

Abstract
The extracellular adherence protein (Eap) of Staphylococcus aureus participates in a wide range of protein–protein interactions that facilitate the initiation and dissemination of Staphylococcal disease. In this report, we describe the use of a multidisciplinary approach to characterize the solution structure of full-length Eap. In contrast to previous reports suggesting that a six-domain isoform of Eap undergoes multimerization, sedimentation equilibrium analytical ultracentrifugation data revealed that a four-domain isoform of Eap is a monomer in solution. In vitro proteolysis and solution small angle X-ray scattering studies both indicate that Eap adopts an extended conformation in solution, where the linkers connecting sequential EAP modules are solvent exposed. Construction of a low-resolution model of full-length Eap using a combination of ab initio deconvolution of the SAXS data and rigid body modeling of the EAP domain crystal structure suggests that full-length Eap may present several unique concave surfaces capable of participating in ligand binding. These results also raise the possibility that such surfaces may be held together by additional interactions between adjacent EAP modules. This hypothesis is supported by a comparative Raman spectroscopic analysis of full-length Eap and a stoichiometric solution of the individual EAP modules, which indicates the presence of additional secondary structure and a greater extent of hydrogen/deuterium exchange protection in full-length Eap. Our results provide the first insight into the solution structure of full-length Eap and an experimental basis for interpreting the EAP domain crystal structures within the context of the full-length molecule. They also lay a foundation for future studies into the structural and molecular bases of Eap-mediated protein–protein interactions with its many ligands.
Keywords: protein stucture/folding, molecular mechanics, correlation of structure and spectra, structure
Staphylococcus aureus is a widespread and persistent human pathogen that causes a remarkable range of community-acquired and nosocomial diseases in humans. While its infections vary in both clinical presentation and severity (Lowy 1998), the initial stages of an S. aureus infection necessarily require contact with discrete areas within the host prior to their colonization (Patti et al. 1994; Lowy 1998; Chavakis et al. 2005). This process depends directly on the function of a broad spectrum of adhesive proteins. Many of these proteins have the ability to recognize one or several of the large glycoproteins found in the host plasma and extracellular matrix. The functional diversity of these adhesins contributes to the ability of S. aureus to adapt efficiently to such biologically distinct microenvironments as connective tissue and bone to the bloodstream and vascular tissue. A majority of the adhesive properties displayed by S. aureus reside within the cell surface tethered MSCRAMM proteins (for Microbial Surface Components Recognizing Adhesive Matrix Molecules) (Patti et al. 1994); however, several important adhesins are also formally secreted from the bacterial cell, and the term SERAM (for Secreted Expanded Repertoire Adhesive Molecules) (Chavakis et al. 2005) has been designated to reflect the function of these proteins. A unique feature of SERAMs in promoting S. aureus pathogenesis is that these molecules appear to use their adhesive properties not solely to anchor the bacterial cell to a target surface, but also to interfere with host defense mechanisms such as coagulation and immunity (Chavakis et al. 2005).
The SERAM extracellular adherence protein (Eap) is nearly ubiquitously distributed among S. aureus strains and appears to function as a virulence determinant in animal models of chronic S. aureus infection (Lee et al. 2002). In addition to its well-established roles in promoting adhesion-based processes such as bacterial aggregation (Palma et al. 1999) and invasion of eukaryotic cells (Hussain et al. 2002; Haggar et al. 2003), Eap has also been shown to interfere directly with complex, signaling-dependent events such as leukocyte recruitment (Chavakis et al. 2002; Lee et al. 2002; Xie et al. 2006) and both wound healing and angiogenesis (Athanasopoulos et al. 2006). Current evidence suggests that this remarkable diversity in Eap functions is related to its unique ability to form protein–protein interactions with an array of nearly a dozen ligands, including a bacterial cell surface-retained phosphatase (Flock and Flock 2001), host extracellular matrix molecules such as collagen, fibronectin, and laminin (Boden and Flock 1992; McGavin et al. 1993; Palma et al. 1999; Chavakis et al. 2002), and the pro-inflammatory mammalian surface adhesin ICAM-1 (Chavakis et al. 2002). However, the structural adaptations and biochemical features of Eap that allow specific interactions with so many different ligands remain largely unexplored.
Depending on the S. aureus strain, the mature Eap molecule is ∼50–70 kDa and comprises four to six tandem repeats of an ∼97 residue EAP domain (Jonsson et al. 1995; Geisbrecht et al. 2005). These are joined by short, 9–12 residue linker regions of unknown structure. We recently reported the high-resolution crystal structures of three individual EAP domains (Geisbrecht et al. 2005). These studies revealed an unexpected structural homology with a family of immunotoxins expressed by pathogenic gram-positive cocci, including S. aureus (Papageorgiou and Acharya 1997; Haas et al. 2005); in particular, EAP domains share a striking similarity to the superantigen C-terminal domain, as both classes of proteins adopt a β-grasp fold where a single α-helix lies diagonally across a five-stranded mixed β-sheet (Geisbrecht et al. 2005). Further sequence/structure analysis of EAP domains along with examination of superantigen co-crystal structures (Fields et al. 1996) suggest that the loop region connecting β5 and β6 is likely to be important to the formation of EAP domain-mediated protein–protein interactions (Geisbrecht et al. 2005). Nevertheless, the role of individual EAP domains in forming any of the numerous Eap–ligand interactions has not been established, and this represents a major hurdle in understanding the structural and molecular bases of Eap functional diversity. Furthermore, the physical and conformational relationships between the individual domains that comprise full-length Eap are currently unknown. Such knowledge may be critical to understanding the contributions of individual EAP domains to these interactions and whether full-length Eap can bind to multiple ligands simultaneously.
We have addressed these issues by employing a multidisciplinary approach to characterize the solution structure of full-length Eap. Our results suggest that Eap adopts an elongated, but structured conformation in solution. Importantly, we provide evidence that additional contacts are likely to be present between adjacent EAP modules in the context of the full-length protein. The potential implications of these observations for understanding the structural basis of Eap–ligand interactions are discussed.
Results
Eap from S. aureus strain Mu50 is a monodisperse monomer in solution
Previous studies on native and recombinant Eap from S. aureus strain Newman have shown that functional Eap self-associates into insoluble, higher-order structures (Palma et al. 1999). The isoform of Eap expressed by this strain comprises six tandem repeats of the EAP domain, in contrast to S. aureus strain Mu50, which expresses a shorter, four EAP domain variant (Kuroda et al. 2001; Geisbrecht et al. 2005). Recent evidence suggests strongly, however, that the four- and six-domain forms of Eap are functionally equivalent (Xie et al. 2006). Further, the four-domain isoform does not undergo spontaneous aggregation like its larger counterpart. This feature makes it preferable for structure/function studies on the Eap protein.
To determine the oligomeric state of Eap from S. aureus strain Mu50, we performed sedimentation equilibrium experiments on purified, recombinant Eap at three different concentrations (0.33, 0.66, and 1.32 mg mL−1). The molecular weight of Eap was fit to the experimental data for three different speeds (20, 30, and 40 k.r.p.m.) for the three concentrations listed above (Fig. 1). Extrapolation of these data to an infinite dilution yields observed molecular weights of 53.0, 50.5, and 49.3 kDa at the concentrations shown. Overall, the experimental molecular weight of full-length Eap (50.9 ± 1.5 kDa) is in excellent agreement with the predicted molecular weight (50.3 kDa) of the mature Eap polypeptide within the 68% confidence interval. This indicates that the four-domain isoform of Eap is a monomer in solution.
Figure 1.
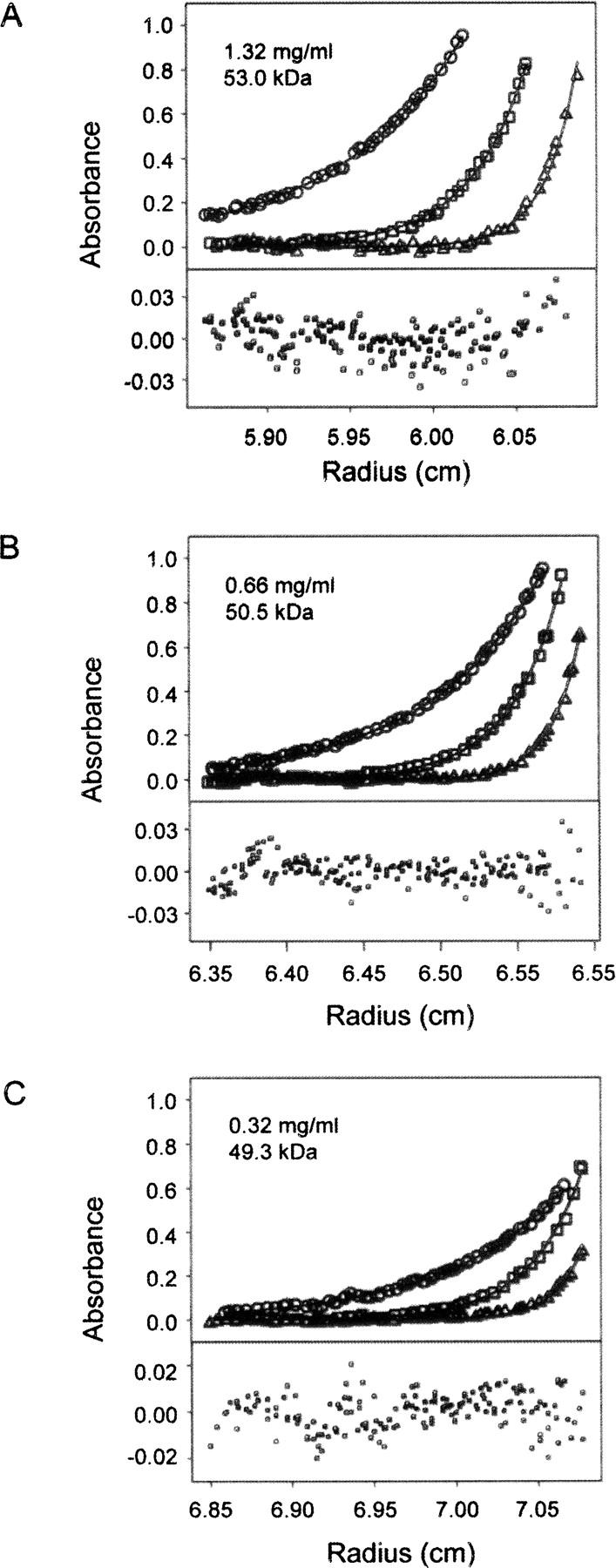
Sedimentation equilibrium analysis of recombinant full-length Eap protein. The individual panels (A–C) show experimental data obtained at 20 (circles), 30 (squares), and 40 k.r.p.m. (triangles) at the Eap concentrations indicated. Equilibrium profiles were fit separately for each concentration (black lines) according to the single particle model described in Materials and Methods to yield the observed molecular mass of the protein. The determined molecular mass of 50.9 ± 1.5 kDa corresponds to monomeric Eap within the 68% confidence interval. Bottom plots show random residuals of the fits.
Evidence for an extended conformation of Eap in solution
Proteolytic susceptibility has long been used as a biochemical probe of protein structure and conformation. Therefore, to begin addressing these issues for full-length Eap, we subjected purified, recombinant Eap to limiting proteolysis by both Subtilisin A and trypsin (Fig. 2A). A series of identical 5 μg samples were incubated with increasing mass ratio dilutions of each protease individually at 25°C for 60 min. Following incubation, the entire proteolysis reaction was separated by gradient SDS polyacrylamide gel electrophoresis and the distinct protein species contained therein were visualized by Coomassie Blue staining. Nine gel bands representing distinct, subtilisin-generated Eap fragments were excised and processed for in-gel trypsin digestion. The resulting tryptic peptides were extracted, separated by capillary liquid chromatography (LC), and analyzed by tandem mass spectrometry (MS/MS). Peptides containing subtilisin-sensitive sites (nontryptic ends) were initially identified using the “no enzyme” search option (Sequest, BioWorks 3.1 SR1) and inspected manually for subsequent validation (MacCoss et al. 2002; Starkweather et al. 2007).
Figure 2.

Protease sensitivity of residues connecting adjacent EAP domains. (A) Purified, recombinant Eap was treated with increasing concentrations of either subtilisin A or trypsin as shown labeled alphabetically. Following separation of each digest by SDS-PAGE, nine distinct species were isolated, denatured, digested with trypsin, and processed for proteomic analysis by LC-MS/MS (Kinter and Sherman 2000). Note that the proteolytic fragments occurred predominantly in multiples of ∼10–13 kDa, which corresponds well with the size of the EAP domain (Geisbrecht et al. 2005). Identification of the products revealed that the sites of proteolysis were localized in the proximity of the linker regions that connect adjacent EAP domains (see also Supplemental Fig. 1). (B) Sequence alignment and residue positions for each EAP module from S. aureus strain Mu50 Eap are shown along with secondary structure assignment from the EAP2 crystal structure (PDB accession code 1YN3) (Geisbrecht et al. 2005). Invariant residues are shown in reversed cyan typeface while residues identical in three of four sequences are shown in reversed purple typeface. The linker regions are shown in a light blue box. This figure was drawn using ESPript (espript.ibcp.fr/).
The results of this proteolytic study reveal several important features of the Eap protein. First, the sequence of each characterized band corresponds to a well-defined combination of the individual EAP domains, ranging from fragments containing three domains (Bands 1, 2, and 3), to those comprising two domains (Bands 4–8), and finally to that of a single EAP domain (Band 9). This suggests that the EAP domains themselves are relatively stable in the presence of protease and, consequently, that the major sites of proteolytic processing lie within the proximity of the individual domain boundaries (sequence shown in Fig. 2B and in Supplemental Fig. 1A with subtilisin-sensitive regions labeled). Second, the enhanced proteolytic sensitivity near the linker regions in Eap implies that these regions of the protein must be relatively solvent accessible in the structure of the full-length molecule. The similar nature of the results obtained with both subtilisin and trypsin suggests these properties are general features of the Eap protein and do not simply reflect the specificity of the proteolytic enzymes. Together, these results suggest that the proteolytic sensitivity observed near the domain linking regions reflects an overall solution conformation of Eap that is elongated rather than compact (Supplemental Fig. 1B).
Analysis of Eap particle behavior by small angle X-ray scattering
To gain additional insight into the extended solution conformation of Eap, we collected small angle X-ray scattering (SAXS) data on monodisperse solutions of the recombinant protein. SAXS studies were performed on four Eap samples within a concentration range from 1.6 to 7.7 mg/mL. Inspection of 20 time frames for each concentration studied during data acquisition showed no radiation damage effects nor was significant interference attributable to polydispersity observed.
The Guinier fits for each of the four concentrations studied are linear throughout the appropriate qRG range up to 1.0 (Fig. 3A). The radii of gyration RG calculated for the various protein concentrations (RG = 40.2, 41.6, 42.2, 42.9 for 1.6, 3.3, 3.9, 7.7 mg mL−1) display only slight concentration dependence, which likely arises from either minor particulate interference or minor aggregation at the highest protein concentrations employed. An interference-free SAXS profile was estimated by extrapolating the measured scattering curves to an infinite dilution (Fig. 3B). The mean value for the radius of gyration, RG = 40.5 ± 0.6 Å, as deduced by extrapolation to zero concentration (c = 0) is substantially increased with respect to the theoretical value (RG ∼ 20 Å) for a globular protein containing the same number of amino acids (Millett et al. 2002). This suggests that the average conformation of Eap in solution is extended and is consistent with the proteolytic sensitivity data presented above (Fig. 2).
Figure 3.

Experimental analysis of Eap by small angle X-ray scattering. SAXS data were collected on solutions of recombinant Eap at 1.6, 3.3, 3.9, and 7.7 mg mL−1 as described in Materials and Methods. (A) Guinier plot of the SAXS data. Filled circles correspond to I(q) data used to obtain experimental radii of gyration, RG. The solid lines and accompanying extrapolations correspond to the best fit through these points above. Guinier plots for each protein concentration are shown from highest concentration to lowest, top to bottom, respectively. (B) The final experimental SAXS curve (black circles) is depicted along with the theoretical scattering profiles for both the average ab initio model (black line; Fig. 4B) and the atomic models revealed by MD (thin black line; Fig. 5B). The scattering profiles representing the theoretical curves for the extremely elongated or compacted conformation of Eap shown inset in Figure 5A, are labeled accordingly. (C) Real-space distance distribution function computed from the experimental SAXS data. The maximum dimension of Eap revealed by P(r) analysis is denoted as D max. The local maxima of the P(r) function (labeled M1 and M2) highlight the most frequently occurring molecular dimensions within full-length Eap. (D) A linear relationship between the molecular weight and the I(0)/c values for five distinct proteins measured in a SAXS experimental session at APS beamline 18-ID. The data correspond to the following proteins: Efb-C and Efb (the C-terminal domain and full-length protein of S. aureus Efb) (Lee et al. 2004; Hammel et al. 2007), chicken lysozyme, recombinant Eap, and Fg-D (the tryptic D-fragment of human fibrinogen) (Mihalyi and Godfrey 1963).
To gain further information on the geometry of Eap, we calculated the pair-distance distribution function, P(r) (Fig. 3C), from the SAXS curve. The P(r) function provides a real space interpretation of the scattering data and revealed local maxima at M 1 = 25 Å and M 2 = 45 Å. These values correspond to the most frequently occupied interatomic distances in full-length Eap and are consistent with the thickness and length of the individual EAP domains as judged by three independent crystal structures of proteins adopting this fold (Geisbrecht et al. 2005). Furthermore, the RG value of 42.7 ± 0.8 Å derived from the final distance distribution function is likewise internally consistent with the RG calculated from the Guinier approximation described above. Interestingly, the profile of the P(r) function exhibits a long tail with a maximal particle dimension, D max, of ∼176 ± 9 Å (Fig. 2C). The simplest interpretation of this feature is that the intact Eap protein is substantially elongated in solution (Glatter 1982).
Restoration of the averaged low-resolution shape for Eap by ab initio modeling
An important advantage of SAXS is that low-resolution information on the overall three-dimensional shape of the protein examined is implicit within the scattering data. Consequently, we used ab initio modeling to derive several independent reconstructions of the overall shapes of Eap. Repetitive runs for each calculation yielded superimposable models with similar overall structural features (Fig. 4A) and similar qualities of fit (χ = 0.2). Since repetition of the modeling process substantially decreases the risk of inferring an erroneous low-solution structure, 10 individual restored models were selected randomly and subjected to least-squares superposition and averaging by the program DAMAVER (Volkov and Svergun 2003).
Figure 4.
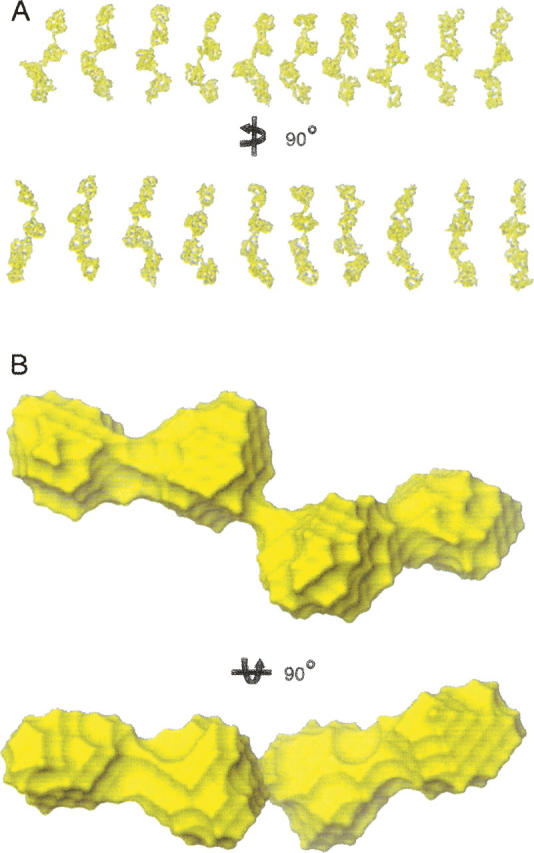
Reconstructed three-dimensional ab initio models of Eap. (A) Ten single models restored by the program GASBOR (Svergun et al. 2001). The models in the bottom panel are the same models rotated counterclockwise by 90° about the Y-axis as indicated. (B) The averaged shape for the molecular envelope of Eap as calculated from the 10 independent GABSOR models shown above via the program DAMAVER (Volkov and Svergun 2003). The shape in the bottom represents the shape rotated clockwise by 90° about the X-axis as indicated. All models are displayed as representative molecular surfaces (yellow) and were rendered using GASBOR (Svergun et al. 2001) and DAMAVER (Volkov and Svergun 2003).
Several important global structural features of the Eap protein are evident in the averaged model (Fig. 4B). First, the overall shape consists of four globular units, where each adjacent pair is connected by a narrow, stretched region. As might be expected, the overall surface shape calculated from the EAP domain crystal structures compares very favorably to that of these discrete globular units. Second, the overall shape of full-length Eap is significantly elongated, which is again consistent with analysis of the Guinier plots, P(r) function, and proteolysis data. Such overall shape characteristics were highly reproducible for all ab initio modeling trials conducted.
Atomic models for Eap derived from rigid body modeling
Considering the extended overall conformation of full-length Eap and the presence of three interdomain linkers with an average length of 12 residues, it was plausible that the intact Eap protein might sample a range of similar conformations in time and space. Furthermore, the conformational space explored by such a protein might reasonably be expected to increase with the absolute length of the linker. Given this possibility, it was not possible to describe accurately the entire scattering curve of full-length Eap through one single conformation. Furthermore, the observed low-resolution shape shown in Figure 4B only represented the average probable conformational space attained by Eap (Hammel et al. 2004, 2005).
Molecular dynamics (MD) simulations have been widely used to explore conformational space in proteins. A common strategy in these scenarios is to perform the simulation at very high temperature (∼1500 K), where the additional kinetic energy prevents the molecule from becoming trapped in a local minimum (Leach 2001). We therefore used molecular modeling on the interdomain linkers to determine the conformational distribution of full-length Eap that best explained the experimental SAXS data. Different conformations of the Eap protein were produced at regular intervals along the MD trajectories, and these conformations were validated by subsequent calculations of the theoretical SAXS profiles for the respective Eap conformations. The initial model for Eap used in our MD simulations was constructed by connecting unique homology models for each individual EAP domain that were generated by virtue of the high sequence identity between the respective EAP domains (Fig. 2B; Geisbrecht et al. 2005). The individual EAP domain homology models were then superimposed into the ab initio calculated shape (Fig. 4B) and then connected with idealized peptide linkers that consisted of the appropriate number of residues (Fig. 2B) (see Materials and Methods).
Without applying RG constraints to MD-simulated Eap conformations, the resulting Eap models adopt both highly stretched (D max > 200 Å, RG ∼ 63 Å) and compacted structures (D max < 100 Å, RG ∼ 34 Å); in general such models result in a poor fit to the experimental scattering curves, where χ > 10 (Figs. 3B, 5A). Incorporation of the empirical RG constraints during the MD simulation, however, eliminates such unrealistic conformations, saves computational time, and produces conformational ensembles whose calculated RG values come close to those observed experimentally. Comparison of the theoretical SAXS profiles for each calculated MD model with the experimental data using the discrepancy function (χ) allows for selection of a finite number of structures that are most reconcilable with both the experimental scattering curve (Fig. 3B) and the experimentally derived RG value. The correlation between fit quality (χ) for 15,000 generated conformations and RG is displayed in Figure 5A. This analysis reveals that the best attainable modeled conformations have RG ∼ 42 Å and result in fit quality of χ ∼ 1.4; importantly, these models have RG values comparable to the value 42.7 ± 0.8 Å obtained by analysis of the P(r) function (Fig. 3C).
Figure 5.
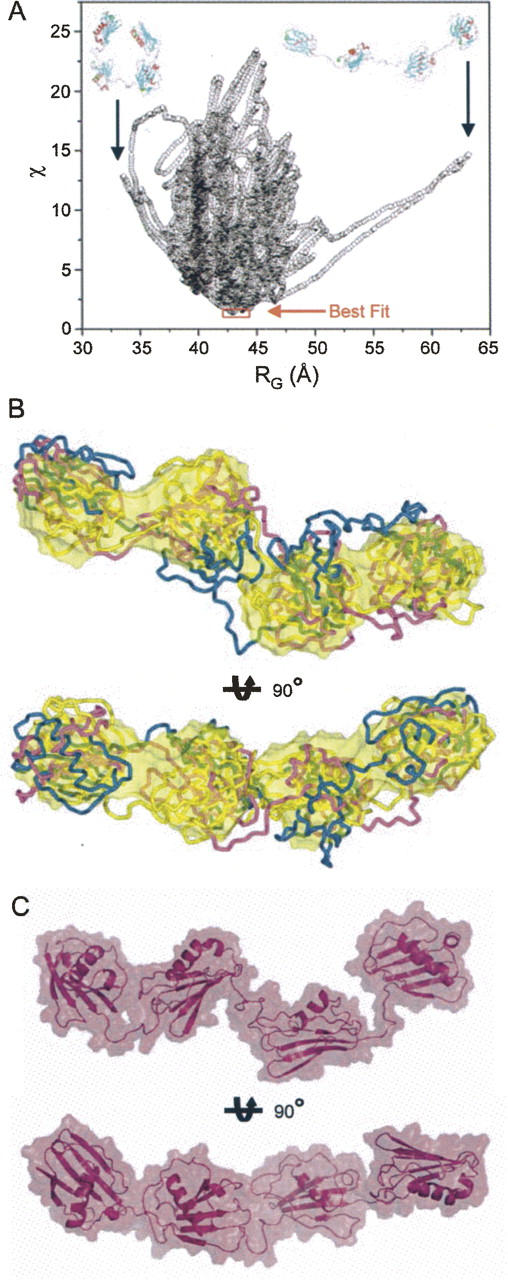
Rigid body molecular dynamics models for Eap. Molecular models for the individual EAP domains were generated on the basis of homology with the EAP2 crystal structure (PDB accession code 1YN3) (Geisbrecht et al. 2005). The resulting homology models were considered as rigid bodies in MD simulations to generate potential solution structures of full-length Eap that were consistent with the molecular parameters determined experimentally by SAXS. (A) A graphical comparison of discrepancy values (χ) for 15,000 rigid body models of Eap is shown along with their calculated RG values. The best fitting models are indicated by red frame. Note that the small subset of best fitting models all lay within the region of experimentally determined RG values (Fig. 3). For the sake of comparison, rigid body solutions for the Eap solution structure that adopt extremely stretched (D max > 200 Å, RG ∼ 63 Å) or collapsed conformations (D max < 100 Å, RG ∼ 34 Å−1), respectively, are shown in inset. (B) Three best fit models derived from independent solutions were superimposed onto the surface of the ab intio model shown in Figure 4B. The secondary structural elements of the different models are shown in Cα tube representations and colored blue, purple, and gold, respectively. The bottom models are rotated clockwise by 90° around the X-axis, as indicated. (C) Two orthogonal views of the best fit model in surface representation superimposed on the secondary structure elements.
Three of the best fitting Eap conformations (RG values of 42.7, 42.9, and 43.7 Å, respectively) are superimposed and shown in Figure 5B. These ensembles can be considered as the most frequent conformations adopted by Eap in solution. A thorough inspection of the commonalities among these best fitting structures highlights several important features of the Eap protein. First, the ab initio shape shown in Figure 4B overlaps exceedingly well with the best fitting structures determined by MD simulation. The similar results achieved from these two distinct approaches suggest strongly that these modeling studies reflect real structural features of Eap. Second, while the overall structure of Eap is typified by an elongated arrangement within the protein, there is nevertheless a distinct bend, or kink, in the linker region that connects the EAP2 and EAP3 subdomains. This suggests that Eap may adopt a somewhat ordered quaternary structure that is characterized by defined spatial arrangements between adjacent EAP domains. Finally, and along similar lines, the individual EAP domains remain in close proximity with one another in the final models. Here, the individual interdomain distances range between 5 and 10 Å. This arrangement is reflected in a depiction of the best fit model (Fig. 5C).
Spectroscopic evidence for interdomain interactions in Eap
The Eap conformations obtained by ab initio and MD modeling of the experimental SAXS data raise the possibility that the individual globular EAP domains may interact with one another within the context of the full-length Eap protein. However, while SAXS can provide important information on the overall three-dimensional shape of a protein in solution, this technique is of neither sufficient resolution nor sensitivity to detect such interactions directly.
To test for the possibility of interdomain interactions in Eap, we employed Raman spectroscopy to probe structural differences between full-length Eap and an equivalent stoichiometric mixture in its composite EAP domains. The Raman difference signature obtained by subtraction of the spectral sum of EAP domains from the spectrum of the full-length Eap protein (Fig. 6A) is consistent with a more highly ordered secondary structure for the full-length protein. Specifically, we observe positive difference peaks for the secondary structure markers at 1677 cm−1 (amide I) and 1238 cm−1 (amide III), as well as for the related structural markers at 712 (amide V) and 945 cm−1 (Cα-C stretching) (Carey 1982; Thomas Jr. 1999; Benevides et al. 2003). The difference bands are diagnostic of greater β-strand structure in full-length Eap (Carey 1982) and are consistent with the expected β-sheet content of the domains (Geisbrecht et al. 2005).
Figure 6.
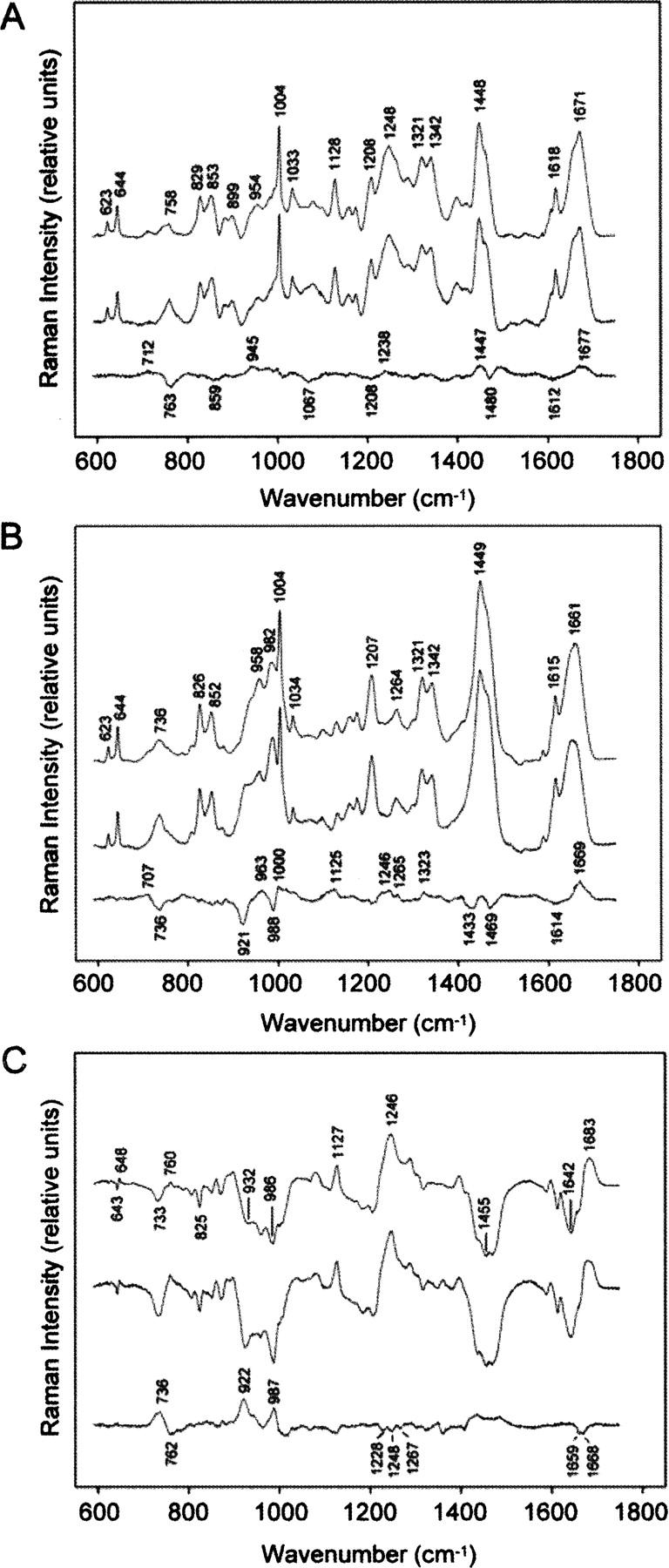
Comparative Raman spectroscopy of full-length Eap and a stoichiometric mixture of its composite domains. To analyze the potential for additional interactions between adjacent modules in full-length Eap, Raman spectra were collected and analyzed for the intact protein as well as a stoichiometric mixture of its purified EAP domains. (A) Difference Raman spectroscopy of Eap in H2O (pH 7.5). Raman spectra of full-length Eap (top trace) and a stoichiometric mixture of EAP domains (middle trace) show similar spectral signatures. The difference spectrum between the top and middle traces (bottom trace) reveals markers of β-stranded structure (1238, 1677 cm−1), which indicate greater structural order in the full-length protein. (B) Difference Raman spectroscopy of Eap in D2O (pD 7.5). Raman spectra were collected after 20 h equilibration in D2O at 4°C. Differences in hydrogen/deuterium (H/D) exchange protection in full-length Eap (top trace) and separate EAP domains (middle trace) are shown in a difference spectrum constructed as in A (bottom trace). Additional H/D exchange protection in the full-length protein is supported by peaks at 1246 and 1669 cm−1 and troughs at 921 and 988 cm−1. (C) Raman difference spectra showing H/D exchange in full-length Eap (top trace) and separate EAP domains (middle trace) are compared in a second-difference spectrum (bottom trace) constructed as in A. Troughs in the regions of amide I (1659 and 1668 cm−1) and amide III (1228, 1248, and 1267 cm−1) markers originate from greater H/D exchange protection in full-length Eap, while peaks in the amide III′ region (922, 987 cm−1) reflect greater H/D exchange in the mixture of EAP domains.
We also measured the Raman difference signature between D2O solutions of full-length Eap and the spectral sum of domains after equilibrium of hydrogen/deuterium (H/D) exchange had been reached under native conditions. The results (Fig. 6B) show that peptide exchange (NH → ND) is more protected in full-length Eap. Thus, the positive difference peaks at 1246 (amide III) and 1669 cm−1 (amide I) are diagnostic of exchange-protected β-strand regions, while those at 1265 and 1323 cm−1 are diagnostic of exchange-protected α-helices in full-length Eap (Benevides et al. 1996). The corresponding Raman markers for the deuterium-exchanged (unprotected) peptides, which are designated as amide III′ and amide I′ bands, are expected to contribute troughs to the difference spectrum, as labeled in Figure 6B. The amide II′ vibration of full-length Eap is also labeled (1449 cm−1), which is expected to exhibit significant Raman intensity in the deuterated protein (Tuma et al. 1998a). The remaining difference features in Figure 6B are due to side chains that may have different local environments in the full-length and constituent domain structures (Thomas Jr. 1999; Benevides et al. 2003).
Detailed differences in the H/D exchange behavior of both samples were revealed by calculating second-difference spectra for data collected in both H2O and D2O (Fig. 6C). Here, the amide I/I′ region exhibits a negative doublet at 1659 and 1668 cm−1, consistent with greater H/D exchange protection in both α-helix and β-strand structures of full-length Eap. Troughs in the amide III region (1220–1330 cm−1) also reflect this effect (Tuma et al. 1998b). The second-difference analysis also allows for interpretation of the amide III′ peaks. Accordingly, the peak at 921 cm−1 reflects greater H/D exchange of nonrepetitive structure in individual EAP domains, while the positive peak at 1005 cm−1 may reflect H/D exchange of the domain linking regions of full-length Eap (Tuma and Thomas 1997; Geisbrecht et al. 2005) as well as uncompensated Raman intensity of phenylalanyl side chains that differ in their local environments in the two protein samples (Tuma and Thomas 1997). Overall, the observed spectral differences are consistent with the presence of ordered structure in full-length Eap that is absent from the stoichiometric ensemble of separated EAP domains.
Discussion
The extracellular adherence protein of Staphylococcus aureus participiates in a wide range of protein–protein interactions that culminate in the disruption of leukocyte and endothelial cell functions during the initial stages and propagation of Staphylococcal infections (Harraghy et al. 2003; Chavakis et al. 2005; Athanasopoulos et al. 2006). The structural and biochemical basis for these many interactions remains poorly defined, however. One limiting factor in furthering our understanding of these processes experimentally has been a lack of structural information vis-à-vis full-length Eap. The multidomain nature of Eap, in conjunction with the presence of apparently flexible linker regions between the domains, suggested that full-length Eap might be unsuitable for traditional approaches of structural characterization used on proteins of this size; indeed, Eap has remained refractory to crystallization in our hands despite repeated efforts toward this goal. As an alternative, we have described here a characterization of the solution structure of full-length Eap through a multidisciplinary approach of analytical ultracentrifugation, proteolytic sensitivity, small angle X-ray scattering combined with MD based rigid body modeling and Raman spectroscopy. This work has yielded an atomic model for an active, four-domain isoform Eap that has been validated using a number of the biophysical parameters inherent to full-length Eap. The model reported here can now serve as a structural framework for design of additional experiments to probe the role of various Eap substructures in direct biochemical studies of Eap interactions. In addition, the importance of these structures to the remarkable physiological effects of Eap can now be assessed.
Our studies are consistent with an elongated, yet structured conformation of Eap in solution. This conclusion was reached by consideration and analysis of several independent lines of evidence. First, Eap could be readily digested by proteolytic enzymes into various EAP domain substructures (Fig. 2; Supplemental Fig. 1). This indicates that linker regions connecting the EAP modules must be solvent exposed. Along this line, the relatively small size of these linkers in comparison to the rest of the Eap polypeptide also suggests that it is unlikely that Eap folds back upon itself based upon steric constraints. Second, the experimental constants determined by SAXS are more consistent with an elongated shape for Eap when compared to a globular protein of the same mass (Fig. 3). Third, the constrained ab initio shape reconstructions from SAXS data consistently reveal models with similar qualities of fit. These show comparable global shape characteristics that are conserved in the final averaged model (Fig. 4). The most striking feature of these models is the roughly linear rearrangement of the terminal pairs of EAP domains that are interrupted by a distinct bend between the EAP2 and EAP3 domains. In addition, the combination of SAXS studies with the known atomic structures of the EAP module (Geisbrecht et al. 2005) allow the use of rigid body modeling to propose a plausible conformation for full-length Eap atomic resolution (Fig. 5). The data show that the most likely models adopt a similar structure in which a small conformational space is sampled relative to the interdomain linkers. This observation is consistent with Eap models having limited conformational flexibility or restricted interdomain motion. The atomic models generated by this strategy are typified by interdomain distances between 5 and 10 Å and raise the possibility that a degree of the rigidity in Eap might be derived from interdomain interactions. Indeed, comparisons of Raman spectra between full-length Eap and a stoichiometric mixture of the individual EAP domains strongly suggest that this is the case (Fig. 6). The observation of additional structured regions in full-length Eap relative to the domain mixture and changes in both the nature and extent of H/D protection in the full-length protein provide experimental evidence for stable interdomain contacts over the timescale of the measurements (hours). The data imply that Eap is not simply a disordered ensemble of linked domains, but an ordered assembly of domains into a well formed, yet elongated protein architecture.
As noted previously, the models reported here should serve as a useful framework for probing the specific structure/function relationships of Eap. In terms of Eap-mediated protein–protein interactions, the roles of the individual domains or combinations thereof can now be explored systematically. While the previously reported EAP domain crystal structures suggest a common ligand-binding site for all domains (Geisbrecht et al. 2005), it is not obvious how Eap might bind specifically to over a dozen different ligands through only one kind of site in each EAP module. The low-resolution and modeled structures presented here (Figs. 4, 5) suggest to the contrary that the full-length Eap protein presents an array of different binding surfaces and interfaces requiring context-specific interactions or orientations involving multiple domains.
We believe that this model suggests a number of important and experimentally testable hypotheses. First, it should be possible to isolate discrete substructures of Eap that retain only subsets of the full array of Eap/ligand interactions. For example, the production of an Eap variant that lacks either the first two or last two EAP domains should help to identify the locus of the anti-inflammatory functions. Second, the order in which the domains of full-length Eap are linked is likely to be critical to retaining native ligand interactions. The generation of EAP domain-permuted mutants of full-length Eap (e.g., where the fourth EAP domain has been moved to the N terminus, etc.) should be useful for testing this hypothesis. Finally, by exploring and defining the low-resolution structural requirements for Eap/ligand interactions, we will be better able to approach high-resolution structural analysis of active Eap substructures bound to their respective ligands. Whereas multidomain forms of proteins are generally less amenable to crystallographic analysis, it is often the case that simpler substructures can produce diffraction-quality crystals or interpretable NMR spectra. In the particular case of Eap complexes, high-resolution structural analysis would provide an important first glimpse into the detailed structural properties of this protein that allow it to form specific complexes with such an impressive array of ligands.
Materials and Methods
Recombinant protein expression and purification
Recombinant S. aureus (strain Mu50) Eap was produced in a derivative of E. coli strain BL21(DE3) that had been transformed with the prokaryotic overexpression vector pT7HMT-Eap (Geisbrecht et al. 2006; Xie et al. 2006). The overexpression, extraction, solubilization, and purification of Eap were each performed according to the previously published method (Xie et al. 2006), the lone exception being that the gel-filtration chromatography step was performed prior to cation exchange. This modification provided superior resolution of full-length Eap from truncated contaminants. The individual EAP modules were overexpressed and purified according to the same procedure employed in our previous crystallization study (Geisbrecht et al. 2005).
Analytical ultracentrifugation
Analytical ultracentrifugation was performed at 293 K using a Beckman XL-A ultracentrifuge. Sedimentation equilibrium was measured for three samples of recombinant Eap at concentrations of 0.33, 0.66, and 1.32 mg mL−1, respectively, that had been dialyzed against a standard buffer (20 mM Tris [pH 8.0], 200 mM NaCl). Data were acquired over 54 h using an AnTi50 rotor equipped with six-sector cells and solution column heights of 12 mm at speeds of 20, 30, and 40 k.r.p.m. Equilibrium at each speed was achieved when sequential measurements over 3-h intervals resulted in experimental absorbance curves that were indistinguishable from one another.
From the absorbance at 280 nm, the observed molecular weight, Mw, was derived according to the following single particle model using the program Heteroanalysis (Cole 2004):
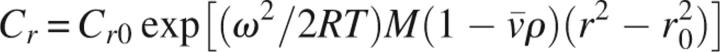 |
In this formalism, cr is the concentration at radius r, cr 0 is the concentration of the monomer at the reference radius r 0, ω is the angular velocity, R is the gas constant, T is the absolute temperature, and ρ is the solvent density. A specific volume of 0.77 mL g−1 for Eap was inferred from its sequence.
Proteolytic assays and mass spectrometry
Purified, recombinant Eap was subjected to limiting proteolysis by both subtilisin A (ICN Biomedical) and trypsin (Sigma-Aldrich, Inc.). Five micrograms of Eap (dissolved in double-deionized water in a total reaction volume of 10 μL) were incubated with a series of mass ratio dilutions of each protease at 25°C for 60 min. Following the incubation, the entire proteolysis reaction was separated by 4%–20% gradient SDS polyacrylamide gel electrophoresis and the distinct protein species contained therein were visualized by Coomassie Blue staining.
Nine gel bands representing distinct, subtilisin-generated Eap fragments (as shown in Fig. 2A) were excised and processed for in-gel trypsin digestion as described by Kinter and Sherman (2000). The resulting tryptic peptides were extracted, separated by capillary liquid chromatography (LC), and analyzed by tandem mass spectrometry (Thermo Finnigan LTQ linear ion trap) as described (Keightley et al. 2004), but with eight data-dependent scans rather than three. The LC-MS/MS data were searched for peptides with nontryptic ends by using the “no enzyme” option (Sequest, BioWorks 3.1 SR1) to identify distinguishable (nontryptic) subtilisin-sensitive sites. Identified peptides were initially filtered by charge state/Xcorr and then manually inspected for validation (MacCoss et al. 2002; Starkweather et al. 2007).
Collection of small angle X-ray scattering data
SAXS measurements were carried out at 20°C using BioCAT beamline 18-ID of the Advanced Photon Source of Argonne National Laboratory. Samples of Eap were prepared in a buffer of 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, and 5% (v/v) glycerol. All samples were filtered through a Millex Microfilter membrane (pore size 0.22 μm) to eliminate any existing aggregates immediately before each measurement. All experiments were performed using a wavelength of 1.0 Å and a sample-detector distance of 2.35 m, which resulted in scattering vectors, q, ranging from 0.006 to 0.21 Å−1. The scattering vector is defined as q = 4π / λ sin θ, where 2θ is the scattering angle. The detector was an X-ray intensified optically coupled CCD camera, and 20 successive frames of 2 s exposure time were recorded for each sample. To avoid radiation-induced protein damage, the samples were continuously recirculated through a quartz capillary. Each frame was carefully checked for possible bubble formation or radiation-induced aggregation in the protein sample. If such effects were not observed, the individual frames were averaged. Background scattering was quantified before or after measurement of the protein sample and then subtracted from the protein patterns.
Analysis of SAXS data
Background-subtracted scattering data were subjected to indirect Fourier transformation using the program GNOM (Svergun 1992). Radii of gyration (RG) were determined by Guinier approximation using the low q-regions of the scattering profiles, which also allowed extrapolation of the scattering intensity to the zero angle, I(0). The relative I(0)/c values measured from samples prepared in an identical buffer during a single data collection session gave the relative molecular mass of the proteins when referenced against a suitable standard (Mylonas and Svergun 2007). Regression analysis of 5 I(0)/c values measured during a single session at beamline 18-ID for proteins in the range of 9–81 kDa showed a linear relationship that was used to derive experimental values for molecular mass, as presented in Figure 3D. The GNOM program was also used to compute the pair-distance distribution function, P(r), which represents the probability of finding a point within the observed particle at a distance, r, from a defined point of reference. Integration of this probability over the surface of a sphere with radius, r, and over the volume, V, yielded the P(r) function as expressed by
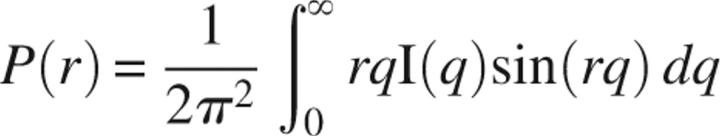 |
This approach offered an alternative calculation of RG and I(0) (Glatter 1982), which is based on the entire scattering curve and gives the maximum dimension of the macromolecule, D max, as the P(r) function approaches zero.
Ab initio deconvolution of the SAXS profiles
The low-resolution shape of full-length Eap was restored from the experimental data using the program GASBOR (Svergun et al. 2001). Scattering profiles up to q max = 0.21 Å−1 were used for the fit. GASBOR provides a chain-compatible spatial distribution of an exact number of dummy residues corresponding to the distribution of Cα atoms within a given polypeptide chain. In this approach, the number of amino acid residues for Eap was specified at 479, which includes three additional residues at the N terminus that are an artifact of the subcloning and expression protocol. The optimal configuration for each model was achieved by minimizing the following discrepancy function:
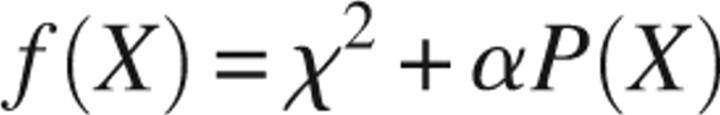 |
between the calculated and experimental curves using simulated annealing methods. In this equation, X represents the discrepancy between calculated and experimental curves, and αP(X) defines a quality of fit penalty with positive weight for all α > 0. The aim of this method is to modify the coordinates of the dummy residues randomly while favoring configurations that decrease the overall energy, f(X). Ten low-resolution models obtained from independent runs were averaged using the program DAMAVER (Volkov and Svergun 2003) to construct an average model representing the general structural features of full-length Eap.
Atomic modeling of Eap
An atomic model for the entire Eap molecule was generated using a three-step approach. First, model structures for the individual domains EAP1, EAP3, and EAP4 were generated using the Swiss Modeler program (Guex and Peitsch 1997) and the 1.35 Å resolution crystal structure of EAP2 (RCSB accession code 1YN3) as a template (Geisbrecht et al. 2005). The high levels of sequence identity between the modeled EAP domains and EAP2 (e.g., EAP1 86%, EAP3 53%, and EAP4 65%) indicate that these proteins have highly similar tertiary structures. Next, the resulting models for all four individual EAP domains were positioned into the low-resolution deconvolution of full-length Eap by GASBOR. Finally, atomic coordinates for the missing linker residues (Fig. 5) located between the individual modules were modeled using the program TURBO (Roussel and Cambillau 1989). This atomic model of Eap was used as an initial model for rigid body modeling by the molecular dynamics approach described below.
Rigid body modeling by molecular dynamics
Conformational sampling by molecular dynamics simulation (MD) was used to conduct a search for potential Eap conformations that most closely matched the experimental SAXS data. The program CHARMM, version 28.b1 (Brooks et al. 1983) with the all-atom CHARMM force field version 22.0 (MacKerell et al. 1998) was employed for MD simulations. The initial atomic model of full-length Eap, as described above, was taken as the starting point of the simulations. In all cases, only the atoms of the linker were allowed to move relative to each other, while the EAP modules were treated as rigid bodies with no internal motion. The crystal structures (Geisbrecht et al. 2005) of three distinct EAP modules suggest that the EAP modules themselves can be considered rigid globular bodies because the amplitudes of their internal motions are not detectable at the resolution achievable by SAXS.
Simulations were performed in implicit aqueous solvent by setting the relative dielectric constant to 80.0. A time interval of 1 fs was employed for simulations according to the following protocol. First, the system was subjected to energy minimization with harmonic constraints on the protein atoms, followed by 30 ps of molecular dynamic heating to 1500 K wherein the protein atoms were restrained in a fixed position. During the subsequent “production phase” (7.5 ns), the resulting structures of the protein were recorded every 0.5 ps in a trajectory file. MD simulations of full-length Eap were limited by constraints in RG max and min (36 < RG < 47 Å) to eliminate unrealistic conformations of Eap having RG values remote from those observed experimentally. To ensure the accuracy of the simulation for each registered conformation, the theoretical SAXS profile, the RG values, and the corresponding fit to the experimental scattering data were calculated using the program CRYSOL (Svergun et al. 1995).
Raman spectroscopy
Samples of both purified full-length Eap and a stoichiometric mixture of EAP domains 1, 2, 3, and 4 were prepared as described above, dialyzed exhaustively against Milli-Q H2O, and lyophilized for long-term storage until needed. Prior to collection of Raman spectra, each sample was dissolved in 10 mM Tris buffer (in pure H2O, pH 7.5 or pure D2O, pD 7.5) to a final protein concentration of 10 mg/mL as determined by UV absorption spectrophotometry. When applicable, the dissolved proteins were stored at 4°C for ∼20 h prior to acquisition of Raman spectra in order to achieve H/D exchange equilibrium. A 10-μL aliquot of the protein solution was sealed in a glass capillary (Kimax No. 34507), and the Raman spectrum was excited with the 532 nm line of a NdY:VO4 laser (Verdi5, Coherent Inc.). Radiant power at the sample was ∼100 mW. All Raman spectra were collected in the 90° scattering geometry using a Spex 500M single spectrograph (1200 groove/mm grating) equipped with a liquid nitrogen cooled CCD detector of 2000 × 800 pixel resolution. The wave numbers (cm−1 units) of all protein Raman bands were calibrated to ±1 cm−1 accuracy using toluene as the standard. Protein Raman spectra were corrected for both buffer and background contributions and the observed Raman intensities were normalized to the 1003 cm−1 band of phenylalanine as the internal standard (Benevides et al. 2003).
Electronic supplementary material
Supplemental Figure 1 is designed to accompany Figure 2 of the main text. This electronic figure displays the subtilisin-sensitive sites of Eap as they are found within the primary and secondary structure of the full-length protein. A brief interpretation of the proteolytic study is included in the legend. (Hammel_etal_SupplementalFig1.pdf).
Acknowledgments
We thank David Gore and the staff of Sector 18 of the Advanced Photon Source for technical assistance with collection of SAXS data. Access to the Advanced Photon Source was provided by the United States Department of Energy (GUP-4261 to B.V.G.). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. W-31-109-ENG-38. BioCAT is a National Institutes of Health-supported Research Center RR-08630. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Center for Research Resources or the National Institutes of Health. This research was supported by grant no. 2509 from the University of Missouri Research Board to B.V.G. and by the School of Biological Sciences at the University of Missouri–Kansas City. Support of the National Institutes of Health (Grant GM50776 to G.J.T.) is also gratefully acknowledged.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Brian V. Geisbrecht, Division of Cell Biology and Biophysics, School of Biological Sciences, University of Missouri–Kansas City, 5100 Rockhill Road, Kansas City, MO 64110, USA; e-mail: GeisbrechtB@umkc.edu; fax: (816) 235-1503.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073170807.
References
- Athanasopoulos A.N., Economopoulou, M., Orlova, V.V., Sobke, A., Schneider, D., Weber, H., Augustin, H.G., Eming, S.A., Schubert, U., Linn, T., et al. 2006. The extracellular adherence protein (Eap) of Staphylococcus aureus inhibits wound healing by interfering with host defense and repair mechanisms. Blood 107: 2720–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benevides J.M., Terwilliger, T.C., Vohnik, S., and Thomas Jr, G.J. 1996. Raman spectroscopy of the Ff gene V protein and complexes with poly(dA): Nonspecific DNA recognition and binding. Biochemistry 35: 9603–9609. [DOI] [PubMed] [Google Scholar]
- Benevides J.M., Overman, S.A., and Thomas Jr, G.J. 2003. Raman spectroscopy of proteins. In Current protocols in protein science (eds. J.E. Coligan et al.), pp. 17.18.11–17.18.35. Wiley, New York. [DOI] [PubMed]
- Boden M.K. and Flock, J.-I. 1992. Evidence for three different fibrinogen-binding proteins with unique properties from Staphylococcus aureus strain Newman. Microb. Pathog. 12: 289–298. [DOI] [PubMed] [Google Scholar]
- Brooks B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4: 187–217. [Google Scholar]
- Carey P.R. 1982. Biochemical applications of Raman and resonance Raman spectroscopies. Academic Press, London, UK.
- Chavakis T., Hussain, M., Kanse, S.M., Peters, G.B., Flock, J.-I., Herrmann, M., and Preissner, K.T. 2002. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat. Med. 8: 687–693. [DOI] [PubMed] [Google Scholar]
- Chavakis T., Wiechmann, K., Preissner, K.T., and Herrmann, M. 2005. Staphylococcus aureus interactions with the endothelium: The role of bacterial “secreteable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems. Thromb. Haemost. 94: 278–285. [DOI] [PubMed] [Google Scholar]
- Cole J.L. 2004. Analysis of heterogeneous interactions. Methods Enzymol. 384: 212–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields B.A., Malchiodi, E.L., Li, H., Ysern, X., Stauffacher, C.V., Schlievert, P.M., Karjalainen, K., and Mariuzza, R. 1996. Crystal structure of a T-cell receptor β-chain complexed with a superantigen. Nature 384: 188–192. [DOI] [PubMed] [Google Scholar]
- Flock M. and Flock, J.-I. 2001. Rebinding of extracellular adherence protein Eap to Staphylococcus aureus can occur through a surface-bound neutral phosphatase. J. Bacteriol. 183: 3999–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbrecht B.V., Hamaoka, B.Y., Perman, B., Zemla, A., and Leahy, D.J. 2005. The crystal structures of EAP domains from Staphylococcus aureus reveal an unexpected homology to bacterial superantigens. J. Biol. Chem. 280: 17243–17250. [DOI] [PubMed] [Google Scholar]
- Geisbrecht B.V., Bouyain, S., and Pop, M. 2006. An optimized system for the expression and purification of secreted bacterial proteins. Protein Expr. Purif. 46: 23–32. [DOI] [PubMed] [Google Scholar]
- Glatter O. 1982. Interpretation. In Small angle X-ray scattering (eds. O. Glatter and O. Kratky), pp. 167–196. Academic Press, London, UK.
- Guex N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18: 2714–2723. [DOI] [PubMed] [Google Scholar]
- Haas P.J., de Haas, C.J., Poppelier, M.J., van Kessel, K.P., van Strijp, J.A., Dijkstra, K., Scheek, R.M., Fan, H., Kruijtzer, J.A., Liskamp, R.M., et al. 2005. The structure of the C5a receptor-blocking domain of chemotaxis inhibitory protein of Staphylococcus aureus is related to a group of immune evasive molecules. J. Mol. Biol. 353: 859–872. [DOI] [PubMed] [Google Scholar]
- Haggar A., Hussain, M., Lonnies, H., Herrmann, M., Norrby-Teglund, A., and Flock, J.-I. 2003. Extracellular adherence protein from Staphylococcus aureus enhances internalization into eukaryotic cells. Infect. Immun. 71: 2310–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M., Walther, M., Prassl, R., and Kuhn, H. 2004. Structural flexibility of the N-terminal β-barrel domain of 15-lipoxygenase-1 probed by small angle X-ray scattering. Functional consequences for activity regulation and membrane binding. J. Mol. Biol. 343: 917–929. [DOI] [PubMed] [Google Scholar]
- Hammel M., Fierobe, H.P., Czjzek, M., Kurkal, V., Smith, J.C., Bayer, E.A., Finet, S., and Receveur-Brechot, V. 2005. Structural basis of cellulosome efficiency explored by small angle X-ray scattering. J. Biol. Chem. 280: 38562–38568. [DOI] [PubMed] [Google Scholar]
- Hammel M., Sfyroera, G., Ricklin, D., Magotti, P., Lambris, J.D., and Geisbrecht, B.V. 2007. A structural basis for complement inhibition by Staphylococcus aureus . Nat. Immunol. 8: 430–437. [DOI] [PubMed] [Google Scholar]
- Harraghy N., Hussain, M., Haggar, A., Chavakis, T., Sinha, B., Herrmann, M., and Flock, J.-I. 2003. The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap. Microbiol. 149: 2701–2707. [DOI] [PubMed] [Google Scholar]
- Hussain M., Haggar, A., Heilmann, C., Peters, G., Flock, J.-I., and Herrmann, M. 2002. Insertional inactivation of Eap in Staphylococcus aureus strain Newman confers reduced staphylococcal binding to fibroblasts. Infect. Immun. 70: 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson K., McDevitt, D., Homonylo, M., Patti, J.M., and Hook, M. 1995. Staphylococcus aureus expresses a major histocompatibility complex class II analog. J. Biol. Chem. 270: 21457–21460. [DOI] [PubMed] [Google Scholar]
- Keightley J.A., Shang, L., and Kinter, M. 2004. Proteomic analysis of oxidative stress-resistant cells. Mol. Cell. Proteomics 3: 167–175. [DOI] [PubMed] [Google Scholar]
- Kinter M. and Sherman, N.E. 2000. Protein sequencing and identification using tandem mass spectrometry. Wiley-Interscience, New York.
- Kuroda M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., Nagai, Y., et al. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus . Lancet 357: 1225–1240. [DOI] [PubMed] [Google Scholar]
- Leach A.R. 2001. Exploring conformational space using simulation methods. In Molecular modelling; Principles and applications (ed. P. Hall), 2nd ed, pp. 457–508. Pearson Education, Harlow, UK.
- Lee L.Y., Miyamoto, Y.J., McIntyre, B.W., Hook, M., McCrea, K.W., McDevitt, D., and Brown, E.L. 2002. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J. Clin. Invest. 110: 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L.Y.L., Liang, X., Hook, M., and Brown, E.L. 2004. Identification and characterization of the C3 binding domain of the Staphylococcus aureus extracellular fibrinogen-binding protein (Efb). J. Biol. Chem. 279: 50710–50716. [DOI] [PubMed] [Google Scholar]
- Lowy F.D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339: 520–532. [DOI] [PubMed] [Google Scholar]
- MacCoss M.J., McDonald, W.H., Saraf, A., Sadygov, R., Clark, J.M., Tasto, J.J., Gould, K.L., Wolters, D., Washburn, M., Weiss, A., et al. 2002. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. 99: 7900–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell J.A.D., Bashford, D., Bellott, M., Dunbrack, R.L.J., Evenseck, J.D., Field, M.J., Ficher, S., Gao, J., Guo, H., Ha, S., et al. 1998. All-atom empirical potential for molecular modeling an dynamics studies of proteins. J. Phys. Chem. B 102: 3586–3616. [DOI] [PubMed] [Google Scholar]
- McGavin M.H., Krajewska-Pietrasik, D., Ryden, C., and Hook, M. 1993. Identification of a Staphylococcus aureus extracellular matrix-binding protein with broad specificity. Infect. Immun. 61: 2479–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalyi E. and Godfrey, J.E. 1963. Digestion of fibrinogen by trypsin. II. Characterization of the large fragment obtained. Biochim. Biophys. Acta 67: 90–103. [DOI] [PubMed] [Google Scholar]
- Millett I.S., Doniach, S., and Plaxco, K.W. 2002. Toward a taxonomy of the denatured state: Small angle scattering studies of unfolded proteins. Adv. Protein Chem. 62: 241–262. [DOI] [PubMed] [Google Scholar]
- Mylonas F. and Svergun, D.I. 2007. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 40: Suppl: s245–s249. [Google Scholar]
- Palma M., Haggar, A., and Flock, J.-I. 1999. Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J. Bacteriol. 181: 2840–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou A.C. and Acharya, K.R. 1997. Superantigens as immunomodulators: Recent structural insights. Structure 5: 991–996. [DOI] [PubMed] [Google Scholar]
- Patti J.M., Allen, B.L., McGavin, M.J., and Hook, M. 1994. MSCRAMM—Mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48: 585–617. [DOI] [PubMed] [Google Scholar]
- Roussel A. and Cambillau, C. Silcon Graphics Committee. 1989. TURBO-FRODO. In Silicon Graphics Geometry Partner Directory, pp. 77–78. Silicon Graphics, Mountain View, CA.
- Starkweather R., Barnes, C.S., Wyckoff, G.J., and Keightley, J.A. 2007. Virtual polymorphism: Finding divergent peptide matches in mass spectrometry data. Anal. Chem. 79: 5030–5039. [DOI] [PubMed] [Google Scholar]
- Svergun D. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25: 495–503. [Google Scholar]
- Svergun D., Baraberato, C., and Koch, M.H. 1995. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28: 768–773. [Google Scholar]
- Svergun D.I., Petoukhov, M.V., and Koch, M.H. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80: 2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G.J. 1999. Raman spectroscopy of protein and nucleic acid assemblies. Annu. Rev. Biophys. Biomol. Struct. 28: 1–27. [DOI] [PubMed] [Google Scholar]
- Tuma R. and Thomas Jr, G.J. 1997. Mechanisms of virus assembly probed by Raman spectroscopy: The icosahedral bacteriophage P22. Biophys. Chem. 68: 17–31. [DOI] [PubMed] [Google Scholar]
- Tuma R., Parker, M.H., Weigele, P., Sampson, L., Sun, Y., Krishna, N.R., Casjens, S., Thomas Jr, G.J., and Previlige Jr, P.E. 1998a. A helical coat protein recognition domain of the bacteriophage P22 scaffolding protein. J. Mol. Biol. 281: 81–94. [DOI] [PubMed] [Google Scholar]
- Tuma R., Prevelige Jr, P.E., and Thomas Jr, G.J. 1998b. Mechanism of capsid maturation in a double-stranded DNA virus. Proc. Natl. Acad. Sci. 95: 9885–9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V.V. and Svergun, D.I. 2003. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36: 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C., Alcaide, P., Geisbrecht, B.V., Schneider, D., Herrmann, M., Preissner, K.T., Luscinskas, F.W., and Chavakis, T. 2006. Suppression of experimental autoimmune encephalomyelitis by extracellular adherence protein of Staphylococcus aureus . J. Exp. Med. 203: 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]