised of T82, T137-G140, N169-G171, and E195-T199 structural segments arranged to recognize the N-acetylglucosamine moiety. It is important to note that the β-phosphate group of UDP is in a different torsional conformation compared with the β-phosphate group of UDP-GlcNAc. It extends away from the GlcNAc-binding subsite into the solvent region. The O7 and N2 of the glucose moiety interact with T82-OG1 and E154-OE2, respectively, in a fashion similar to that in the ecGlmU and spGlmU crystal structures (Kostrewa et al. 2001; Olsen and Roderick 2001; Sulzenbacher et al. 2001). The O3 forms a hydrogen bond with E154-OE1 and N169-ND2. Finally, oxygen atom O4 of the glucose moiety interacts with G140-N and N169-N (Table 2). The region undergoes a substantial structural rearrangement upon the UDP-GlcNAc-binding event when the entire region encompassing residues L133-G140 and V150-K166 tilts ∼20 degrees to anchor the GlcNAc moiety of the product (Fig. 1B). Our analysis of the conformational change shows that residue E154 can act as a structural switch, driving the uridyltransferase active site into the closed conformation upon binding of UDP-GlcNAc. An additional product-induced structural rearrangement includes a 100-degree rotation of the Y197 side chain around the χ2 torsion angle to accommodate the N-acetyl group of the product as well as a closure of a tunnel formed by residues E154, K156 and Q193, Y197, and D200. This structural tunnel has not been reported earlier and exists only in the GlmU structures found in an open conformation. The tunnel connects the GlcNAc-binding subsite to a concave protein surface area filled with solvent water molecules. In its open form, the tunnel is ∼7.3 Å wide (distance measured from E154 to Y197). In the closed form, the distance between residues E154 and Y197 is 3.5–4.0 Å. Interestingly, in the spGlmU apo structure, the residues that form the wall of the tunnel are disordered, as the surface loop consisting of residues 188–193 (spGlmU numbering) was not detected in the electron density maps due to its flexibility (Kostrewa et al. 2001).
Lipophilic pocket
Further analysis of the uridyltransferase active sites of the hiGlmU crystal structures led to identification of a lipophilic pocket that has not been previously characterized (Table 1; Fig. 2B,C). The lipophilic site is adjacent to the GlcNAc-binding subsite and consists of A217-V223, L129-L133, and V168-T170 structural arrays that are partially shielded from solvent by the Y139 and E224 residues. In the apo-like crystal structure, the distance between Y139-OH and E224-OE1 is 5.24 Å, and the lipophilic pocket is partially occupied by a polyethylene glycol (PEG) solvent molecule. Structural rearrangements caused by the binding of UDP-GlcNAc resulted in tightening of the lipophilic pocket by forming a direct hydrogen bond between Y139-OH and E224-OE1 (2.92 Å). Sequence alignment of the residues in the lipophilic pocket indicates that the residues are not highly conserved among the GlmU orthologs (Table 1). Mutations of E224 and V223 in this region did not have impact on catalytic activity, further underscoring the nonessentiality of these residues for substrate or kinetic use (Fig. 3). Given the potential area and properties of this pocket, this region could be used for designing compound inhibitors to gain a better selectivity profile.
Discussion
Sequential ordered catalytic mechanism of GlmU
While the mechanism of the transphosphorylation reaction of GlmU has been previously suggested (Kostrewa et al. 2001), the binding order of UTP and GlcNAc-1-P and the driving mechanism of the conformational change at the uridyltransferase active site, to our knowledge, have not been proposed. The determined GlmU crystal structures provide structural information into a mechanistic model of the uridyltransferase reaction of the enzyme and allow us to propose the following reaction scheme (Fig. 4A–C): UTP binds first to the apo form of the GlmU enzyme, preserving the uridyltransferase active site in the open conformation. As the structures indicated, the side chains of all key residues at the active site in the open apo-like conformation are optimally positioned for binding of uridine, UMP, or UDP. Based on the UDP-bound structure, in which the α-phosphate group occupies the same position as the corresponding phosphate of UDP-GlcNAc (within 0.7 Å) and the β-phosphate group extends into the solvent region away from the N-acetylglucosamine-1-P-binding subsite, we propose that both β- and γ-phosphate groups of UTP are oriented into solvent, allowing the phosphate group of GlcNAc-1-P to attack the phosphorus group of the α-phosphate group of UTP upon binding of N-acetylglucosamine-1-P, which induces large conformational changes at the GlcNAc-binding subsite to create a proper stereochemical environment for the enzymatic reaction to occur upon binding of the cofactor Mg2+ detected in the other GlmU structures (Kostrewa et al. 2001). As our attempt to co-crystallize GlmU with both UMP and GlcNAc-1-P resulted in the UMP-only bound structure, we have modeled GlcNAc-1-P in the UMP-bound GlmU structure. After protein-ligand minimization using an in-house implementation of the AMBER force field with the option to allow the protein active-site atoms to move (movable shell thickness was 6.0 Å), the O7 of the glucose moiety interacted with T82-OG1 and the oxygen atoms O3′ and O4′ formed hydrogen bonds with N169-ND2 and N169-N, respectively. A comparison of protein–ligand interactions of the GlcNAc group modeled as GlcNAc-1-P with those observed in the UDP-GlcNAc-bound crystal structure indicates that both N-acetyl and hydroxyl O3′ of the glucose moiety are important structural elements for inducing the conformational change. The N-acetyl group recognizes E154-OE2 and T82-OG1, while O3 forms a direct hydrogen bond to E154-OE1 (Table 2). This is consistent with the GlmU kinetic preference for N-acetylglucosamine-1-P over glucosamine-1-P (Gehring et al. 1996; Pompeo et al. 1998) and emphasizes that both E154 and T82 play an important role in regulating the conformational changes activated by the binding of GlcNAc-1-P at the GlcNAc-binding subsite. With both substrates bound, the phosphate group of GlcNAc-1-P attacks the phosphorus atom of the α-phosphate group of UTP, releasing the pyrophosphate and inversing the configuration of the α-phosphate group (Kostrewa et al. 2001). To summarize, based on analysis of the crystal structures, we propose that the reaction of condensation of UTP and GlcNAc-1-P at the GlmU uridyltransferase active site follows a sequential mechanism in which the substrates bind in a specific order. Conformation of the proposed sequential-binding order using biophysical studies such as equilibrium dialysis of radiolabeled substrates to determine whether binding of GlcNAc-1-P is dependent on UTP binding is being planned. This mechanism is found in many other nucleotidyltransferases, including glucose-1-phosphate thymidylyltransferase (Blankenfeldt et al. 2000; Zuccotti et al. 2001; Sivaraman et al. 2002) and cytidylyltransferase (Kwak et al. 2002).
Figure 4.

Structural insights into the mechanism of uridylation. (A) View of the GlmU uridyltransferase active site (open conformation) in the UDP-bound crystal structure. The GlcNAc-1-P-binding subsite is circled with a white dotted line. (B) Schematic diagram of the mechanism of the uridyltransferase reaction catalyzed by GlmU. (C) View of the GlmU uridyltransferase active site (closed conformation) in the UDP-GlcNAc bound crystal structure. Connolly binding-site surface is colored by hydrophobicity (brown, hydrophobic; blue and red, hydrophilic).
Implication for structure-based drug discovery
The overall comparison of the active-site relationships between hiGlmU and the other GlmU orthologs reveals striking similarities both at the structural and kinetic levels. Mutagenesis of conserved and variable amino acid residues support the essential nature of residues involved in uracil recognition (Q76) and those involved in catalytic activity with respect to coordination of magnesium binding (D105 and K25) as defined by lack of enzyme activity. In this respect, to develop broad-specificity inhibitors that target both Gram-negative and Gram-positive bacteria from a wide range of species, we should consider intermolecular interactions with strictly conserved residues of the GlmU active site (Table 1). Those residues mutated in nonsubstrate-binding regions, such as in the presently defined lipophilic pocket (V223 and E224) and Y103, did not influence catalytic activity. However, from the structural viewpoint, these residues can be key differentiators between bacterial Gram species.
Materials and Methods
Protein expression and purification of hiGlmU
The hiGlmU gene was cloned by PCR from genomic DNA inserted into a pPW2 expression vector (Affinium Pharmaceuticals) and overexpressed in BL21 AI cells (Invitrogen) using a proprietary rich medium supplemented with 100 μg/mL ampicillin. Protein expression was induced in mid-log phase with the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. The culture was incubated for an additional 18 h at 15°C, and the cells were concentrated and collected by centrifugation at 8900g for 25–30 min at 4°C. The protein was purified to homogeneity using a DE52 column followed by IMAC chromatography. The molecular mass of purified hiGlmU was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-MS) (data not shown) and was in agreement with the calculated mass of the full-length protein.
Site-directed mutagenesis and purification of mutant proteins
Site-directed mutagenesis of hiGlmU was carried out using the QuikChange Site-Directed Mutagenesis Kit from Stratagene, Inc. to construct the following mutants: K25A, Q76A, Y103A, D105A, V223A, and E224A (Supplemental material). Briefly, primers containing the desired mutations were annealed to denatured pPW2-hiGlmU plasmid DNA. Pfu Turbo DNA polymerase was used to extend and incorporate the mutagenic primers, resulting in nicked circular strands followed by DpnI digestion to remove the nonmutated parental strand. The final product was transformed into XL1-Blue supercompetent cells. Correct incorporation of the mutation was verified by restriction digest of isolated plasmid DNA from transformants and by sequencing.
Plasmid DNAs containing the desired mutations were introduced into BL21-Gold (DE3) cells. Cultures were grown in Terrific Broth and induced by 0.5 mM IPTG at 15°C for 16 h. Cells were lysed using CelLytic Express (Sigma). Removal of cell debris was performed by centrifugation of the lysed cell solution at 11,500 rpm, 16,000g for 15 min at 4°C (JA-25.5, 25,000 rpm Serial # 01U 2494), followed by a second centrifugation run at 3 K rpm for 15 min at 4°C. Lysate containing the 6x His-tagged protein are purified by Ni-NTA spin column. Protein purity was determined by SDS-PAGE.
Crystallization
The crystals of hiGlmU were grown at the ambient temperature using the hanging-drop vapor diffusion method. Four microliter drops of hiGlmU consisted of mixing equal parts of protein solution (14 mg/mL) and the reservoir solution composed of 1.2–1.8 M ammonium sulfate, 2% PEG-400, and 0.1 M MES (pH 5.2–6.1). To obtain the GlmU apo crystals, cobalt (II) chloride (6.25 mM) was added to the crystallization drops. For the UDP-GlcNAc and UDP crystals, the enzyme was incubated at 4°C for 4 h with 20 mM MgCl2 and 10 mM UPD-GlcNAc or UDP. For the UMP crystals, the enzyme was incubated at 4°C for 4 h with 20 mM MgCl2 and 10 mM UMP and GlcNAc-1-P. In all cases, the GlmU crystals were visible after 3–5 d and grew typically to a dimension of 0.20 × 0.15 × 0.07 mm. For the uridine crystals, uridine was introduced into the apo-crystals incrementally within 4 h by solvent diffusion at the final concentration of 5 mM.
X-ray data collection, structure determination, and refinement
X-ray diffraction data from the hiGlmU crystals were collected at the Advanced Photon Source facility on beamline 17-ID operated by the Industrial Macromolecular Crystallography Association. The crystals were mounted in the cryo loops and treated with the cryo-protection solutions that were prepared using 1 μL of 80% PEG-200, 20% ethylene glycol mixture, 2 μL of the crystallization well solution (cryo-solution #1), 1 μL of cryo-solution #1, and 1 μL of the crystallization drop solution (cryo-solution #2). The crystals were moved from the crystallization drop to cryo-solution #2 for 10 sec and then to cryo-solution #1 for 3–5 sec before being flash-frozen in liquid nitrogen. Intensity data were measured at about −180°C. One X-ray data set was collected for each of the apo, UDP-GlcNAc, uridine, UMP, and UDP crystals. Auto-indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinoski and Minor 1997). The intensity data-collection statistics are summarized in Table 4.
Table 4.
X-ray intensity data collection from the hiGlmU crystals

The crystal structure of apo hiGlmU was determined by molecular replacement using the full-length structure of apo ecGlmU as the search model (Olsen and Roderick 2001) (PDB ID 1HV9). The crystal structure of the UDP-GlcNAc co-complex was determined by molecular replacement using the final coordinates of the apo hiGlmU structure. The rotation/translation searches were carried out with the MOLREP program (Collaborative Computational Project Number 4 1994; Vagin and Teplyakov 1997). The molecular replacement solution was further optimized by rigid-body, coordinates, and B-value minimization using CNX 2000 (Brünger et al. 1998). Calculated (2Fo-Fc) and (Fo-Fc) electron density maps were utilized for interactive fitting of protein structures into electron density using the X-BUILD and X-AUTOFIT (Oldfield 2001a) modules implemented in QUANTA (Accelrys, Inc). Protein model building was alternated with coordinate minimization and individual B-factor refinement using REFMAC 5.1 (Collaborative Computational Project Number 4 1994). Placement of the ligands into electron-density maps was carried out with QUANTA X-LIGAND (Oldfield 2001b). Solvent water molecules were added periodically based on examination of difference density maps using X-SOLVATE. Final coordinates were validated using the SQUID VALIDATE (Oldfield 1992) and PROCHECK validation tools (Laskowski et al. 1993). Summary of final refinement parameters and the final R-work and R-free values are listed in Table 5.
Table 5.
Refinement summary of deviations from ideality and final refinement parameters and the final R and R-free values

hiGlmU enzyme kinetic assays and MgCl2 dose curve
GlmU uridyltransferase activity was measured using a modified format of the MESG/PNPase-coupled assay format for the continuous detection of the product inorganic phosphate described in Gehring et al. (1996). Using a 50-μL volume in a 384-well format, the standard assay mix contained the following: 40 mM HEPES (pH 7.5), 2.5 mM MgCl2, 0.3 mM TCEP, 10 μg/mL SipA, 0.05 U/mL PNPase, 0.2 mM MESG, 1 mM Citrate buffer (pH4.2) (to keep MESG stable at room temperature), 0.01% NP-40, 3 nM hiGlmU enzyme, and two substrates (UTP and Glucosamine-1P). Pyrophosphate production was monitored with excess inorganic pyrophosphatase, which hydrolyzes one equivalent of pyrophosphate to two equivalents of inorganic phosphate, and PNPase, which catalyzes the phosphorolysis of MESG with a resulting change in the extinction coefficient at 360 nm of 11 mM-1 cm-1. The reaction was monitored for 5 min at A360 on a SPECTROmax 384 Plus (Molecular Devices) plate reader. Using the same assay format but varying MgCl2 concentrations by 1:2 serial dilutions (80 mM to 0 mM), it shows the dependence of hiGlmU for magnesium ions.
Km and initial reaction rates
Reaction volumes and components are the same as GlmU enzyme assay, but apparent steady-state kinetic parameters were measured keeping one substrate at saturating concentrations between 10 to 15 times the Km value, while varying the second substrate from 0.25 to 10 times the Km values. The reaction was monitored for 5 min at A360 on a SPECTROmax 384 Plus (Molecular Devices) plate reader. Data were analyzed by using KaleidaGraph graphing and data analysis software tools from Synergy, Inc.
Data deposition
Atomic coordinates have been deposited in the RCSB Protein Data Bank (accession codes: 2v0h [apo]; 2v0i [UDP-GlcNAc]; 2v0j [UMP]; 2v0k [UDP]; 2v0l [uridine]).
Acknowledgments
We thank Dhushy Kanagarajah, Bryan Beattie, Matt Tai, Robert Lam, and Akil Dharamsi from Affinium Pharmaceuticals for the pPW2 hiGlmU protein expression construct. We also thank Anne Mulichak, Kevin Battaile, Rong Huang, Irina Koshelev, and Lisa Keefe of the IMCA-CAT beamline at the Advanced Photon Source for their assistance in collecting X-ray diffraction data from the crystals of the GlmU complexes. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. Access to the IMCA-CAT facilities was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Illinois Institute of Technology (IIT) executed through IIT's Center for Synchrotron Radiation Research and Instrumentation.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Igor Mochalkin, Eastern Point Road, Groton, CT 06340, USA; e-mail: Igor.Mochalkin@pfizer.com; fax: (734) 622-2782.
Abbreviations: GlmU, N-Acetylglucosamine-1-phosphate uridyltransferase; UDP-GlcNAc, uridine-diphospho-N-acetylglucosamine; GlcNAc-1-P, N-acetylglucosamine-1-phosphate; PPi, pyrophosphate; hiGlmU, GlmU from Haemophilus influenzae; ecGlmU, GlmU from Escherichia coli; spGlmU, GlmU from Streptococcus pneumoniae; ngGlmU, GlmU from Neisseria gonorrhoeae; shGlmU, GlmU from Staphylococcus haemolyticus; paGlmU, GlmU from Pseudomonas aeruginosa; saGlmU, GlmU from Staphylococcus aureus; LßH, left-handed β-sheet helix.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073135107.
References
- Anderson M.S. and Raetz, C.R.H. 1987. Biosynthesis of lipid A precursors in Escherichia coli. A cytoplasmic acetyltransferase that converts UDP-N-acetylglucosamine to UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine. J. Biol. Chem. 262: 5159–5169. [PubMed] [Google Scholar]
- Blankenfeldt W., Asuncion, M., Lam, J.S., and Naismith, J.H. 2000. The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J. 19: 6652–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K., Pompeo, F., Dixon, S., Mengin-Lecreulx, D., Cambillau, C., and Bourne, Y. 1999. Crystal structure of the bifunctional N-acetylglucosamine-1-phosphate uridyltransferase from Escherichia coli: A paradigm for the related pyrophosphorylase superfamily. EMBO J. 18: 4096–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54: 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D50: 760–763. [DOI] [PubMed] [Google Scholar]
- Gehring A.M., Lees, W.J., Mindiola, D.J., Walsh, C.T., and Brown, E.D. 1996. Acetyltransfer precedes uridyltransfer in the formation of UDP-N-acetylglucosamine in separable active sites of the bifunctional GlmU protein of Escherichia coli . Biochemistry 35: 579–585. [DOI] [PubMed] [Google Scholar]
- Green D.W. 2002. The bacterial cell wall as a source of antibacterial targets. Expert Opin. Ther. Targets 6: 1–19. [DOI] [PubMed] [Google Scholar]
- Han S., Forman, M.D., Loulakis, P., Rosner, M.H., Xie, Z., Wang, H., Danley, D.E., Yuan, W., Schafer, J., and Xu, Z. 2006. Crystal structure of nicotinic acid mononucleotide adenyltransferase from Staphyloccocus aureus: Structural basis for NaAD interaction in functional dimer. J. Mol. Biol. 360: 814–825. [DOI] [PubMed] [Google Scholar]
- Kostrewa D., D'Arcy, A., Takacs, B., and Kamber, M. 2001. Crystal structure of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase, GlmU, in apo form at 2.33 Å resolution and in complex with UDP-N-acetylglucosamine and Mg2+ at 1.96 Å. J. Mol. Biol. 305: 279–289. [DOI] [PubMed] [Google Scholar]
- Krissinel E. and Henrick, K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372: 774–797. [DOI] [PubMed] [Google Scholar]
- Kwak B.-Y., Zhang, Y.-M., Yun, M., Heath, R., Rock, C., Jackowski, S., and Park, H.-W. 2002. Structure and mechanism of CTP:phosphocholine cytidylyltransferase (LicC) from Streptococcus pneumonia . J. Biol. Chem. 277: 4343–4350. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Levy S.B. 2005. Antibacterial resistance—the problem intensifies. Adv. Drug Deliv. Rev. 57: 1446–1450. [DOI] [PubMed] [Google Scholar]
- Mengin-Lecreulx D. and van Heijenoort, J. 1993. Identification of the GlmU gene encoding N-acetylglucomsamine-1-phosphate uridyltransferase in Escherichia coli . J. Bacteriol. 175: 6150–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengin-Lecreulx D. and van Heijenoort, J. 1994. Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase activities of Escherichia coli: Characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UPD-N-acetylglucosamine synthesis. J. Bacteriol. 176: 5788–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield T.J. 1992. SQUID: A program for the analysis and display of data from crystallography and molecular dynamics. J. Mol. Graph. 10: 247–252. [DOI] [PubMed] [Google Scholar]
- Oldfield T.J. 2001a. A number of real-space torsion-angle refinement techniques for proteins, nucleic acids, ligands and solvent. Acta Crystallogr. D57: 82–94. [DOI] [PubMed] [Google Scholar]
- Oldfield T.J. 2001b. X-LIGAND: An application for the automated addition of flexible ligands into electron density. Acta Crystallogr. D57: 696–705. [DOI] [PubMed] [Google Scholar]
- Olsen L.R. and Roderick, S.L. 2001. Structure of the Escherichia coli GlmU pyrophosphorylase and acetyltransferase active sites. Biochemistry 40: 1913–1921. [DOI] [PubMed] [Google Scholar]
- Olsen L.R., Vetting, M.W., and Roderick, S.L. 2007. Structure of the E. coli bifunctional GlmU acetyltransferase active site with substrates and products. Protein Sci. 16: 1230–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinoski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods in enzymology (eds. C.W. Carter Jr and R.M. Sweet), pp. 307–326. Academic Press, New York. 276(A) ed. [DOI] [PubMed]
- Pompeo F., van Heijenhoort, J., and Mengin-Lecreulx, D. 1998. Probing the role of cysteine residues in glucosamine-1-phosphate acetyltransferase activity of the bifunctional glmu protein from Escherichia coli: Site-directed mutagenesis and characterization of the mutant enzymes. J. Bacteriol. 180: 4799–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman J., Sauve, V., Matte, A., and Cygler, M. 2002. Crystal structure of Escherichia coli glucose-1-phosphate thymidylyltransferase (RffH) complexed with dTTP and Mg2+ . J. Biol. Chem. 277: 44214–44219. [DOI] [PubMed] [Google Scholar]
- Sulzenbacher G., Gal, L., Peneff, C., Fassy, F., and Bourne, Y. 2001. Crystal structure of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase bound to acetyl-coenzyme A reveals a novel active site architecture. J. Biol. Chem. 276: 11844–11851. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov, A. 1997. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 30: 1022–1025. [Google Scholar]
- Zuccotti S., Zanardi, D., Rosano, C., Sturla, L., Tonetti, M., and Bolognesi, M. 2001. Kinetic and crystallographic analysis support a sequential-ordered bi bi catalytic mechanism for Escherichia coli glucose-1-phosphate thymidylyltransferase. J. Mol. Biol. 313: 831–843. [DOI] [PubMed] [Google Scholar]