

Abstract
Peptidyl-prolyl isomerase (PPIase) activity is exhibited by many proteins belonging to the PPIase family. However, the catalytic mechanism of this activity remains to be completely elucidated. Here, we selected human FK506-binding protein 12 (FKBP12) as the model PPIase and investigated the nature of amino acid residues essential for the activity. The crystal structures of several complexes of PPIase with short peptides revealed that the residues Asp37, Arg42, Phe46, Val55, Trp59, and Tyr82 in the substrate-binding cavity of FKBP12 appear to play key roles in the PPIase activity. Each of these six residues was substituted by 20 common amino acid residues. The activity of each mutant protein was measured using a peptide analog by the chymotrypsin digestion assay and then compared with wild-type FKBP12. It was found that site-specific interactions by the side chains of amino acid residues constituting the substrate-binding cavity were not essential for the PPIase activity, although the 37th, 55th, and 82nd amino acid residues significantly contributed to the activity. This suggests that the PPIase activity requires only the hydrophobic cavity that captures the Pro-containing peptide.
Keywords: peptidyl-prolyl isomerase, human FK506-binding protein 12, substrate-binding cavity, comprehensive mutational analysis
The ubiquitous protein family peptidyl-prolyl cis/trans isomerase (PPIase) (Fischer 1994) shows a common activity in which it generally accelerates the conformational change between cis and trans of the Xaa-Pro. More than 3000 proteins in the PPIase family are classified into three subfamilies of cyclophilin, FK506-binding protein (FKBP), and parvulin (Fischer and Schmid 1999). Amino acid sequences are highly conserved within the members of each subfamily, although these sequences share a limited similarity among the three subfamilies (Wiederrecht et al. 1991). Similarity in the sequence and structure among the subfamilies, however, is somewhat higher for the substrate-binding cavity than for the entire molecule, thereby suggesting a convergent evolution of the enzyme catalytic site (Denesyuk et al. 1993). PPIase generally accelerates the conformational change between cis and trans forms of the Xaa-Pro peptide bond during the folding of a substrate protein in vitro (Fischer and Schmid 1999; Schmid 2001), whereas in vivo the enzyme functions in various situations, including signaling, mitochondrial function, chaperone activity, RNA splicing, stress response, gene expression, and regulation of kinase activity (Yao et al. 2005; Bell et al. 2006; Sokolskaja and Luban 2006; Shaw 2007). Some of these functions are directly related to the PPIase activity, while others are not (Huse et al. 1999, 2001; Chakraborty et al. 2004). For example, human FK506-binding protein 12 (FKBP12) regulates the activation of type I transforming growth factor β receptor (TβR-I) by binding to its GS region; however, it does not directly bind to any proline residues but only to leucine residues of the GS region (Huse et al. 1999). Thus, the relationship between the PPIase activity and the in vivo function remains unknown.
To date, several catalytic mechanisms have been proposed for the PPIase activity; these include catalysis by the formation of a covalent tetrahedral intermediate (Fischer et al. 1989a,b), catalysis by distortion (Harrison and Stein 1990), protonation of the amide nitrogen (Kofron et al. 1991), solvent-assisted mechanism (Ke et al. 1993), and so on. Most of these mechanisms were deduced on the basis of the peptide-complex structures of cyclophilins, in which the side chains of amino acid residues constituting the substrate-binding cavity directly bound to the peptide. Consequently, the researchers presumed that specific interactions between the PPIase and the substrate occurred during the catalytic reaction, similar to that observed in general enzymatic reactions. Mutagenesis and pH titration studies of candidates of the catalytic center, however, did not support the expected relationship between specific interactions and the PPIase activity (Yu and Fesik 1994; Scholz et al. 1999). Therefore, the amino acid residues essential for the PPIase activity and the reaction pathway remain unclear.
In the present study, we investigated the amino acid residues essential for the PPIase activity by the comprehensive mutational analysis as the first step to elucidate the catalytic mechanism of this activity. Here, FK506-binding protein 12 (FKBP12) was selected as the model PPIase because it is one of the simplest PPIases. If we consider the substrate-binding cavity as a bowl, this bowl is characterized by a hydrophilic edge and a hydrophobic wall and bottom: The former constitutes the main chains of Phe36, Phe46, Val55, and Ile56 and the side chains of Asp37, Arg42, Tyr82, and His87, while the latter constitutes the side chains of Tyr26, Phe36, Phe46, Val55, Ilr56, Trp59, and Phe99 (Fig. 1A; Wilson et al. 1995). We then selected six residues, namely Asp37, Arg42, Phe46, Val55, Trp59, and Tyr82, and investigated how site-specific hydrophilic interactions were affected by mutations of Asp37, Arg42, and Tyr82 and how nonspecific hydrophobic interactions were affected by mutations of Phe46, Val55, and Trp59. The activity of each mutant was measured using the peptide analog N-succinyl-Ala-Ala-Pro-Phe-4-methylcoumaryl-7-amide (AAPF-MCA) by the chymotrypsin digestion assay and then compared with wild-type FKBP12. We found that the PPIase activity of FKBP12 did not require the side chains of the 37th, 42nd, 46th, 55th, 59th, and 82nd amino acid residues. This suggests that any site-specific interactions of the side chains of the residues constituting the substrate-binding cavity are unessential for the PPIase activity, although the side chains of the 37th, 55th, and 82nd amino acid residues significantly affect the activity. This result also suggests that the hydrophobic environment of the substrate-binding cavity and/or site-specific interactions of the main chains of the residues constituting the cavity are sufficient for the PPIase activity. Based on the present results, “catalysis by distortion” in the hydrophobic cavity is the most plausible mechanism of the PPIase activity.
Figure 1.
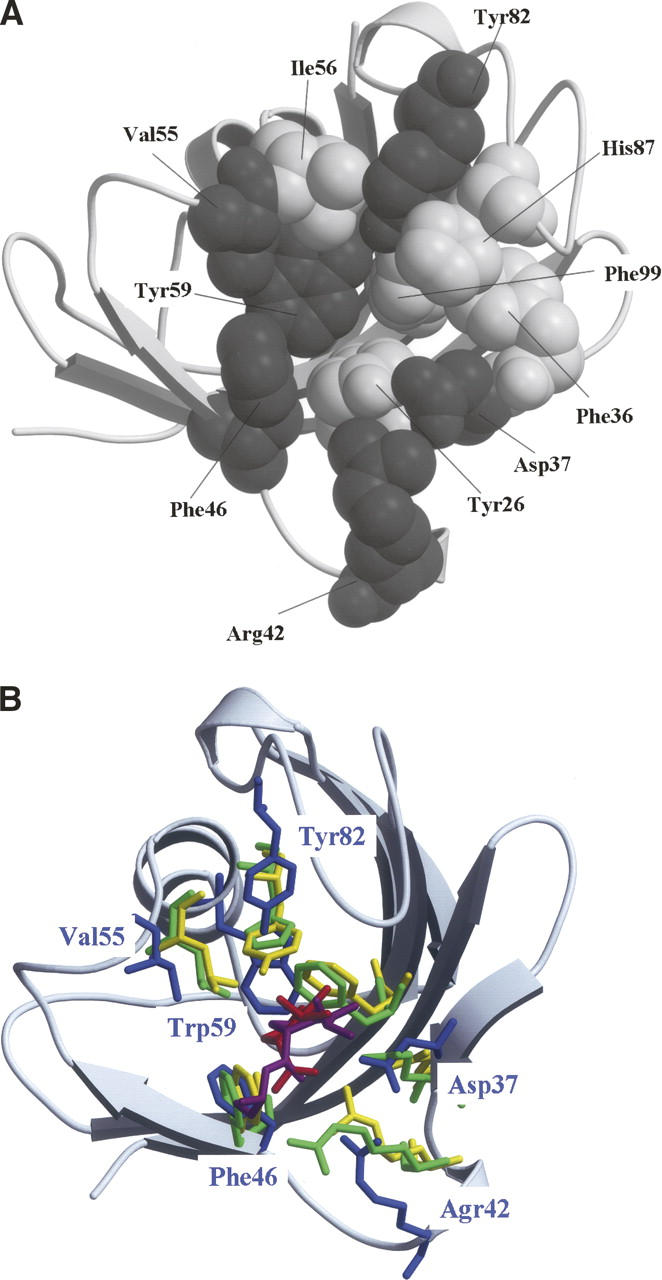
Structures of FKBP12. (A) Eleven amino acid residues constituting the substrate-binding cavity of FKBP12 are drawn using the space-fill model. Gray and white spheres represent the residues investigated in this study (Asp37, Arg42, Phe46, Val55, Trp59, and Tyr82) and the remaining five residues (Tyr26, Phe36, Ile56, His87, and Phe99), respectively. (B) When the substrate-binding cavities of FKBP12 (Wilson et al. 1995), human cyclophilin A (hCyPA) (Ke et al. 1993), and E. coli cyclophilin A (eCyPA) (Konno et al. 1996) were superimposed on each other, the corresponding six amino acid residues were drawn using the stick model: Asp37, Agr42, Phe46, Val55, Trp59, and Tyr82 of FKBP12 (blue); Arg55, Phe60, Gln63, Phe113, Leu122, and His126 of hCyPA (green); Arg43, Phe48, Gln51, Phe99, Leu108, and Tyr120 of eCyPA (yellow). The root-mean-square deviations among the main chain atoms were calculated within 4.0 Å. The residues of FKBP12 are labeled blue. The substrates of hCyPA and eCyPA are also drawn using the stick model: Red and magenta sticks denote the dipeptide Ala-Pro bound to hCyPA and the tripeptide Ala-Pro-Ala bound to eCyPA, respectively. The secondary structure representation of FKBP12 was produced using MolScript and rendered using Raster3D (gray) in both panels.
Results
The aforementioned six residues of FKBP12 were substituted by the rest of 20 amino acid residues. The activity of each mutant protein was then measured using the peptide analog AAPF-MCA by the chymotrypsin digestion assay (Fig. 2; Hayano et al. 1991). This isomerase activity was compared with the activity of wild-type FKBP12.
Figure 2.

Relative PPIase activities of mutants of Asp37 (A), Arg42 (B), Phe46 (C), Val55 (D), Trp59 (E), and Tyr82 (F) to the activity of wild-type FKBP12, whose k cat/Km was 1.98 μM−1s−1 at 30°C. The activity of each mutant protein was determined within an error of 5%. Hatched bars denote wild-type amino acids at the 37th (A), 42nd (B), 46th (C), 55th (D), 59th (E), and 82nd (F) residues.
Mutations of Asp37
As shown in Figure 2A, most of the substitutions of Asp37 with other 19 amino acids hardly affected or unexpectedly increased the PPIase activity, while the remaining substitutions decreased the activity. D37E, D37F, D37G, D37T, and D37V mutants showed activity similar to that of wild-type FKBP12. The D37H, D37K, D37P, and D37R mutations increased the activity by a factor of 3.8–4.4. Of these four substituted residues, three residues, except Pro, have positively charged side chains at neutral pH. This positive charge might contribute to the activity (Fischer et al. 1989a). In contrast, D37A, D37I, and D37S mutants showed 50%–80% of the activity of wild-type FKBP12. The remaining mutations moderately increased the activity.
Mutations of Arg42
Figure 2B shows that most of the substitutions of Arg42 with other 19 amino acids hardly affected the PPIase activity. R42A, R42H, R42I, R42K, R42P, and R42W mutants showed activities similar to that of wild-type FKBP12. The R42C, R42D, R42E, R42L, R42M, R42N, R42S, R42T, and R42V mutants showed 60%–80% of the activity of wild-type FKBP12, while the other mutants—R42F, R42G, R42Q, and R42Y—showed 40%–50% of the activity of wild-type FKBP12. No mutation completely abolished the activity.
Mutations of Phe46
Figure 2C shows that approximately half of the substitutions of Phe46 with other 19 amino acids increased or did not affect the activity. The F46L, F46M, F46V, F46W, and F46Y mutants showed activity similar to that of wild-type FKBP12, while the F46G, F46I, F46N, F46P, and F46R mutants showed an increase in the PPIase activity by a factor of 1.2–2.2. In contrast, the F46A, F46D, F46K, F46Q, F46S, and F46T mutations decreased the activity to 50%–70% of wild-type FKBP12 activity, while the F46C, F46E, and F46H mutations decreased the activity to 30%–40% of wild-type FKBP12 activity. No mutation completely abolished the activity. Furthermore, the F46G mutant showed higher activity than wild-type FKBP12, suggesting that the Phe residue conserved among the three PPIases might be responsible for other functions.
Mutations of Val55
Figure 2D shows that 17 of the 19 substitutions of Val55 increased the activity. The V55C, V55D, V55E, and V55G mutations increased the activity by a factor of 1.4–1.9, while the V55F, V55H, V55K, V55L, V55M, V55N, V55P, V55Q, V55S, V55T, V55W, and V55Y mutations increased the activity by a factor of 2.2–5.7; furthermore, the V55R mutation unexpectedly increased the activity by a factor of 11.4. In contrast, the V55A and V55I mutations decreased the activity to 90% and 40% of wild-type FKBP12 activity, respectively.
Mutations of Trp59
Figure 2E shows that more than half of the substitutions of Trp59 with other 19 amino acids hardly affected or increased the PPIase activity. The W59D, W59E, and W59G mutants showed activity similar to that of wild-type FKBP12, while the W59F, W59K, W59N, W59P, W59Q, W59R, W59S, W59T, and W59Y mutants showed an increase in the activity by a factor of 1.2–1.8. In contrast, the W59H, W59I, W59L, and W59M mutations decreased the activity to 50%–80% of wild-type FKBP12 activity, while the W59A, W59C, and W59V mutations decreased the activity to 20%–30% of wild-type FKBP12 activity.
Mutations of Tyr82
Figure 2F shows that most of the substitutions of Tyr82 by other 19 amino acids drastically decreased the activity. The Y82A, Y82C, Y82D, Y82E, Y82H, Y82I, Y82K, Y82L, Y82M, and Y82V mutations decreased the activity to 20%–30% of wild-type FKBP12 activity. The Y82G, Y82Q, and Y82T mutations decreased the activity to 40% of wild-type FKBP12 activity. The Y82N and Y82S mutations decreased the activity to 60% of wild-type FKBP12 activity. In contrast, the Y82F and Y82R mutants showed activity similar to that of wild-type FKBP12, while the Y82P and Y82W mutants showed an increase in the activity by a factor of 1.2 and 3.0, respectively. The 82nd residue might prefer bulky and aromatic side chain for the PPIase activity because Phe-, Trp-, and Tyr-substituted proteins showed higher activities. Although it is unclear why Arg- and Pro-substituted proteins showed higher activities, the positively charged side chain of Arg might contribute to the activity, as observed for the 37th and 55th residues.
Sextuple mutation D37G/R42G/F46G/V55G/W59G/Y82G
No substitutions of each of the six residues of FKBP12 by 19 amino acids completely abolished the PPIase activity, although some mutations drastically increased or decreased the activity. Furthermore, the Gly mutant of each of the six residues retained the activity, suggesting that the side chains of the six amino acid residues were unessential for the PPIase activity. However, the possibility that the role of the side chain removed by the Gly mutation was compensated by the side chains of the remaining six residues could not be excluded. Therefore, we constructed and measured the isomerase activity of a sextuple mutant protein, namely D37G/R42G/F46G/V55G/W59G/Y82G, to confirm that the side chains of all six residues were unessential for the activity. The sextuple mutant protein showed 90% of the activity of wild-type FKBP12 (Fig. 3). This value is similar to the average value (97%) obtained for the activities of the six single mutant proteins D37G, R42G, F46G, V55G, W59G, and Y82G. This suggests that the six amino acid residues independently contribute to the activity.
Figure 3.

Relative isomerase activities of the Gly mutant proteins of FKBP12. The label All G (hatched bar) denotes the sextuple mutant protein D37G/R42G/F46G/V55G/W59G/Y82G, while the other labels denote six single Gly mutants extracted from Figure 2.
Discussion
The present study clearly showed that the PPIase activity of FKBP12 did not require the side chains of the 37th, 42nd, 46th, 55th, 59th, and 82nd amino acid residues (Fig. 2). These six residues constitute the substrate-binding cavity of FKBP12 along with Tyr26, Phe36, Ile56, His87, and Phe99 (Fig. 1A). As mentioned earlier, the substrate-binding “bowl” is characterized by a hydrophilic edge and a hydrophobic wall and bottom. It is considered that the former is responsible for site-specific interactions, while the latter is responsible for nonspecific interactions. The hydrophilic edge of the bowl consists of the main chains of Phe36, Phe46, Val55, and Ile56 and the side chains of Asp37, Arg42, Tyr82, and His87, while the hydrophobic wall and bottom of the bowl consists of the side chains of Tyr26, Phe36, Phe46, Val55, Ilr56, Trp59, and Phe99. The hydrophilic functional groups of the residues on the edge of the bowl were thought to be responsible for site-specific interactions, including nucleophilic attack on the carbonyl carbon preceding the Pro residue (Fischer et al. 1989a,b; Ke et al. 1993). Among these hydrophilic residues on the edge of the bowl, the side chain of His87 has been reported to be unessential for the PPIase catalysis (Yu and Fesik 1994). In the present study, the remaining three residues in the hydrophilic edge, i.e., Asp37, Arg42, and Tyr82, were investigated to elucidate the contribution of their side chains to site-specific interactions. These residues were thought to be essential for the PPIase catalysis because they were positionally conserved in another subfamily of the PPIase family. However, the side chains of these amino acid residues were found to be unessential for the PPIase catalysis. Therefore, we conclude that the side chains are unessential for site-specific interactions in the PPIase catalysis. It is possible that the main chains of the eight residues in the hydrophilic edge of the bowl contribute to site-specific interactions. However, it is difficult to determine by mutational studies whether these main chains are essential for site-specific interactions in the PPIase catalysis.
On the other hand, mutational studies on Phe46, Val55, and Trp59 revealed the contribution of their side chains to nonspecific hydrophobic interactions. Most of the substitutions of Val55 increased the PPIase activity, while the substitutions of Phe46 and Trp59 did not substantially affect the activity, indicating that their side chains were unessential for nonspecific hydrophobic interactions in the PPIase catalysis. Four other hydrophobic residues—Tyr26, Phe36, Ilr56, and Phe99—are present in the substrate-binding cavity, and they might be sufficient to provide hydrophobicity required for catalysis.
Although the side chains of the six residues studied here were unessential for the activity, the side chains of several residues considerably affected the PPIase activity. Mutations of Asp37, Val55, and Tyr82 residues resulted in considerably more changes in the activity than mutations of other residues. In particular, the effects of Val55 mutation were the most notable; the PPIase activity of the V55R mutant showing the maximum activity was 29 times higher than that of the V55I mutant showing the minimum activity. This suggests that the side chain of the 55th amino acid residue has the largest contribution to the activity among the side chains of the six residues. In the cases of hCyPA and eCyPA, Leu is located at the position corresponding to Val55 of FKBP12 (Fig. 1B). Since the activity of the V55L mutant was 4.6 times higher than that of wild-type FKBP12, the high activities exhibited by the cyclophilins might be partially due to the Leu residue at this position (Bierer et al. 1990; Harrison and Stein 1990; Bergsma et al. 1991; Hayano et al. 1991; Holzman et al. 1991; Levy et al. 1991; Compton et al. 1992). Most of the mutations of Asp37 also increased the activity, suggesting that its side chain significantly contributes to the activity. The positively charged side chains were particularly preferred at this position as well as at the position of the 55th amino acid residue.
Tradler et al. (1997) reported that the D37L mutation decreased the PPIase activity for the peptidyl-prolyl bond of Ala-Pro to 32% of the activity of wild-type FKBP12, while our result revealed that the mutant showed 180% of the activity of wild-type FKBP12. The present study was carried out at 30°C, while the experiment by Tradler and colleagues was conducted at 10°C. Thus, the temperature dependency of the PPIase activity might be responsible for this discrepancy.
Most of the mutations of Tyr82 drastically decreased the activity, suggesting that its side chain also significantly contributes to the activity. Arg, Pro, and aromatic residues were preferred as the 82nd amino acid residue. Tyrosine and His residues are located at the corresponding positions in eCyPA and hCyPA, respectively (Table 1; Fig. 1B). The Y82H mutant retained only 20% of the activity of wild-type FKBP12, suggesting that this His residue might play a role in another function of hCyPA. Yu and Fesik (1994) reported that the side chain of His126 of hCyPA was unessential for the PPIase catalysis.
Table 1.
Corresponding residues in the substrate-binding cavities of FKBP12, hCyPA, and eCyPA

The importance of Trp59 was investigated by Fulton et al. (2003) in the light of protein stability. Both W59F and W59L mutations stabilized the protein by ∼2–3 kcal mol−1, while they hardly affected the entire structure of the FKBP12 molecule. Comparison of the crystal structures of the mutant and wild-type proteins revealed that the average temperature factor of the W59L mutant was 1.4 and 1.8 times greater than those of the W59F mutant and wild-type FKBP12, respectively. This suggests that the W59L mutant fluctuates more than the other mutants. This is a typical example showing the positive correlation between stability and fluctuation. In contrast, the W59L mutation in this study decreased the PPIase activity to 30% of the activity of wild-type FKBP12, while the W59F mutation increased the activity by a factor of 1.2. Thus, the PPIase activity probably does not strongly correlate with the fluctuation in this mutational site.
The effects of mutations of Phe99 on the PPIase activity have been previously investigated (Timerman et al. 1995; Tradler et al. 1997). Tradler et al. (1997) reported that the F99Y mutant drastically decreased the PPIase activity. The level of the activity of this mutant protein, however, was dependent on the experiments. Tradler et al. (1997) observed 37% of the activity of wild-type FKBP12 for the peptidyl-prolyl bond of Ala-Pro, while Timerman et al. (1995) observed a complete loss of activity. Nevertheless, the hydrophobicity of Phe99 probably plays an important role in the PPIase activity.
Although we used a tetrapeptide (AAPF-MCA) as a substrate in the present study, the amplitude of the PPIase activity often depended on the amino acid residue preceding a Pro residue in the substrate: As the residue preceding Pro, FKBP12 strongly preferred Leu to Ala, whereas hCyPA strongly preferred Ala to Trp (Harrison and Stein 1990). Such substrate specificity in the PPIase activity should be caused by site-specific interactions of the side chain atoms of the substrate-binding cavity. Thus, it seems plausible that the side chain positions under study might influence PPIase activity for substrates with different amino acid sequences.
The present results provide some new insights into the catalytic mechanism of the PPIase activity. To date, several mechanisms have been proposed for the PPIase activity as mentioned earlier (Fischer et al. 1989a,b; Harrison and Stein 1990; Kofron et al. 1991; Ke et al. 1993). Based on the structure of cyclophilin bound to its substrate, most of these researchers presumed that the site-specific interactions of the side chains of the residues constituting the substrate-binding cavity were essential for the PPIase catalysis. However, our study showed that, even if site-specific interactions occurred, these interactions were independent of the side chains. Since the mutational study could not reveal the role of carbonyl oxygen and amide nitrogen of the main chain in site-specific interactions, it was considered that the main chain atoms of the substrate-binding cavity may be involved in these interactions. On the other hand, since the substrate-binding cavity of the sextuple mutant appears to be a hydrophobic pocket, it is more plausible that the hydrophobic nature of this cavity plays an essential role in the PPIase catalysis, although we cannot completely rule out a direct role of other side chains in the substrate-binding cavity. Thus, the present results support the mechanism of “catalysis by distortion” proposed previously by Harrison and Stein (1990). In this mechanism, geometric, desolvation, or electrostatic destabilization results in distortion of the substrate in the complex form. The hydrophobic nature of the cavity must facilitate such destabilization.
Materials and Methods
Materials
The cloned gene of wild-type human FKBP12 was a generous gift from Professor N. Takahashi, Tokyo University of Agriculture and Technology. Restriction endonucleases, T4-DNA ligase, and JM109 competent cells were purchased from Takara Bio, Inc.
Candidates of the residues required for the PPIase activity
Since the complex structure of FKBP12 with an Xaa-Pro peptide was not available, we selected the candidates of amino acid residues required for the PPIase activity as follows. All the members of the PPIase family exhibit PPIase activity; as a result, the nature of amino acid residues essential for the PPIase activity should be highly conserved among the subfamilies. Hence, human and Escherichia coli cyclophilin A (hCyPA and eCyPA, respectively) were selected to compare their substrate-binding cavities with that of FKBP12 because the biochemical and structural properties of both these cyclophilins are well characterized (Ke et al. 1993; Wilson et al. 1995; Konno et al. 1996). Although structural variations were observed among the complexes of FKBP12 with different substrates (Wilson et al. 1995), the root-mean-square deviations among the structures were sufficiently small (1.0 Å at the most) to ignore in the scope of the present study. When the substrate-binding cavities of the aforementioned cyclophilins were superimposed with that of FKBP12, several amino acid residues were found to be well fitted as supported partly by Denesyuk et al. (1993) (Fig. 1B), and these residues were selected as candidate residues required for the activity: Asp37, Agr42, Phe46, Val55, Trp59, and Tyr82 of FKBP12 (Table 1). These six residues of FKBP12 corresponded to Gln63, Arg55, Phe60, Leu122, Phe113, and His126 of hCyPA and Gln51, Arg43, Phe48, Leu108, Phe99, and Tyr120 of eCyPA, respectively. Interestingly, Tyr82 of FKBP12 and His126 of hCyPA are suggested to have similar contribution to the PPIase activity (Fischer 1994).
If an Xaa-Pro peptide binds to FKBP12 in a manner similar to that observed in the peptide-complex structures of hCyPA and eCyPA, the Xaa-Pro peptide will be located at the position shown in Figure 1B. In the hCyPA-dipeptide Ala-Pro complex (Ke et al. 1993), the guanidinium group of Arg55 and the side chain carboxyl group of Gln63 were directly bound to the carboxyl group of Pro2 of the peptide, while the main chain carboxyl group of Asn102 was directly bound to the amide group of Ala1 of the peptide. In the eCyPA-tripeptide Ala-Pro-Ala complex (Konno et al. 1996), the guanidinium group of Arg43 was directly bound to the carboxyl group of Pro2 of the peptide, while the carboxyl group of Arg87 was directly bound to the amide group of Ala1 of the peptide. Arg42 of FKBP12 and Arg55 of hCyPA or Arg43 of eCyPA might bind to the carboxyl group of Pro, while His87 of FKBP12 might be regarded as an amino acid residue corresponding to Asn102 of hCyPA or Arg87 of eCyPA (Fig. 1A). However, the pH titration study of His87 of FKBP12 revealed that the side chain of His87 of FKBP12 was unessential for the PPIase catalysis (Yu and Fesik 1994). Thus, if His87 participates in the PPIase catalytic reaction, its main chain might contribute to the catalysis.
Expression plasmid and site-directed mutagenesis
The gene of wild-type FKBP12 was amplified by polymerase chain reaction (PCR) using two specific primers with either NdeI or BamHI restriction site. After subjecting the fragment to an NdeI/BamHI double-digestion, it was cloned into the expression vector pET14b (Novagen, EMD Biosciences, Inc.). The PCR product, the double-digested product, and the expression plasmid were purified by the PCR purification kit (Qiagen K.K.), gel extraction kit (Qiagen K.K.), and plasmid miniprep kit (Qiagen K.K.), respectively. The expression plasmids containing 19 different types of mutations of Asp37, Arg42, Phe46, Val55, Trp59, or Tyr82 were constructed using the QuikChange site-directed mutagenesis kit (Stratagene Co.) with specific mutagenic primers. The sextuple Gly mutant D37G/R42G/F46G/V55G/W59G/Y82G was constructed by a step-by-step procedure. The DNA sequence of all the expression plasmids was confirmed by nucleotide sequencing.
Overexpression and purification of the proteins
The expression plasmids were transformed into competent E. coli C41(DE3) cells (Miroux and Walker 1996). Two milliliters of overnight preculture of the cells supplemented with 50 μg mL−1 ampicillin was inoculated into 1 L of 2 × YT medium supplemented with 50 μg mL−1 ampicillin, and this culture was grown to the log phase (OD at 600 nm was 0.3–0.4). Protein overproduction was initiated by adding 0.5 mM isopropyl β-D-thiogalactopyranoside. The culture was grown for an additional 4 h, and the cells were collected by centrifugation. The cell pellet was once frozen at −80°C and then thawed slowly at room temperature. The cell pellet was suspended in 35 mM HEPES-Na buffer (pH 7.8) containing 5 mM β-mercaptoethanol (buffer A). The cells were then lyzed by sonication using a Branson sonicator. The cell debris was removed by centrifugation. The supernatant was filtered through a membrane filter with a pore size of 0.22 μm and then passed through a Superdex 75 column (GE Healthcare) equilibrated with buffer A. The column was washed with 1 column volume of the same buffer, and the eluent was fractionated by a fraction collector. After analysis by SDS-PAGE, the fractions containing the protein were collected and stored at 4°C. The concentration of FKBP12 and its mutant proteins was determined by UV absorption at 280 nm using molar extinction coefficients estimated with the method developed by Pace et al. (1995).
Assay of the isomerase activity
The cis/trans isomerization of the Ala-Pro peptide bond in the synthetic peptide AAPF-MCA was measured by a coupled assay with chymotrypsin, based on the ability of this protease to cleave the synthetic peptide only when the Ala-Pro bond, which is in equilibrium between cis and trans, is in trans configuration (Hayano et al. 1991). Twenty microliters of 1.68 mM peptide was preincubated with the appropriate concentrations of FKBP12 in buffer A, and the assay was started at 30°C by manual mixing with 20 μL of 0.76 mM chymotrypsin (Sigma) in a spectrophotometer cell mounted on an Hitachi spectrophotometer (U-1800) with a temperature controller (NesLab). Hydrolysis of the MCA in the cis peptide was monitored by an increase in the absorbance at 354 nm, whereas the trans peptide was cleaved within the dead time of the experiment. The kinetic curves were analyzed using Kaleida Graph (Hulinks).
Acknowledgments
We thank Professor Nobuhiro Takahashi for providing the cloned gene of human FKBP12. This work was supported by Grants-in-Aid for Scientific Research (Nos. 16041210 and 18031009) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Reprint requests to: Teikichi Ikura, Laboratory of Structural Biology, School of Biomedical Science, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan; e-mail: ikura.str@tmd.ac.jp; fax: 81-3-5803-4594.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073203707.
References
- Bell A., Monaghan, P., and Page, A.P. 2006. Peptidyl-prolyl cis-trans isomerases (immunophilins) and their roles in parasite biochemistry, host-parasite interaction and antiparasitic drug action. Int. J. Parasitol. 36: 261–276. doi: 10.1016/j.ijpara.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Bergsma D.J., Eder, C., Gross, M., Kersten, H., Sylvester, D., Appelbaum, E., Cusimano, D., Livi, G.P., McLaughlin, M.M., Kasyan, K., et al. 1991. The cyclophilin multigene family of peptidyl-prolyl isomerases. Characterization of three separate human isoforms. J. Biol. Chem. 266: 23204–23214. [PubMed] [Google Scholar]
- Bierer B.E., Mattila, P.S., Standaert, R.F., Herzenberg, L.A., Burakoff, S.J., Crabtree, G., and Schreiber, S.L. 1990. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. 87: 9231–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A., Sen, B., Datta, R., and Datta, A.K. 2004. Isomerase-independent chaperone function of cyclophilin ensures aggregation prevention of adenosine kinase both in vitro and under in vivo conditions. Biochemistry 43: 11862–11872. [DOI] [PubMed] [Google Scholar]
- Compton L.A., Davis, J.M., Macdonald, J.R., and Bachinger, H.P. 1992. Structural and functional characterization of Escherichia coli peptidyl-prolyl cis-trans isomerases. Eur. J. Biochem. 206: 927–934. [DOI] [PubMed] [Google Scholar]
- Denesyuk A.I., Vihinen, M., Lundell, J., Zav'yalov, V.P., and Korpela, T. 1993. Structural similarity of the binding sites of cyclophilin A-cyclosporin A and FKBP-FK506 systems. Biochem. Biophys. Res. Commun. 192: 912–917. [DOI] [PubMed] [Google Scholar]
- Fischer G. 1994. Peptidyl-prolyl cis/trans isomerases and their effectors. Angew. Chem. Int. Ed. Engl. 33: 1415–1436. [Google Scholar]
- Fischer G. and Schmid, F.X. 1999. Peptidyl-prolyl cis/trans isomerases. In Molecular chaperones and folding catalysis: Regulation, cellular function, and mechanisms (ed. B. Bukau), pp. 461–489. Harwood Academic Publishers, Amsterdam, Netherlands.
- Fischer G., Berger, E., and Bang, H. 1989a. Kinetic β-deuterium isotope effects suggest a covalent mechanism for the protein folding enzyme peptidylprolyl cis/trans-isomerase. FEBS Lett. 250: 267–270. [DOI] [PubMed] [Google Scholar]
- Fischer G., Wittmann-Liebold, B., Lang, K., Kiefhaber, T., and Schmid, F.X. 1989b. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337: 476–478. [DOI] [PubMed] [Google Scholar]
- Fulton K.F., Jackson, S.E., and Buckle, A.M. 2003. Energetic and structural analysis of the role of tryptophan 59 in FKBP12. Biochemistry 42: 2364–2372. [DOI] [PubMed] [Google Scholar]
- Harrison R.K. and Stein, R.L. 1990. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: Evidence for the existence of a family of distinct enzymes. Biochemistry 29: 3813–3816. [DOI] [PubMed] [Google Scholar]
- Hayano T., Takahashi, N., Kato, S., Maki, N., and Suzuki, M. 1991. Two distinct forms of peptidylprolyl-cis-trans-isomerase are expressed separately in periplasmic and cytoplasmic compartments of Escherichia coli cells. Biochemistry 30: 3041–3048. [DOI] [PubMed] [Google Scholar]
- Holzman T.F., Egan, D.A., Edalji, R., Simmer, R.L., Helfrich, R., Taylor, A., and Burres, N.S. 1991. Preliminary characterization of a cloned neutral isoelectric form of the human peptidyl prolyl isomerase cyclophilin. J. Biol. Chem. 266: 2474–2479. [PubMed] [Google Scholar]
- Huse M., Chen, Y.G., Massague, J., and Kuriyan, J. 1999. Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell 96: 425–436. [DOI] [PubMed] [Google Scholar]
- Huse M., Muir, T.W., Xu, L., Chen, Y.G., Kuriyan, J., and Massague, J. 2001. The TGFβ receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 8: 671–682. [DOI] [PubMed] [Google Scholar]
- Ke H., Mayrose, D., and Cao, W. 1993. Crystal structure of cyclophilin A complexed with substrate Ala-Pro suggests a solvent-assisted mechanism of cis-trans isomerization. Proc. Natl. Acad. Sci. 90: 3324–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofron J.L., Kuzmic, P., Kishore, V., Colon-Bonilla, E., and Rich, D.H. 1991. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 30: 6127–6134. [DOI] [PubMed] [Google Scholar]
- Konno M., Ito, M., Hayano, T., and Takahashi, N. 1996. The substrate-binding site in Escherichia coli cyclophilin A preferably recognizes a cis-proline isomer or a highly distorted form of the trans isomer. J. Mol. Biol. 256: 897–908. [DOI] [PubMed] [Google Scholar]
- Levy M.A., Brandt, M., Livi, G.P., and Bergsma, D.J. 1991. Purification and characterization of human T-cell cyclophilin expressed in Escherichia coli . Transplant. Proc. 23: 319–322. [PubMed] [Google Scholar]
- Miroux B. and Walker, J.E. 1996. Overproduction of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260: 289–298. [DOI] [PubMed] [Google Scholar]
- Pace C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4: 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid F.X. 2001. Prolyl isomerases. Adv. Protein Chem. 59: 243–282. [DOI] [PubMed] [Google Scholar]
- Scholz C., Maier, P., Dolinski, K., Heitman, J., and Schmid, F.X. 1999. R73A and H144Q mutants of the yeast mitochondrial cyclophilin Cpr3 exhibit a low prolyl isomerase activity in both peptide and protein-folding assays. FEBS Lett. 443: 367–369. [DOI] [PubMed] [Google Scholar]
- Shaw P.E. 2007. Peptidyl-prolyl cis/trans isomerases and transcription: Is there a twist in the tail? EMBO Rep. 8: 40–45.17203101 [Google Scholar]
- Sokolskaja E. and Luban, J. 2006. Cyclophilin, TRIM5, and innate immunity to HIV-1. Curr. Opin. Microbiol. 9: 404–408. [DOI] [PubMed] [Google Scholar]
- Timerman A.P., Wiederrecht, G., Marcy, A., and Fleischer, S. 1995. Characterization of an exchange reaction between soluble FKBP-12 and the FKBP.ryanodine receptor complex. Modulation by FKBP mutants deficient in peptidyl-prolyl isomerase activity. J. Biol. Chem. 270: 2451–2459. [DOI] [PubMed] [Google Scholar]
- Tradler T., Stoller, G., Rucknagel, K.P., Schierhorn, A., Rahfeld, J.U., and Fischer, G. 1997. Comparative mutational analysis of peptidyl prolyl cis/trans isomerases: Active sites of Escherichia coli trigger factor and human FKBP12. FEBS Lett. 407: 184–190. [DOI] [PubMed] [Google Scholar]
- Wiederrecht G., Brizuela, L., Elliston, K., Sigal, N.H., and Siekierka, J.J. 1991. FKB1 encodes a nonessential FK 506-binding protein in Saccharomyces cerevisiae and contains regions suggesting homology to the cyclophilins. Proc. Natl. Acad. Sci. 88: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K.P., Yamashita, M.M., Sintchak, M.D., Rotstein, S.H., Murcko, M.A., Boger, J., Thomson, J.A., Fitzgibbon, M.J., Black, J.R., and Navia, M.A. 1995. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr. D Biol. Crystallogr. 51: 511–521. [DOI] [PubMed] [Google Scholar]
- Yao Q., Li, M., Yang, H., Chai, H., Fisher, W., and Chen, C. 2005. Roles of cyclophilins in cancers and other organ systems. World J. Surg. 29: 276–280. [DOI] [PubMed] [Google Scholar]
- Yu L. and Fesik, S.W. 1994. pH titration of the histidine residues of cyclophilin and FK506 binding protein in the absence and presence of immunosuppressant ligands. Biochim. Biophys. Acta 1209: 24–32. [DOI] [PubMed] [Google Scholar]