

Abstract
Pathogens have evolved numerous strategies to infect their hosts, while hosts have evolved immune responses and other defenses to these foreign challenges. The vast majority of host–pathogen interactions involve protein–protein recognition, yet our current understanding of these interactions is limited. Here, we present and apply a computational whole-genome protocol that generates testable predictions of host–pathogen protein interactions. The protocol first scans the host and pathogen genomes for proteins with similarity to known protein complexes, then assesses these putative interactions, using structure if available, and, finally, filters the remaining interactions using biological context, such as the stage-specific expression of pathogen proteins and tissue expression of host proteins. The technique was applied to 10 pathogens, including species of Mycobacterium, apicomplexa, and kinetoplastida, responsible for “neglected” human diseases. The method was assessed by (1) comparison to a set of known host–pathogen interactions, (2) comparison to gene expression and essentiality data describing host and pathogen genes involved in infection, and (3) analysis of the functional properties of the human proteins predicted to interact with pathogen proteins, demonstrating an enrichment for functionally relevant host–pathogen interactions. We present several specific predictions that warrant experimental follow-up, including interactions from previously characterized mechanisms, such as cytoadhesion and protease inhibition, as well as suspected interactions in hypothesized networks, such as apoptotic pathways. Our computational method provides a means to mine whole-genome data and is complementary to experimental efforts in elucidating networks of host–pathogen protein interactions.
Keywords: host–pathogen interactions, protein–protein interactions, comparative modeling, protein interaction prediction, neglected tropical diseases
Genome sequencing has changed the scale and diversity of biomedical problems amenable to investigation as complete sequences are now available for many species, including human and a number of biomedically relevant microbes (Guttmacher and Collins 2005). Functional insights into the proteins encoded by these genomes are emerging from technical advances such as three-dimensional structure determination and the detection of genetic and physical interactions (Westbrook et al. 2002; Bader et al. 2003). However, in general, the wealth of genomic information available for both human host and pathogens remains unmined due to the lack of whole-genome protocols that can predict host–pathogen interactions.
Pathogens have evolved numerous strategies to successfully invade their hosts, acquire nutrients, and evade their immune defenses (Munter et al. 2006). These strategies often involve direct interactions between host and pathogen molecules, including the formation of protein complexes (Stebbins 2005). Much remains to be learned about the network of interactions between host and pathogen proteins. If the intraspecies interaction network of Saccharomyces cerevisiae is a guide, several independent large-scale studies are likely required for a comprehensive mapping of host–pathogen interactions (Collins et al. 2007).
Interactions between host and pathogen proteins are typically studied using traditional small-scale biochemical and genetic experiments, which focus on one protein or pathway at a time. Large-scale interaction discovery methods, such as tandem affinity purification and yeast–two-hybrid experiments, enable more comprehensive detection but at the cost of significant false-negative and false-positive error rates (Hart et al. 2006). Computational methods have demonstrated utility in improving the coverage, accuracy, and efficiency of identifying protein–protein interactions in combination with experimental data sets (Jansen et al. 2003; Lee et al. 2004) and are likely to similarly complement large-scale experimental efforts to characterize host–pathogen interaction networks.
Here we hypothesize that host–pathogen protein interactions, knowledge of which is severely lacking, can be inferred from the growing body of experimentally observed interactions, which is reaching saturation in some species. We previously showed that this approach can be useful in predicting intraspecies interactions (Davis et al. 2006). We now provide three additional lines of evidence that suggest the hypothesis is a valid one and that the developed protocol can predict functionally relevant host–pathogen protein interactions. The protocol identifies pairs of host and pathogen proteins with similarity to proteins known to interact, assesses the likelihood of interaction based on structural modeling, and then identifies those pairs with a greater chance of encounter as suggested by their subcellular location and expression properties. The result of the protocol is an enriched candidate set that is suitable for subsequent experimental study. We have applied the protocol to 10 human pathogens, including species of mycobacteria, kinetoplastida, and apicomplexa, which are responsible for “neglected” human diseases. These pathogens cause tropical diseases with a significant global burden, infecting over 1 billion people and incurring over 1 million annual deaths (World Health Organization 2003).
We first describe the protocol, detailing the data sources, the computations used, and its performance on intraspecies protein interactions in S. cerevisiae. We then present the predictions made for the 10 pathogens and assess them by three independent computational procedures. We then discuss the observed performance of the method and potential future improvements. We present several specific predictions that warrant experimental follow-up. Finally, we conclude by discussing the implications of these results for understanding the molecular mechanisms of pathogenesis.
Results
The protocol begins with the target set of host and pathogen protein sequences (Materials and Methods) (Fig. 1).
Figure 1.

Prediction protocol. The protocol begins with the set of host and pathogen proteins. Sequence matching procedures are then used to identify similarities between the host or pathogen proteins and proteins with known structure or known interaction partners. A structure-based statistical potential assessment, or a sequence similarity score in the absence of structure, is then used to predict interacting partners. Finally, this set of potential interactions is filtered using the biological contexts of the host and pathogen proteins and a network-level filter. The protocol reduces the number of potential P. falciparum–human protein interactions by about five orders of magnitude (Table 2).
Detecting sequence and structure similarities and identifying pairs of proteins with similarity to known complexes
Similarities were first detected between the target sequences and components of known protein complexes, using an automated comparative protein structure modeling pipeline. The fraction of the pathogen proteomes for which a suitable interaction template was identified varied from 16% of Trypanosoma cruzi sequences to 25% of Cryptosporidium parvum sequences, while the human proteome coverage was 34% (Table 1).
Table 1.
Interaction template and biological data coverage of the genomes analyzed

Pairs of host and pathogen proteins that each had detectable similarity to components of a known interaction were then identified. The number of these pairs varied widely among the pathogens, with the prokaryotes having far fewer pairs than the eukaryotes (Table 2, column 2). For example, 43,528 host–pathogen protein pairs were identified for Mycobacterium tuberculosis (3954 sequences, 18% template coverage), while 160,952 pairs were identified for Cryptosporidium hominis with approximately the same proteome size and interaction template coverage (3886 sequences, 20% template coverage). Among the eukaryotic pathogens, the number of pairs varied approximately in proportion to the proteome sizes (Tables 1, 2).
Table 2.
Potential interaction set reduction by assessment and filtering

Assessing the sequence or structural basis of the potential interactions
Next, the sequence or structural basis of interaction between the identified pairs was assessed using sequence similarity and statistical potential scores, respectively. This step identified ∼5% of the host–pathogen pairs identified in the previous step as possible interacting partners (Table 2), almost all (99.5%) of which were based on structural templates. The minimal contribution of sequence-based templates to the predictions is due to the stringent joint sequence identity threshold (≥80%; Materials and Methods) required to reliably transfer interactions (Yu et al. 2004; Mika and Rost 2006). The reduction in the number of pairs by the assessment step was greatest for the Toxoplasma gondii–human pairs, of which only 3.4% passed the scoring thresholds. As expected from the number of host–pathogen protein pairs with interaction templates, fewer predictions were made for the prokaryotic than for the eukaryotic pathogens.
Applying biological and network-level filters
The interactions were then filtered by the biological context of their component proteins, such as life-cycle stage and tissue expression, and by network-level information regarding the template usage frequencies. Interactions that met at least one host and one pathogen biological criterion were considered to pass the biological context filter (Materials and Methods) (Table 1; see Table 4 and Supplemental Table S3). Next, the network-level filter flagged those predictions based on templates that were used for more than 1% of the total predictions, as these predictions exhibited a low level of interaction specificity. For example, many pairs of G-protein subunits α and β were predicted to interact based on the crystal structure of the G-protein Gi heterotrimer (Protein Data Bank [PDB] 1GG2).
Table 4.
Host–tissue filters used for each pathogen

The filters resulted in a wide range of reductions in predicted interactions (Table 2), due to the different levels of biological annotation used for the genomes. For example, Plasmodium falciparum had the highest biological annotation coverage (88%) and, as expected, the highest fraction of interactions that passed the biological and network-level filters (13%). This final set of P. falciparum–human interactions is five orders of magnitude smaller than the initial set of all possible protein pairs. The low coverage of biological annotation for other pathogens was also evident, as filtering the predictions for two pathogens, Trypanosoma brucei and T. gondii, resulted in removal of all interactions. The type of annotation available for the pathogen proteins is particularly important. For example, both T. brucei and T. cruzi have biological annotation for 45% of their proteomes (Table 1); however, filtering results in zero interactions for the former and 914 for the latter. This difference occurs because life cycle annotation is available for 1930 (10%) of T. cruzi proteins but only 120 (1%) of T. brucei proteins (Supplemental Table S3). The majority of the biological annotations are GO terms that do not pass the filtering criteria.
Assessment
Next, the predictions were assessed to characterize the coverage and accuracy of the method. Coverage refers to the fraction of interactions that are accessible by the method, and accuracy refers to the fraction of the covered interactions that were correctly identified. The structure- and sequence-based prediction methods have both been previously benchmarked in the context of intraspecies interactions (Yu et al. 2004; Davis et al. 2006), and the results are briefly described in Materials and Methods. In contrast to interspecies interactions, large experimental data sets of thousands of intraspecies interactions are available and ideal for benchmarking prediction methods. These benchmarking results remain informative in the host–pathogen context as the underlying physichochemistry remains the same. We assessed the quality of the protocol in the host–pathogen context in three additional ways.
Assessment I: Comparison of predicted and known host–pathogen protein interactions
The predicted interactions were first compared with the set of known host–pathogen interactions (Supplemental Table S1), which although too small to rigorously assess the method, still allow insight into the performance of the method. Our protocol recovered four of the 33 host–pathogen protein interactions published in the literature for the 10 pathogen species. Other known interactions were not identified because of the lack of available templates. None of these latter cases was due to incorrect assessment by our method (Fig. 1, step 3). As expected, this result suggests that currently, a limitation of the protocol's coverage is the restriction to interactions with an appropriate template.
No interactions have been previously identified for three of the species we studied, Leishmania major, C. hominis, and C. parvum. The method recovered 67% (n = 2) of the known T. brucei–human interactions. One of these interactions, an ornithine decarboxylase (ODC) interspecies dimer whose physiological relevance has not been established, was later filtered out of the predictions because it was based on a homodimer template. For the species with the most observed interactions, P. falciparum and T. cruzi, the method recovered 9% (n = 1) and 8% (n = 1) of the previously observed interactions, respectively. In both cases, the interactions were protease–protease inhibitor interactions.
Assessment II: Comparison to gene expression and essentiality data
Next, we compared our prefiltered predictions to genome-scale data sets describing pathogen genes involved in M. tuberculosis infection and human genes involved in L. major, M. tuberculosis, and T. gondii infections. These comparisons were performed because genomic studies are, so far, the only source of large-scale data sets describing host–pathogen interactions, even though only weak correlation has been observed between physical protein interactions and expression data (Mrowka et al. 2001; Jansen et al. 2002).
Previous studies have identified 194 M. tuberculosis genes that are essential for in vivo infection (Sassetti and Rubin 2003) and 286 genes that are up-regulated in granuloma, pericavity, or distal lung infection sites compared with in vitro conditions (Rachman et al. 2006). Comparison of these two sets of genes to the set of M. tuberculosis proteins predicted to interact with human proteins revealed minimal overlap (Supplemental Table S2). In fact, only one gene occurs in both experimental data sets and our predictions: Rv3910 (GI 15611046), a probable conserved trans-membrane protein. The overlap of our predictions with the set of genes up-regulated during infection (23 genes) is greater than that between the two experimental sets of up-regulated genes and genes essential for infection (18 genes).
Previous studies have identified human genes that are differentially regulated in response to a variety of protozoal infections, in particular within the macrophage and dendritic cells of the immune system (Chaussabel et al. 2003). The human proteins predicted to interact with L. major, M. tuberculosis, and T. gondii include, respectively, 231, 78, and 169 proteins encoded by genes differentially expressed in macrophages and dendritic cells upon infection by these pathogens (Supplemental Table S2B) (Chaussabel et al. 2003).
Assessment III: Functional overview of predicted interactions
Finally, we evaluated the functional relevance of the predicted interactions by searching for functional annotations of proteins that were significantly enriched in the human proteins predicted to interact with pathogens, compared with the whole human proteome. This analysis was done before the application of the biological filters to prevent introduction of filter bias into the functional profile of the predictions.
The human proteins predicted to interact with pathogen proteins were significantly enriched in several gene ontology terms (Table 3). For example, the human proteins predicted to potentially interact with M. tuberculosis are enriched in cellular component terms that make sense in light of known mechanisms of tuberculosis infection including immunological synapse (7.7-fold enrichment, P = 10−3), T-cell receptor complex (8.5-fold enrichment, P = 1.6 × 10−2), and autophagic vacuole (17.1-fold enrichment, P = 3 × 10−4). These terms all reflect the known immunobiology of this pathogen, which elicits a T-cell response and was recently found to be eliminated through autophagy (Gutierrez et al. 2004; Deretic 2006; Singh et al. 2006; Vergne et al. 2006). Similarly, the human proteins predicted to interact with P. falciparum proteins are enriched in terms such as extrinsic to plasma membrane (5.2-fold enrichment, P = 9.2 × 10−15) and homophilic cell adhesion (4.2-fold enrichment, P = 2.8 × 10−21).
Table 3.
Functional annotation of human proteins predicted to interact with M. tuberculosis

The enriched functional terms that have not been previously implicated in infection represent either novel biological insights or false positives. Distinguishing between these two possibilities requires experiments beyond the scope of this paper. However, some of the enriched terms suggest that false positives could be identified and discarded if they arise from conservation of core cellular components. For example, the conservation of core translation machinery across all divisions of life (Tatusov et al. 1997) could result in erroneously predicted interactions causing the enrichment in the human–P. falciparum network for eukaryotic translation elongation factor (7.4-fold, P = 8.4 × 10−4). Similarly, terms such as pyruvate deydrogenase activity (25.6-fold, P = 2.2 × 10−2) and asparate-tRNA ligase activity (24.4-fold, P = 5.3 × 10−5), which are enriched in the human proteins predicted to interact with M. tuberculosis, may also be false positives caused by the conservation of core cellular components, and could be filtered.
Discussion
We presented a protocol that reduces the number of host–pathogen protein pairs to an experimentally tractable set of predicted interactions, by a series of assessments: (1) identifying template interactions; (2) assessing the putative interaction, using structure if available; and, finally, (3) filtering using biological context and network-level information (Fig. 1; Tables 1, 2, 4; Supplemental Table S3). For example, the procedure resulted in a five order of magnitude reduction in the number of possible human–P. falciparum protein interactions (Table 2). Although it is not possible to directly assess the enrichment of true interactions in the predictions, previous assessment in the context of S. cerevisiae interactions found an enrichment of about two orders of magnitude (Materials and Methods). In addition, assessment of the method by comparison to known host–pathogen interactions (Supplemental Table S1), genomics data (Supplemental Table S2), and functional analysis (Table 3) suggests that the method is capable of enriching for functionally relevant interactions.
We now discuss the observed performance of the method, present several specific predictions and their support in the literature, and close by discussing future developments and applications of the method to characterize host–pathogen and other types of interspecies interactions.
Limitations in coverage
The performance of the method can be characterized by two factors: coverage, describing the fraction of all interactions covered by the method, and accuracy, describing the fraction of the covered interactions that were correctly identified.
The main factor that limits the coverage of our method is that, like all comparative approaches, it depends on previous experimental observations of similar interactions. Despite the limited coverage, reflected in the low number of known interactions recovered by the method (four of 33), the availability of structure enables a more rigorous assessment of the interactions than that allowed by sequence alone (Fig. 2; Davis et al. 2006). As experimental efforts identify more interactions and further characterize the biology of host and pathogen proteins, the increased number of templates and expanded biological context data will increase the coverage and accuracy of our method, respectively.
Figure 2.

Example of a validated prediction: falcipain-2–cystatin-A. (A) An interaction was predicted between falcipain-2 and cystatin-A based on a template structure of cathepsin-H (orange) bound to cystatin-A (teal) (PDB 1NB3). (B) The structure of falcipain-2 bound to chicken cystatin was recently experimentally determined (PDB 1YVB). Although the interaction is experimentally verified, the question remains whether it would occur in vivo. Figures were generated by PyMOL (http://www.pymol.org).
Another factor that limits the coverage of our method is that the template identification procedure is primarily restricted to domain-mediated interactions, although peptide-mediated interactions are also known to contribute to protein interaction networks (Neduva and Russell 2006). Peptide motifs that mediate protein interactions are being identified through a combination of computational and experimental methods (Tong et al. 2002; Neduva et al. 2005), and application of these motif-based methods will likely expand the coverage of host–pathogen protein interactions.
Errors in accuracy
Several factors affect the accuracy of the method. These include errors in the comparative modeling process (Marti-Renom et al. 2000), the coarse-grained nature of the statistical potential used to assess the interface residue contacts (Davis et al. 2006), and consideration of only interactions between individual domains (i.e., incorrectly predicting interactions that are unfavorable in the context of the full-length proteins). While these three sources of error affect both intraproteomic and host–pathogen protein interactions, an additional type of error uniquely affects interspecies interactions. As the pathogen and host species are both eukaryotic for eight of the 10 pathogens studied, many of the predicted interactions are between core cellular components, such as translation machinery, metabolic enzymes, and ubiquitin-signaling components (Table 3). Although these interactions could potentially occur if the host and pathogen proteins encountered one another, their availability for such an encounter is not guaranteed. We used biological data, such as known exported pathogen proteins and known host–tissue targets, to address the “accessibility” issue. However, the precise spatial and temporal locations of these proteins are generally difficult to characterize. We expect this last source of errors to be diminished when the evolutionary distance between pathogen and host is greater, such as between bacterial or viral pathogens and their human hosts.
Specific examples of validated predictions
We now describe two examples of predicted interactions that have been previously observed experimentally. We predicted several interactions between proteases and protease inhibitors, the best scoring of which occurred between P. falciparum falcipain-2 protease and the human cystatin-A inhibitor based on a template structure of human cathepsin-H bound to cystatin-A (PDB 1NB3) (Fig. 2A). This prediction was recently experimentally validated, with chicken cystatin (PDB 1YVB) (Fig. 2B; Wang et al. 2006). This crystal structure was not present in our template set, because it has not yet been classified by the SCOP domain annotation database (Materials and Methods) (Murzin et al. 1995). Thus, the predicted complex was a true blind prediction. The experimentally determined structure provides direct validation of our prediction, although it does not demonstrate relevance to infection. However, the known involvement of cysteine proteases in malaria pathogenesis and experimentally established cross-talk between host and pathogen protease and inhibitors (Pandey et al. 2006) suggests that the interaction may play a role during infection. This case is an example where structure is important both in making the prediction and in highlighting its potential relevance as a potential pharmacologic target. Falcipain-2 and cathepsin-H share only 34% sequence identity, beyond the threshold of the sequence-based method required for a reliable prediction of interaction (Yu et al. 2004). However, comparison of the experimental falcipain-2–cystatin structure with the template cathepsin-H–cystatin-A structure reveals a high degree of structural similarity at the interface (Cα RMSD of 0.43 Å). In addition, this structure can be used to search for small-molecules that may disrupt or mimic the target interaction.
We predicted several interspecies enzyme dimerizations, such as T. brucei ornithine decarboxylase (ODC) binding to human ODC. Functional dimerization of parasitic and host enzyme subunits have been previously observed, such as in T. brucei and mouse ODC (Osterman et al. 1994). Although both host and pathogen ODCs have been implicated in viral and protozoal infections (Kierszenbaum et al. 1987; Das Gupta et al. 2005; Singh et al. 2007), the in vivo relevance of these homodimer-like complexes is not clear, and thus, we generally removed predictions based on homodimer sequence templates or template structures of subunits classified in the same domain family (Materials and Methods). This restriction also facilitates visualization and analysis of the networks, although some true positive predictions may be lost.
Specific examples of predicted interactions
We now describe two specific examples of predicted interactions whose indirect support in the literature warrants experimental follow-up. Two additional examples are discussed in the Supplemental material.
We predicted that P. falciparum thrombospondin-related adhesive protein (TRAP, SSP2, PF13_0201) interacts with human Toll-like receptor 4 (TLR4, ENSP00000346893), based on a template structure of Glycoprotein IBα bound to Von Willenbrand factor (PDB 1M10) (Fig. 3A; Huizinga et al. 2002). TRAP, an immunogenic protein used as a component of several vaccine candidates (Hill 2006), was also predicted to interact with three other leucine-rich repeat proteins; however, the interaction with TLR4 had the most support from the biological filters. Single nucleotide polymorphisms have been observed in TLR4, a “pattern recognition module” involved in the innate immune response. These mutations are associated with an increased severity of malaria, although they fall outside of the region that was modeled here (Mockenhaupt et al. 2006). Analysis of TRAP sequence data from a Gambian P. falciparum population indicates that the gene is under strong selection for variation in the sequence, with peaks in this variation occurring in the A-domain that we predicted to interact with TLR4 (Weedall et al. 2007). The possible encounter of these two proteins is also supported by the known expression of TRAP on the parasite surface during the sporozoite stage of the plasmodium life cycle and of TLR4 in the liver. While alternative explanations are possible, the biological evidence and the structural predictions made here suggest that a TRAP–TLR4 interaction may play an in vivo role in infection.
Figure 3.

Examples of predicted interactions. (A) P. falciparum thrombospondin-related adhesive protein (TRAP) was predicted to interact with human Toll-like receptor 4 (TLR4) based on a structure of glycoprotein IBα (orange) bound to von Willenbrand factor (teal), respectively (PDB 1M10). (B) M. tuberculosis probable exported protein Rv0888 was predicted to interact with actin based on a structure of DNAse-I (orange) bound to actin (teal), respectively (PDB 1ATN). Figures were generated by PyMOL (http://www.pymol.org).
We predicted that M. tuberculosis probable exported protein Rv0888 (GI 15608028) may interact with several human α-actins (ENSP00000295137) based on the template structure of DNAse I bound to actin (PDB 1ATN) (Fig. 3B; Kabsch et al. 1990). The interaction between DNAse and actin is known to be strong enough to depolymerize actin (Kabsch et al. 1990), and so the predicted interaction could be involved in the observed M. tuberculosis rearrangement of host actin (Guerin and de Chastellier 2000), which has been hypothesized to be triggered by a secreted pathogen factor (Garcia-Perez et al. 2003).
Future developments
The identification of protein–protein interactions is an important problem that has inspired the development of numerous algorithms to predict them (Shoemaker and Panchenko 2007). Several of these methods rely on information such as genomic proximity, gene fission/fusion, phylogenetic tree similarity, gene co-occurrence, colocalization, co-expression, and other features that only make sense or are currently feasible in the context of a single genome. However, comparative approaches that infer interactions based on previously observed interactions remain applicable to host–pathogen protein interactions, including the sequence and structure-based methods we have used here (Yu et al. 2004; Davis et al. 2006). Other applicable methods include those that identify peptide motifs (Neduva and Russell 2006) or sequence signatures (Sprinzak and Margalit 2001) that mediate interactions.
Another possible extension of the presented method that may aide in the interpretation of the predictions is an analysis of the genetic polymorphisms at loci encoding for the proposed interacting proteins. If the host gene exhibits polymorphisms associated with infection severity or the pathogen gene exhibits a pattern of polymorphisms suggesting antigenic variation, for example, human TLR4 and P. falciparum TRAP (Fig. 3A), there may be greater reason to believe that the interaction is relevant to infection.
Potential impact
We developed a computational whole-genome method to study potential host–pathogen protein interactions and presented four lines of evidence that suggest it is a valid approach to enrich for these interactions. The method, like any experimental or computational method, has limitations in coverage and accuracy, as we have quantified to the best of our ability. Despite these limitations, our resource is valuable as it is the first attempt to provide large data sets enriched for host–pathogen protein interactions.
Knowledge of host–pathogen interactions is useful in the development of strategies to treat and prevent infectious diseases. These interactions may serve as pharmacologic targets, both for traditional drug discovery efforts aimed at disrupting individual pathogen proteins and for small molecule or antibody inhibitors of protein–protein interactions. The proposed interactions also highlight pathogen proteins that may be potential immunization targets.
We have also applied our method to 10 pathogens involved in human infectious diseases. The predictions are available on the Internet at http://salilab.org/hostpathogen and can be viewed and filtered according to criteria of interest to an investigator, such as particular host tissues or pathogen life-cycle stages. We hope that the predictions serve the larger biomedical research community in moving toward the goal of treating infectious diseases, in the “open source” model of the Tropical Disease Initiative, a decentralized, Web-based, community-wide effort where scientists from laboratories, universities, institutes, and corporations work together for a common cause (http://www.tropicaldisease.org) (Maurer et al. 2004). In closing, we expect our method to complement experimental methods in providing insight into the basic biology of host–pathogen systems, as well as other interspecies relationships that fall elsewhere on the mutualism–parasitism continuum.
Materials and Methods
The protocol began with the host and pathogen protein sequences: CryptoDB (Heiges et al. 2006), GeneDB (Hertz-Fowler et al. 2004), OrthoMCL-DB (Chen et al. 2006), PlasmoDB (Stoeckert Jr. et al. 2006), ToxoDB (Kissinger et al. 2003), TubercuList (http://genolist.pasteur.fr/TubercuList/) (Table 1).
Detecting sequence and structure similarities
First, protein structure models were calculated for all sequences using MODPIPE, our automated software pipeline for large-scale protein structure modeling (Eswar et al. 2003). MODPIPE relies on MODELLER (Sali and Blundell 1993) for its functionality and calculates comparative models for a large number of sequences using different template structures and sequence-structure alignments. Sequence-structure matches are established using a variety of fold-assignment methods, including sequence–sequence (Smith and Waterman 1981), profile–sequence (Altschul et al. 1997) (BUILD_PROFILE, a module for calculating sequence profiles in MODELLER), and profile–profile alignments (Marti-Renom et al. 2004) (PROFILE_SCAN, a module for fold-assignment using profile-–profile scanning in MODELLER). Increased sensitivity of the search for known template structures is achieved by using an E-value threshold of 1.0. Ten models are calculated for each of the sequence-structure matches to achieve a reasonable degree of conformational sampling (Sali and Blundell 1993). The best scoring model for each alignment is then chosen using a statistical potential (Shen and Sali 2006). Finally, all models generated for a given input sequence are evaluated for the correctness of the fold using a composite model quality criterion that includes the coverage of the model, sequence identity of the sequence-structure alignment, the fraction of gaps in the alignment, the compactness of the model, and statistical potential Z-scores (Melo et al. 2002; Eramian et al. 2006; Shen and Sali 2006). Only models that are assessed to have the correct fold were included in the final data sets. The models have been deposited in our database of comparative models, MODBASE (Pieper et al. 2006) (http://salilab.org/modbase), as publicly accessible data sets.
The detected structural similarities were then used to assign structural domain boundaries to the modeled sequences, according to the SCOP classification system (Murzin et al. 1995), as previously described (Davis et al. 2006). Briefly, domain boundaries were assigned to the target proteins when the putative domain contained at least 70% of the residues in the template domain. If the template-target domain similarity was more than 30% sequence identity, the target domain was classified at the family level of the template's domain classification. If the sequence identity was more than 30% and a reliable model was built or if the sequence identity was more than 30% but MODBASE deemed only a reliable fold assignment, the superfamily was assigned. The remaining target domains received the template domains SCOP classification at the fold level, and were not used in the interaction prediction.
Identifying pairs of proteins with similarity to known interactions and assessing the sequence or structural basis of the potential interactions
Next, pairs of host and pathogen proteins were searched for similarity to known interactions collected in PIBASE (Davis and Sali 2005) and IntAct (Kerrien et al. 2007). PIBASE (release 1.69) is a comprehensive relational database of structurally defined protein interfaces that currently includes 209,961 structures of interactions between 2613 SCOP domain families. As previously described, these structures were clustered and then filtered to remove potential crystallographic artifacts, resulting in a set of template binary interfaces of 5275 structures (Davis and Sali 2005). IntAct (release 2006-08-18) is an open source database of protein interaction data and contains 63,276 binary protein interactions (Kerrien et al. 2007).
Putative interactions between pairs of host and pathogen proteins that contained domains classified in the same superfamily as those previously observed to interact (PIBASE) were assessed by alignment of their comparative structure models onto the corresponding domains of the template complexes and by subsequent assessment of the putative interface by a statistical potential, as previously described (Davis et al. 2006). Briefly, pairs of residues from the host and pathogen protein models whose side chains occurred within a distance of 8 Å of one another were identified and their scores summed according to a statistical potential derived from binary interface structures in PIBASE. A Z-score was calculated to assess the significance of this raw statistical potential score, by consideration of the mean and standard deviation of the statistical potential scores for 1000 sequences where all amino acid residues in the target domain sequences were shuffled.
The ability of the statistical potential to discriminate a set of 100 true protein interfaces from a background set of 100,000 sequence-randomized decoys was previously assessed using a receiver-operator-curve (ROC) analysis (Davis et al. 2006). This ROC analysis exhibited an area under the curve (AUC) of 0.993 and suggested an optimal statistical potential Z-score threshold of −1.7, which gave true-positive and false-positive rates of 97% and 3%, respectively. Interactions predicted based on template complexes formed by protein domains from the same SCOP family were omitted from the analysis, because these predictions primarily consisted of multimeric enzyme complexes formed by both host and pathogen proteins, as well as core cellular components such as ribosome subunits and proteasome subunits.
Sequence profiles, built by MODPIPE, were searched for proteins that participate in binary protein interactions (IntAct) (Kerrien et al. 2007). Host and pathogen sequences were predicted to interact when each aligned to at least 50% of the sequence of members of a template complex with a joint sequence identity of 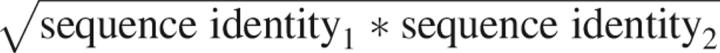 ≥ 80% (Yu et al. 2004). This threshold has been previously shown to correctly predict true protein–protein interactions (Yu et al. 2004). Interactions predicted based on homodimer templates were omitted from the analysis, because the predictions primarily consisted of complexes formed between corresponding core cellular components of host and pathogens (e.g., histones).
≥ 80% (Yu et al. 2004). This threshold has been previously shown to correctly predict true protein–protein interactions (Yu et al. 2004). Interactions predicted based on homodimer templates were omitted from the analysis, because the predictions primarily consisted of complexes formed between corresponding core cellular components of host and pathogens (e.g., histones).
Applying biological and network-level filters
The predicted interactions were filtered using biological context and network-level information. The biological context filter was imposed at two levels, individual proteins and their interactions (Table 4; Supplemental Table S3). The host proteins were filtered by expression in tissues known to be targeted by the pathogen (GNF Tissue Atlas [Su et al. 2004], Harrison's Principles of Internal Medicine [Kasper et al. 2004]), known expression on cell surface, and known immune system involvement (ENSEMBL [Hubbard et al. 2007], Gene Ontology Annotation [GOA] [Camon et al. 2004], IRIS [Abbas et al. 2005]) (Table 4). The pathogen proteins were filtered by known or predicted secretion, known expression on cell surface, infective life-cycle stage, and functional annotation to defense response mechanisms (PlasmoDB [Stoeckert Jr. et al. 2006], ToxoDB [Kissinger et al. 2003], CryptoDB [Heiges et al. 2006], GeneDB [references in Supplemental Table S1] [Hertz-Fowler et al. 2004]). The GO terms for human protein involvement in immune system were GO:0051707, GO:0002376, and GO:0006955. The GO terms for pathogen protein involvement in host–pathogen interactions were GO:00044419 (involved in defense response), GO:0043657 (cellular component: host cell), and GO:0009405 (pathogenesis). Potential interactions between human and pathogen proteins that each met at least one biological criterion were considered to pass the biological filter.
The second level of biological filters was applied simultaneously to both human and pathogen proteins, as follows: M. tuberculosis, pairs of human proteins expressed in lung tissue or bronchial epithelial cells and pathogen proteins up-regulated in granuloma, pericavity, or distal infection sites (Rachman et al. 2006); L. major, pairs of human proteins expressed in skin and pathogen proteins expressed in the promastigote or metacyclic life-cycle stage and human proteins expressed in blood and pathogen proteins expressed in amastigote life-cycle stage; T. brucei, pairs of human proteins expressed in blood and pathogen proteins expressed in the bloodstream life-cycle stage; P. falciparum, pairs of human proteins expressed in erythrocytes and pathogen proteins expressed in the merozoite life-cycle stage, known or predicted to be secreted, and found on the surface of infected erythrocytes and human proteins expressed in liver and pathogen proteins expressed in the sporozoite life-cycle stage; and Plasmodium vivax, pairs of human proteins expressed in erythrocyte and pathogen proteins predicted to be secreted.
The network-level filter removed predictions based on templates used for more than 1% of the total number of predictions in each host–pathogen network. This filter was imposed due to the lack of specificity in the predictions based on these highly used templates. On average, 15 interaction templates were removed from each run.
The filtering step was performed after the initial modeling and interaction prediction steps so that the filters could be easily updated to include biological annotation resulting from future experiments, without requiring re-calculation of models and interactions.
Assessment: Intraspecies interactions benchmark
The sequence- and structure-based prediction methods have both been previously benchmarked in the context of intraspecies S. cerevisiae protein interactions. For the sequence-based method, all of the interactions transferred from Caenorhabditis elegans, Drosophila melanogaster, and Helicobacter pylori onto S. cerevisiae were correct at a joint sequence identity threshold of 80% (Yu et al. 2004).
For the structure-based method, 270 of 3387 (8%) predicted S. cerevisiae interactions overlapped with experimentally observed interactions, 90% of which exhibited less than 80% sequence identity to the their interaction template (Davis et al. 2006). The use of orthogonal biological information as filters was found to provide a significant (threefold) enrichment of previously observed interactions. The method could not predict the correct specificities in families of homologous receptor-ligand networks, such as the epidermal growth factor receptor and tumor necrosis factor-β network of ligand receptor interactions. In total, 19,424 interactions have been experimentally observed out of the possible 21,776,700 pairs of yeast proteins (0.09%; Jan 2006) (Davis et al. 2006). Thus, the number of protein pairs was reduced by about four orders of magnitude, while the enrichment was increased by about two orders of magnitude. The analysis suggested that the method was applicable as a first pass for genome-wide predictions of protein complexes.
Assessment: Functional overview of predicted complexes
The human proteins predicted to interact with pathogen proteins were analyzed for significant enrichment of gene ontology function terms using GO∷TermFinder (Boyle et al. 2004). The analysis was done on the interactions before application of the biological filters to prevent introduction of filter bias into the functional profile of the predictions. The enrichment for a given GO term was computed as the ratio of the fraction of proteins in the predicted set annotated with the GO term to the fraction in the entire human genome. The significance of this enrichment was computed as a P-value with Bonferroni correction for multiple hypothesis testing (Sokal and Rohlf 1995).
Assessment: Comparison to gene expression and essentiality data
Human genes differentially regulated (two-tailed t-test, P < 0.05) in macrophages and dendritic cells during infection by L. major, M. tuberculosis, and T. gondii were retrieved from GEO Omnibus (GDS2600) (Edgar et al. 2002; Chaussabel et al. 2003). Lists of M. tuberculosis genes essential for in vivo infection (Sassetti and Rubin 2003) and genes that are up-regulated in granuloma, pericavity, or distal lung infection sites compared with in vitro conditions (Rachman et al. 2006) were obtained from literature.
Acknowledgments
We thank J. Cox (UCSF), E. Brown (UCSF), K. Kim (AECOM), and J. Boothroyd (Stanford) for their help in building the set of known host–pathogen protein interactions. We thank D. Shanmugam (U. Penn) for collating the protein sequences analyzed and U. Pieper (UCSF) for assistance with MODBASE. We thank T. Kortemme (UCSF) and members of the Sali laboratory for valuable comments and suggestions. We are also grateful for the support of the U.S. National Institutes of Health grants P01-A135707, R01-GM54762, U54 RR022220, U.S. National Science Foundation grant EIA-0325004, Human Frontier Science Program, The Sandler Family Supporting Foundation, Hewlett-Packard, NetApps, IBM, and Intel. F.P.D. acknowledges support from a Howard Hughes Medical Institute predoctoral fellowship.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Fred P. Davis, Janelia Farm Research Campus, 19700 Helix Drive, Ashburn, VA 20147, USA; e-mail: davisf@janelia.hhmi.org; fax: (571) 209-4943; or Andrej Sali, QB3 at Mission Bay, Suite 503B, University of California at San Francisco, 1700 4th Street, San Francisco, CA 94158, USA; e-mail: sali@salilab.org; fax: (415) 514-4231.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073228407.
References
- Abbas A.R., Baldwin, D., Ma, Y., Ouyang, W., Gurney, A., Martin, F., Fong, S., van Lookeren Campagne, M., Godowski, P., Williams, P.M., et al. 2005. Immune response in silico (IRIS): Immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 6 319–331. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G.D., Betel, D., and Hogue, C.W. 2003. BIND: The biomolecular interaction network database. Nucleic Acids Res. 31 248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle E.I., Weng, S., Gollub, J., Jin, H., Botstein, D., Cherry, J.M., and Sherlock, G. 2004. GO∷TermFinder-open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 20 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camon E., Magrane, M., Barrell, D., Lee, V., Dimmer, E., Maslen, J., Binns, D., Harte, N., Lopez, R., and Apweiler, R. 2004. The gene ontology annotation (GOA) database: Sharing knowledge in Uniprot with gene ontology. Nucleic Acids Res. 32 D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaussabel D., Semnani, R.T., McDowell, M.A., Sacks, D., Sher, A., and Nutman, T.B. 2003. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 102 672–681. [DOI] [PubMed] [Google Scholar]
- Chen F., Mackey, A.J., Stoeckert Jr, C.J., and Roos, D.S. 2006. OrthoMCL-DB: Querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34 D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S.R., Kemmeren, P., Zhao, X.C., Greenblatt, J.F., Spencer, F., Holstege, F.C., Weissman, J.S., and Krogan, N.J. 2007. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae . Mol. Cell. Proteomics 6 439–450. [DOI] [PubMed] [Google Scholar]
- Das Gupta R., Krause-Ihle, T., Bergmann, B., Muller, I.B., Khomutov, A.R., Muller, S., Walter, R.D., and Luersen, K. 2005. 3-Aminooxy-1-aminopropane and derivatives have an antiproliferative effect on cultured Plasmodium falciparum by decreasing intracellular polyamine concentrations. Antimicrob. Agents Chemother. 49 2857–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis F.P. and Sali, A. 2005. PIBASE: A comprehensive database of structurally defined protein interfaces. Bioinformatics 21 1901–1907. [DOI] [PubMed] [Google Scholar]
- Davis F.P., Braberg, H., Shen, M.Y., Pieper, U., Sali, A., and Madhusudhan, M.S. 2006. Protein complex compositions predicted by structural similarity. Nucleic Acids Res. 34 2943–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. 2006. Autophagy as an immune defense mechanism. Curr. Opin. Immunol. 18 375–382. [DOI] [PubMed] [Google Scholar]
- Edgar R., Domrachev, M., and Lash, A.E. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramian D., Shen, M.Y., Devos, D., Melo, F., Sali, A., and Marti-Renom, M.A. 2006. A composite score for predicting errors in protein structure models. Protein Sci. 15 1653–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., John, B., Mirkovic, N., Fiser, A., Ilyin, V.A., Pieper, U., Stuart, A.C., Marti-Renom, M.A., Madhusudhan, M.S., Yerkovich, B., et al. 2003. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31 3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez B.E., Mondragon-Flores, R., and Luna-Herrera, J. 2003. Internalization of Mycobacterium tuberculosis by macropinocytosis in non-phagocytic cells. Microb. Pathog. 35 49–55. [DOI] [PubMed] [Google Scholar]
- Guerin I. and de Chastellier, C. 2000. Pathogenic mycobacteria disrupt the macrophage actin filament network. Infect. Immun. 68 2655–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez M.G., Master, S.S., Singh, S.B., Taylor, G.A., Colombo, M.I., and Deretic, V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119 753–766. [DOI] [PubMed] [Google Scholar]
- Guttmacher A.E. and Collins, F.S. 2005. Realizing the promise of genomics in biomedical research. JAMA 294 1399–1402. [DOI] [PubMed] [Google Scholar]
- Hart G.T., Ramani, A.K., and Marcotte, E.M. 2006. How complete are current yeast and human protein-interaction networks? Genome Biol. 7 120. doi: 10.1186/gb-2006-7-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiges M., Wang, H., Robinson, E., Aurrecoechea, C., Gao, X., Kaluskar, N., Rhodes, P., Wang, S., He, C.Z., Su, Y., et al. 2006. CryptoDB: A Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 34 D419–D422. doi: 10.1093/nar/gkj078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Fowler C., Peacock, C.S., Wood, V., Aslett, M., Kerhornou, A., Mooney, P., Tivey, A., Berriman, M., Hall, N., Rutherford, K., et al. 2004. GeneDB: A resource for prokaryotic and eukaryotic organisms. Nucleic Acids Res. 32 D339–D343. doi: 10.1093/nar/gkh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A.V. 2006. Pre-erythrocytic malaria vaccines: Towards greater efficacy. Nat. Rev. Immunol. 6 21–32. [DOI] [PubMed] [Google Scholar]
- Hubbard T.J., Aken, B.L., Beal, K., Ballester, B., Caccamo, M., Chen, Y., Clarke, L., Coates, G., Cunningham, F., Cutts, T., et al. 2007. Ensembl 2007. Nucleic Acids Res. 35 D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizinga E.G., Tsuji, S., Romijn, R.A., Schiphorst, M.E., de Groot, P.G., Sixma, J.J., and Gros, P. 2002. Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297 1176–1179. [DOI] [PubMed] [Google Scholar]
- Jansen R., Greenbaum, D., and Gerstein, M. 2002. Relating whole-genome expression data with protein–protein interactions. Genome Res. 12 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N.J., Chung, S., Emili, A., Snyder, M., Greenblatt, J.F., and Gerstein, M. 2003. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302 449–453. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Mannherz, H.G., Suck, D., Pai, E.F., and Holmes, K.C. 1990. Atomic structure of the actin:DNase I complex. Nature 347 37–44. [DOI] [PubMed] [Google Scholar]
- Kasper D.L., Braunwald, E., Fauci, A., Hauser, S., Longo, D., and Jameson, J.L. 2004. Harrison's principles of internal medicine. McGraw-Hill Professional, New York.
- Kerrien S., Alam-Faruque, Y., Aranda, B., Bancarz, I., Bridge, A., Derow, C., Dimmer, E., Feuermann, M., Friedrichsen, A., Huntley, R., et al. 2007. IntAct-open source resource for molecular interaction data. Nucleic Acids Res. 35 D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierszenbaum F., Wirth, J.J., McCann, P.P., and Sjoerdsma, A. 1987. Impairment of macrophage function by inhibitors of ornithine decarboxylase activity. Infect. Immun. 55 2461–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissinger J.C., Gajria, B., Li, L., Paulsen, I.T., and Roos, D.S. 2003. ToxoDB: Accessing the Toxoplasma gondii genome. Nucleic Acids Res. 31 234–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I., Date, S.V., Adai, A.T., and Marcotte, E.M. 2004. A probabilistic functional network of yeast genes. Science 306 1555–1558. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M.A., Stuart, A.C., Fiser, A., Sanchez, R., Melo, F., and Sali, A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29 291–325. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M.A., Madhusudhan, M.S., and Sali, A. 2004. Alignment of protein sequences by their profiles. Protein Sci. 13 1071–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer S.M., Rai, A., and Sali, A. 2004. Finding cures for tropical diseases: Is open source an answer? PLoS Med. 1 e56. doi: 10.1371/journal.pmed.0010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo F., Sanchez, R., and Sali, A. 2002. Statistical potentials for fold assessment. Protein Sci. 11 430–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika S. and Rost, B. 2006. Protein-protein interactions more conserved within species than across species. PLoS Comput. Biol. 2 e79. doi: 10.1371/journal.pcbi.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockenhaupt F.P., Cramer, J.P., Hamann, L., Stegemann, M.S., Eckert, J., Oh, N.R., Otchwemah, R.N., Dietz, E., Ehrhardt, S., Schroder, N.W., et al. 2006. Toll-like receptor (TLR) polymorphisms in African children: Common TLR-4 variants predispose to severe malaria. Proc. Natl. Acad. Sci. 103 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowka R., Patzak, A., and Herzel, H. 2001. Is there a bias in proteome research? Genome Res. 11 1971–1973. [DOI] [PubMed] [Google Scholar]
- Munter S., Way, M., and Frischknecht, F. 2006. Signaling during pathogen infection. Sci. STKE 2006 re5. doi: 1031126/stke.3352006re5. [DOI] [PubMed] [Google Scholar]
- Murzin A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Neduva V. and Russell, R.B. 2006. Peptides mediating interaction networks: New leads at last. Curr. Opin. Biotechnol. 17 465–471. [DOI] [PubMed] [Google Scholar]
- Neduva V., Linding, R., Su-Angrand, I., Stark, A., de Masi, F., Gibson, T.J., Lewis, J., Serrano, L., and Russell, R.B. 2005. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 3 e405. doi: 10.1371/journal.pbio.0030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman A., Grishin, N.V., Kinch, L.N., and Phillips, M.A. 1994. Formation of functional cross-species heterodimers of ornithine decarboxylase. Biochemistry 33 13662–13667. [DOI] [PubMed] [Google Scholar]
- Pandey K.C., Singh, N., Arastu-Kapur, S., Bogyo, M., and Rosenthal, P.J. 2006. Falstatin, a cysteine protease inhibitor of Plasmodium falciparum, facilitates erythrocyte invasion. PLoS Pathog. 2 e117. doi: 10.1371/journal.ppat.0020117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini E.M., Kirkegaard, M., Mortensen, P., Lutz, H.U., Thomas, A.W., and Mann, M. 2006. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood 108 791–801. [DOI] [PubMed] [Google Scholar]
- Pieper U., Eswar, N., Davis, F.P., Braberg, H., Madhusudhan, M.S., Rossi, A., Marti-Renom, M., Karchin, R., Webb, B.M., Eramian, D., et al. 2006. MODBASE: A database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 34 D291–D295. doi: 10.1093/nar/gkj059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachman H., Strong, M., Ulrichs, T., Grode, L., Schuchhardt, J., Mollenkopf, H., Kosmiadi, G.A., Eisenberg, D., and Kaufmann, S.H. 2006. Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 74 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Sassetti C.M. and Rubin, E.J. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. 100 12989–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.Y. and Sali, A. 2006. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker B.A. and Panchenko, A.R. 2007. Deciphering protein–protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comput. Biol. 3 e43. doi: 10.1371/journal.pcbi.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.B., Davis, A.S., Taylor, G.A., and Deretic, V. 2006. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313 1438–1441. [DOI] [PubMed] [Google Scholar]
- Singh S., Mukherjee, A., Khomutov, A.R., Persson, L., Heby, O., Chatterjee, M., and Madhubala, R. 2007. Antileishmanial effect of 3-aminooxy-1-aminopropane is due to polyamine depletion. Antimicrob. Agents Chemother. 51 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Sokal R.R. and Rohlf, F.J. 1995. Biometry: The principles and practice of statistics in biological research, 3rd ed. W.H. Freeman, New York.
- Sprinzak E. and Margalit, H. 2001. Correlated sequence-signatures as markers of protein-protein interaction. J. Mol. Biol. 311 681–692. [DOI] [PubMed] [Google Scholar]
- Stebbins C.E. 2005. Structural microbiology at the pathogen–host interface. Cell. Microbiol. 7 1227–1236. [DOI] [PubMed] [Google Scholar]
- Stoeckert C.J., Fischer, S., Kissinger, J.C., Heiges, M., Aurrecoechea, C., Gajria, B., and Roos, D.S. 2006. PlasmoDB v5: New looks, new genomes. Trends Parasitol. 22 543–546. [DOI] [PubMed] [Google Scholar]
- Su A.I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K.A., Block, D., Zhang, J., Soden, R., Hayakawa, M., Kreiman, G., et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. 101 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R.L., Koonin, E.V., and Lipman, D.J. 1997. A genomic perspective on protein families. Science 278 631–637. [DOI] [PubMed] [Google Scholar]
- Tong A.H., Drees, B., Nardelli, G., Bader, G.D., Brannetti, B., Castagnoli, L., Evangelista, M., Ferracuti, S., Nelson, B., Paoluzi, S., et al. 2002. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295 321–324. [DOI] [PubMed] [Google Scholar]
- Vergne I., Singh, S., Roberts, E., Kyei, G., Master, S., Harris, J., de Haro, S., Naylor, J., Davis, A., Delgado, M., et al. 2006. Autophagy in immune defense against Mycobacterium tuberculosis . Autophagy 2 175–178. [DOI] [PubMed] [Google Scholar]
- Wang S.X., Pandey, K.C., Somoza, J.R., Sijwali, P.S., Kortemme, T., Brinen, L.S., Fletterick, R.J., Rosenthal, P.J., and McKerrow, J.H. 2006. Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. Proc. Natl. Acad. Sci. 103 11503–11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedall G.D., Preston, B.M., Thomas, A.W., Sutherland, C.J., and Conway, D.J. 2007. Differential evidence of natural selection on two leading sporozoite stage malaria vaccine candidate antigens. Int. J. Parasitol. 37 77–85. [DOI] [PubMed] [Google Scholar]
- Westbrook J., Feng, Z., Jain, S., Bhat, T.N., Thanki, N., Ravichandran, V., Gilliland, G.L., Bluhm, W., Weissig, H., Greer, D.S., et al. 2002. The Protein Data Bank: Unifying the archive. Nucleic Acids Res. 30 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. 2003. The world health report 2003: Shaping the future. World Health Organization, Geneva, Switzerland.
- Yu H., Luscombe, N.M., Lu, H.X., Zhu, X., Xia, Y., Han, J.D., Bertin, N., Chung, S., Vidal, M., and Gerstein, M. 2004. Annotation transfer between genomes: Protein–protein interologs and protein-DNA regulogs. Genome Res. 14 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]