

Abstract
The p90 ribosomal S6 kinases (RSKs) also known as MAPKAP-Ks are serine/threonine protein kinases that are activated by ERK or PDK1 and act as downstream effectors of mitogen-activated protein kinase (MAPK). RSK1, a member of the RSK family, contains two distinct kinase domains in a single polypeptide chain, the regulatory C-terminal kinase domain (CTKD) and the catalytic N-terminal kinase domain (NTKD). Autophosphorylation of the CTKD leads to activation of the NTKD that subsequently phosphorylates downstream substrates. Here we report the crystal structures of the unactivated RSK1 NTKD bound to different ligands at 2.0 Å resolution. The activation loop and helix αC, key regulatory elements of kinase function, are disordered. The DFG motif of the inactive RSK1 adopts an “active-like” conformation. The β-PO4 group in the AMP–PCP complex adopts a unique conformation that may contribute to inactivity of the enzyme. Structures of RSK1 ligand complexes offer insights into the design of novel anticancer agents and into the regulation of the catalytic activity of RSKs.
Keywords: RSK1, kinase, cancer, crystal structure, kinase inhibitor
Protein kinases are considered important small molecule-based therapeutic targets, due to their role in a number of cellular functions. The chemical interplay of kinases together with phosphatases that affects signal transduction pathways through phosphorylation/dephosphorylation of appropriate substrates in the signaling cascade has been associated with a number of human diseases. The p90 ribosomal S6 kinases (RSK) group of serine/threonine (S/T) protein kinases are downstream effectors of mitogen-activated protein kinase (MAPK) signaling pathways that are responsible for coordination and regulation of cellular growth and differentiation (Shaeffer and Webber 1999). In humans, RSK group consists of four paralogs (RSK1–4), each being a product of a distinct gene. All of the RSK paralogs have similar overall organization including two nonidentical kinase domains separated by a linker region of ∼100 amino acids, and short amino, and carboxy-terminal segments (Fig. 1). N-terminal kinase domain (NTKD) is most closely related to p70 S6 kinase (p70S6K), whereas the C-terminal kinase domain (CTKD) is most similar to the calmodulin-dependent protein kinases (Jones et al. 1988). The NTKD of RSK1 is a member of the structurally related AGC family of protein kinases which includes protein kinase A (PKA), protein kinase B (PKB), and 3-phosphoinositide-dependent protein kinase-1 (PDK1).
Figure 1.

Domain organization of RSKs. RSK1 has two kinase domains that can be activated by ERK and PDK1. The current model for activation suggests that, following mitogen stimulation, ERK phosphorylates Thr573, which is located in the activation loop of the CTKD of RSK1, and Thr359 and Ser363, which are in the linker region of RSK1 (Gavin and Nebreda 1999; Smith et al. 1999). Activation of the CTKD by ERK leads to autophosphorylation of Ser380 which is also located in the linker region. Ser380 is located in a hydrophobic motif conserved among a large number of the AGC family kinases. The result is a conformational alteration of RSK1, creating a docking site for PDK1, which then fully activates RSK1 by phosphorylation of Ser221 in the activation loop of the NTKD.
The regulation of RSK requires a cascade of phosphorylation events that involves ERK, the CTKD of RSK, and 3-phosphoinositide-dependent kinase-1 (PDK1) (Fig. 1; Bjorbaek et al. 1995; Fisher and Blenis 1996; Dalby et al. 1998; Jensen et al. 1999; Poteet-Smith et al. 1999; Richards et al. 1999). In RSK1, the NTKD has been shown to phosphorylate substrates (Leighton et al. 1995). The CTKD and the linker region play a role in the regulation of NTKD activity. Phosphorylation of S380 leads to the recruitment of PDK1 to the hydrophobic motif in the linker region that activates the NTKD. Upon phosphorylation of the Ser221, the NTKD is activated to phosphorylate the downstream substrates. Substrates identified for RSKs include transcription factors like estrogen receptor-α, cyclic AMP response element-binding protein (CREB), c-Fos, and IκBα/nuclear factor-κB (Xing et al. 1996; Ghoda et al. 1997; Schouten et al. 1997; De Cesare et al. 1998; Joel et al. 1998), as well as other kinases, such as glycogen synthase kinase-3, the p34cdc2-inhibitory kinase Myt1, and the mitotic checkpoint kinase, Bub1 (Sutherland et al. 1993; Palmer et al. 1998; Schwab et al. 2001).
RSK1 plays a central role in a number of cellular processes, including being a downstream effector of Ras/MAPK pathway that is activated in >50% primary tumors. RSK1 is overexpressed in primary breast and prostate cancers and its activity is highly correlated with MAPK activity in vitro (Clark et al. 2005). RSK1 also transduces signals in other pathways implicated in cancer progression, such as BAD, IκB, Myt1, and mTOR pathway (Roux et al. 2004). Due to its role in mediating anti-apoptotic and anti-proliferative signals, RSK1 is likely to serve as an attractive anticancer target. Development of small molecules that are either ATP competitors or allosteric inhibitors of RSK1 activity would enable evaluation of the value of therapeutic intervention of RSK1 activity. Such an effort would benefit significantly from the knowledge of the three-dimensional structure of the RSK1 catalytic domain.
The ATP-binding pocket of inactive kinases is likely to offer better opportunities for developing kinase selective inhibitors (Fischer 2004). In order to provide the structural information for structure-based inhibitor design for RSK1, we have determined crystal structures of the inactive form of RSK1 NTKD bound to AMP–PCP, Staurosporine, and Purvalnol A. We used a combination of highly parallel cloning, protein expression, purification, and crystallization techniques to obtain crystals of RSK1 NTKD. The structure of RSK1 is the first among the members of the RSK family, and comparison with related kinases of the AGC family provides insights into regulation of RSK1 activity. In addition, knowledge of the structure offers opportunities for rational design of RSK1-specific inhibitors.
Results
Structure determination
A number of RSK1 NTKD open reading frames (ORFs) were expressed in insect cells as a series of molecular constructs designed to influence crystallization properties of the protein. To accelerate the process, we adopted a medium throughput approach using automated and parallel molecular biology procedures (Kornienko et al. 2005) to make 24 expression clones of RSK1 NTKD. The modifications included alternative domain boundaries based on the homology model derived from mitogen- and stress-activated kinase MSK1 (Smith et al. 2004) (PDB entry 1VZO). MSK1 like RSK1 belongs to a family of protein kinases that contain two kinase domains in a single polypeptide chain. The catalytic N-terminal kinase domain of MSK1 shares 49% sequence identity with RSK1 NTKD. In all 24 different expression plasmids were made and screened for expression in Escherichia coli and insect cells. Attempts to express the protein in E. coli were unsuccessful. Of the 24 variants tested in insect cells, only four variants (residues 33–353 WT, 33–387 S221D, 33–735 WT, 33–735 S221D) showed useable protein expression. These variants were scaled up and protein was purified (see Materials and Methods). However, only one variant (residues 33–353 WT) was amenable to crystallization as a complex with ATP competitors. Attempts to crystallize the apo form of these variants were unsuccessful. Activity analysis of the purified protein showed that the crystallized variant, as expected, was catalytically inactive (data not shown). The structure was determined by molecular replacement using an MSK1-based homology model. Data collection and refinement statistics are listed in Table 1.
Table 1.
Data collection and refinement statistics

Overview of the structure of RSK1
The overall topology of the RSK1 is similar to the structures of other known protein kinases. The structure consists of two domains (lobes), the N-terminal lobe, which is composed largely of five-stranded β-sheet, and the C-terminal lobe, which is predominantly α-helical. The expected ATP-binding site is located at the interface of the two lobes. All ligands presented in this study, being ATP analogs, bind in the ATP-binding pocket. Residues 33–55 at the N terminus and residues 341–353 are disordered (Fig. 2A). In addition, residues 97–114 of the putative helices αB and αC connecting strands 3 and 4 and residues 212–223 of the activation loop are disordered. Although the crystallized RSK1 NTKD is catalytically inactive, the conformation of the DFG motif in the activation loop resembles active conformation observed in other kinases.
Figure 2.

Structure of RSK1 and its comparison with PKB and MSK1. Panels B and C are rotated by 180° compared to panel A. (A) Ribbon diagram of the RSK1 NTKD bound to AMP–PCP. (B) Ribbon representation of the superimposed structures of inactive forms of PKB (green) and RSK1 (red). The helices αC and αB and parts of the activation loop are disordered in both structures. The overall rmsd for the two structures is 1.3 Å for 224 atom pairs. (C) Conformation of the DFG motif in RSK1. The magnesium ion is shown in cyan. (D) Ribbon representation of the superimposed structures of inactive forms of MSK1 (green) and RSK1 (red). The overall rmsd for the two structures is 0.93 Å for 224 atom pairs.
Comparison with other inactive AGC family kinases
As expected, the overall structure of RSK1 resembles the catalytic subunit of the members of AGC family of kinases. In particular, structural features of inactive RSK1 resemble the features of inactive PKB structure (Fig. 2B; Huang et al. 2003) with which RSK1 shares 41% sequence identity. In both structures, helices αB and αC and the parts of activation loop are disordered. The structures of RSK1 and inactive PKB superimpose with a root-mean-square deviation (rmsd) of 1.3 Å. Both the lobes superimpose fairly well, with the exception of the P-loop that is tilted at the tip by ∼5 Å in RSK1 toward ATP-binding pocket due to its interaction with the ligand, compared to apo structure of inactive PKB. The conserved aromatic residues of the DFG motif at the beginning of the activation loop in the PKB structure block the ATP-binding pocket (“DFG-out” conformation) and have been implicated in the autoinhibition of the kinase activity. The “DFG-in” conformation is one in which the catalytic aspartic acid points toward the β-phosphate of the bound ATP and helps coordinate the catalytic Mg2+ ion, and phenylalanine points away from the ATP. In the DFG-out conformation the aspartic acid and phenylalanine point in almost opposite directions of DFG-in conformation. The DFG motif in RSK1 swings away from the binding pocket, making room for bound ligand, and adopts a conformation similar to the DFG-in conformation observed in activated AGC kinases (Huse and Kuriyan 2002). This conformation of the DFG motif in RSK1 (Fig. 2C) is also different from what has been observed in crystal structures of some inactive tyrosine kinase domains (Nowakowski et al. 2002; Schubert et al. 2007). The DFG motif in ligand bound structures of inactive IRTK (S. Munshi, P. Darke, M. Kornienko, D. Hall, J. Reid, and L.C. Kuo, unpubl.) and cFMS (colony stimulating factor-1 receptor kinase) swings away from the ATP-binding pocket to accommodate the bound inhibitor while adopting the DFG-out conformation. The conformation of DFG motif in RSK1 closely resembles that observed in the crystal structures of AMP–PNP bound unphosphorylated form of EphA2 (Nowakowski et al. 2002) and SyK (spleen tyrosine kinase) (Atwell et al. 2004), with the equivalent aspartic acid pointing toward the phosphate groups of the bound ligand and the phenylalanine pointing away from the ligand.
The DFG motif in the structure of the inactive NTKD of MSK1, another dual kinase protein, adopts a different and unique conformation (Smith et al. 2004). This is due to the presence of an additional three-stranded β-sheet that contributes to the changed course of the proximal part of the activation loop, resulting in an autoinhibited conformation of the MSK1 (Fig. 2D). In addition the P-loop in MSK1 apo structure is tipped significantly toward the ATP-binding pocket, with residue Tyr60 stereically blocking the ligand binding in this pocket. The P-loop in RSK1 is pulled inward, interacting with the bound ligand.
The ATP-binding site
Although the NTKD of RSK1 protein was catalytically inactive, it does bind ATP analogs at the expected place as is evident from the well-defined electron density of ligands in the structures for AMP–PCP, Staurosporine, and Purvalnol A complexes (see below). Analysis of the RSK1–Mg2+–AMP–PCP complex indicates that AMP–PCP interacts with several residues located at the interface of the two lobes (Fig. 3A). The purine ring of AMP–PCP is anchored by two hydrogen bonds which are conserved among kinases. One bond exists between the atom N1 of adenine and the backbone amide of Leu144, and the second is between adenine atom N6 and the backbone carbonyl of Asp142. The adenine base is positioned in a hydrophobic pocket and the phosphates are held in position by ionic and hydrogen-bonding interactions. An oxygen atom from each of the three phosphates of AMP–PCP contributes to the octahedral coordination of the Mg2+ ion. Side chains of Asn192 and Asp205 also coordinate with the Mg2+ ion. In the AMP–PCP RSK1 complex, the conformation of the triphosphate and the coordination geometry of the Mg2+ are different from that observed in the structures of AMP–PNP bound to unphosphorylated EphA2 or to ATP bound complex of activated PKA (Fig. 3C,D; Zheng et al. 1993; Nowakowski et al. 2002). In comparison, the triphosphate groups adopt a similar conformation in inhibitor bound unphosphorylated EphA2 and PKA (Fig. 3E). In PKA, Lys72 and Asp184 are critical residues involved in phosphate orientation and metal ion coordination. The equivalent residues Lys94 and Asp205 in RSK1 adopt different conformations. However, major differences are in the position of the Mg2+ ion and the conformation of the triphosphate group of AMP–PCP. Positions of the α- and γ-phosphates in RSK1 and PKA are similar, however the position of the β-phosphate is far removed from its position in the ATP–PKA complex. This conformation of the triphosphate group may be a result of the conformation of the DFG motif, in particular Asp205, Asn192, and the position of Mg2+ (Fig. 2C). In the PKA ternary complex, the γ-phosphate of ATP is reasonably well aligned with the substrate and is in the right position to promote in-line transfer to the substrate. However, in RSK1 the alignment of the β- and γ-phosphates is almost perpendicular to that in PKA. The correct alignment of the ATP with respect to the catalytic residues is crucial for catalysis as observed in the structure of active PKA (Zheng et al. 1993). However, based on the complex with AMP–PCP in the inactive RSK1 structure, the altered triphosphate conformation and change in Mg2+ coordination suggests a nonproductive complex. Assuming that the protein substrate binds to RSK1 in roughly the same orientation as it does in PKA, it is unlikely that the triphosphate moiety of ATP would assume a similar conformation in the activated form of RSK1 since in-line phosphoryl transfer to the substrate would not be possible.
Figure 3.
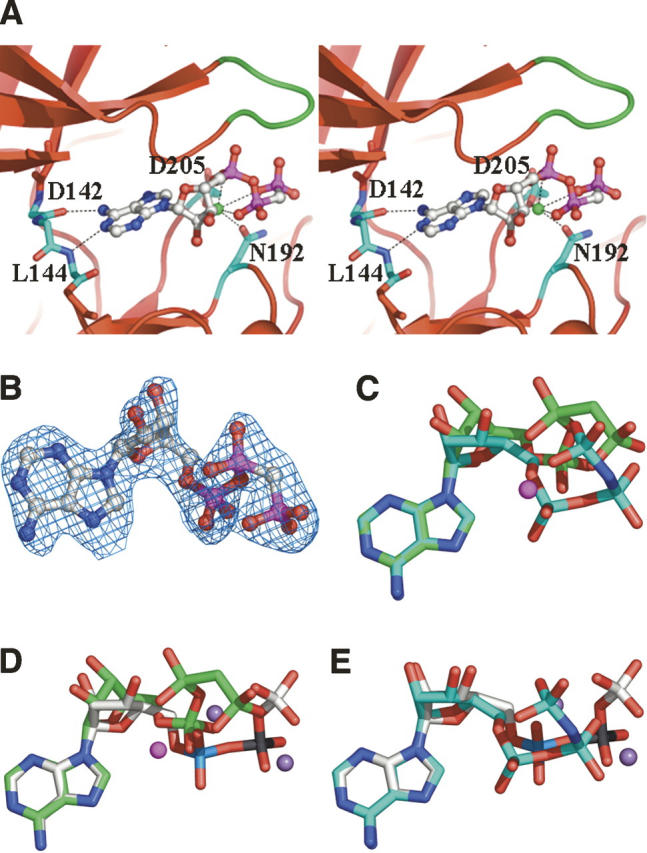
Close-up view of the ATP-binding site. (A) Stereoview of RSK1 with bound AMP–PCP. Magnesium ion is shown as a green sphere. The P-loop is colored green. (B) 2Fo−Fc map contoured at 1.5σ for AMP–PCP bound to RSK1. (C) Superimposed AMP–PCP of RSK1 (green) and AMP–PNP in unphosphorylated EphA2 (cyan) (PDB code 1MQB). The Mg2+ ion bound to RSK1 is shown as a magenta-colored sphere. (D) Superimposed AMP–PCP of RSK1 (green) and ATP in active PKA (gray) (PDB code 1ATP). Two Mg2+ ions bound to PKA are shown as a purple-colored sphere. (E) Superimposed ATP of PKA (gray) and AMP–PNP in unphosphorylated EphA2 (cyan). In the inactive RSK1 structure, the sugar ring is in a different conformation compared to EphA2 and PKA. The phosphate groups in PKA and EphA2 are similarly oriented. However, β-phosphate in RSK1 meanders in an opposite direction.
Flexibility of the αC-helix
Within the N-terminal lobe of RSK1, the five-stranded β-sheet is well ordered; however, residues 97–114, equivalent to the αB-helix and the majority of the αC-helix of PKA, are disordered in AMP–PCP RSK1 structure. As described below, ordered structure of the αB- and αC-helices of RSK1 may be dependent on the presence of phosphorylated C-terminal hydrophobic motif. The αC-helix is an important structural feature, serving to correctly align the catalytic residues in the active conformation of a kinase. In the active conformation of a kinase, a conserved glutamate residue in αC-helix (Glu91 of PKA, Glu112 of RSK1) contributes to its catalytic function by accepting a hydrogen bond from a conserved lysine side chain (Lys72 of PKA, Lys94 of RSK1), which interacts with the β-phosphate of ATP. Both residues are highly conserved over the entire protein kinase family. A key salt bridge expected between residues Lys94 and Glu112 is missing due to conformational disorder. Lack of this interaction may contribute to inactivity of unphosphorylated RSK1 NTKD.
The C-terminal hydrophobic motif and hydrophobic pocket
The crystal structures of active and inactive forms of PKB show how PKB is activated by phosphorylation of Ser474 in the C-terminal hydrophobic motif (Yang et al. 2002; Huang et al. 2003). Unphosphorylated enzyme is characterized by disordered αB- and αC-helices in the N-lobe, accompanied by a disordered activation segment and hydrophobic motif. Phosphorylation of Ser474 results in the interaction of the phosphorylated hydrophobic motif with the hydrophobic pocket/phosphoserine docking site in the N-lobe. This leads to a “disorder-to-order” transition of the αC-helix. The ordered αC-helix, by interacting with pThr309 of the activation segment, restructures and orders the activation segment, generating an active conformation.
In RSK1, a conserved hydrophobic motif (FRGFS[380]F) is found in the linker region between the two kinase lobes (Fig. 1). This motif contains the phosphorylation site Ser380, which is believed to be autophosphorylated by active RSK1 CTKD. In the crystal structure of RSK1, visualization of the interaction of the phosphorylated hydrophobic motif with the hydrophobic pocket in the N-lobe is not possible, because the hydrophobic motif of RSK1 is absent from the expression construct. Absence of the hydrophobic motif in the chosen construct and hence unphosphorylated activation loop leads to the inactive RSK1 NTKD. In addition, electron density for residues 340 to the C-terminal (353) is not visible.
Activation loop
The activation loop of kinases can be defined as the region beginning with the DFG motif and ending with residues APE, which refers to residues 205–232 in RSK1. Phosphorylation of Ser221, catalyzed by PDK1, is necessary to activate RSK1. However, no insights can be gleaned about the conformation of the activation loop in RSK1 structure since the majority of the activation segment (residues 212–224) is disordered.
AGC family kinases are activated by the phosphorylation of regulatory residues, S/T of the activation loop, and S/T of the hydrophobic motif, which are conserved within the AGC family kinases. Structural data indicate that the phosphorylated S/T of the hydrophobic motif interacts with the N-lobe of the kinase domain, ordering the αC-helix, which in turn interacts with phosphorylated S/T of the activation loop, stabilizing the activation segment and resulting in an active kinase conformation. The phosphate group of pThr197 in PKA in the activation loop (pThr309 in PKB) is available to form a salt bridge with His87 in PKA (His196 in PKB) of the αC-helix. In RSK1, the activation loop is disordered probably because of the loss of expected interaction of phosphorylated Ser221 with Arg108 which is likely to be part of the disordered αC-helix. This would result in the misalignment of the catalytic residues of the RSK1 and provides explanation for the inactivity of the RSK1 enzyme. Comparison between RSK1 and PKA suggests significant conformational changes including ordering of the αC-helix and the activation loop upon activation of RSK1.
Interactions between RSK1 and Staurosporine
Staurosporine is a microbial alkaloid that was firs characterized as an inhibitor of protein kinase C (Tamaoki et al. 1986). It has been shown to be a potent, but nonspecific, protein kinase inhibitor, with IC50 values against a number of kinases in the nanomolar range. The IC50 of Staurosporine against RSK1 is 0.3–1 nM. Despite disordering of the helix αC and activation loop, the electron density for Staurosporine in the ATP-binding pocket and most of the surrounding residues is clearly defined. Staurosporine binding results in an almost perfect fit with many van der Waals interactions and characteristic polar contacts, consistent with the high inhibitory potency of Staurosporine.
Staurosporine binds to the ATP-binding pocket with the tetrahydropyran ring in a boat conformation (Fig. 4A). One side of the hydrophobic core of the indole carbazole makes many favorable van der Waals contacts with the backbone and side chains of residues Leu68, Val76, Ala92, Lys94, Leu141, and Phe143 in the N-terminal lobe. The other side of the indole carbazole makes van der Waals interactions with the side chains of Val125, Leu194, and Asp205 from both the N- and C-terminal lobes. The tetrahydropyran ring group sits in the ribose-binding pocket surrounded by Asp148, Asn192, Leu194, Ser204, and Asp205. The N1 atom of Staurosporine on the lactam moiety makes a hydrogen bond (2.64 Å) to the backbone carbonyl oxygen of Asp142, while O5 atom of the inhibitor accepts a hydrogen bond (2.5 Å) from the amide nitrogen of Leu144 in the linker region. This hydrogen-bonding pattern is common in many known protein kinase–ATP complexes. The N4 atom of Staurosporine in the tetrahydropyran ring makes a hydrogen bond to Asp148 of RSK1. The boat conformation of the tetrahydropyran ring is believed to be biologically active (Furet et al. 1995). This conformation allows the N4 atom of Staurosporine to hydrogen bond to Asp148 of RSK1. Comparison of Staurosporine with AMP–PCP bound to RSK1 suggests that the purine ring of the AMP–PCP roughly superimposes with the indole carbazole rings of Staurosporine. The massive indole carbazole rings extend much further than the adenosine moiety, while the position of the triphosphate group is left unoccupied. The purine base of AMP–PCP is anchored to the RSK1 kinase by two hydrogen bonds which appear to be conserved among kinases. One bond exists between the N1 of ATP and the backbone amide of Leu144, the second is between N6 and the backbone carbonyl of Asp142. Both these interactions are mimicked by Staurosporine. In the Staurosporine-bound RSK1, side chains of residues Lys94 and Asp205 appear shifted compared to RSK–AMP–PCP structure. Movement of these residues is required to avoid an unfavorable contact between protein and Staurosporine. Although Staurosporine is a potent inhibitor of most protein kinases, it does display some level of specificity. The IC50 of Staurosporine against RSK1, PKA, CDK2-cyclin A, and Casein kinase I (CK1) are 0.3–1 nM, 8.2 nM, 7 nM, and 164 μM, respectively. Comparison of the crystal structures suggests that the N4 atom of Staurosporine in the tetrahydropyran ring makes a hydrogen bond to Asp148 of RSK1, Glu127 of PKA (Prade et al. 1997), and Asp86 of CDK2 (Lawrie et al. 1997). However, in CK1, this position is occupied by Ser91 that is unable to a hydrogen bond with N4 of Staurosporine. These sequence differences between kinases may explain the specificity of Staurosporine.
Figure 4.

The binding mode of Staurosporine and Purvalnol A. (A, B) Chemical structure of Staurosporine and its interactions with RSK1. Staurosporine maintains van der Waals interactions with residues Leu68, Val76, Ala92, Lys94, Leu141, Phe143, Val125, Leu194, and Asp205. A pair of hydrogen bonds is observed between the N1 atom of Staurosporine on the lactam moiety and the backbone carbonyl of Asp142, and between O5 atom of the inhibitor and the backbone NH of Leu144. The N4 atom of Staurosporine in the tetrahydropyran ring makes a hydrogen bond to carbonyl oxygen of Glu191. (C, D) Chemical structure of Purvalnol A and its interactions with RSK1. Purvalnol A maintains van der Waals interactions with residues Leu68, Val76, Ala92, Leu141, Asp142, Phe143, Leu144, Arg145, Gly147, and Leu194. A pair of hydrogen bonds is observed between the N7 imidazole nitrogen of Purvalnol A and the backbone NH of Leu144, and between the N6 amino group of the inhibitor and the backbone carbonyl of Leu144.
Interactions between RSK1 and Purvalanol A
Purvalanol A and B are known as low nanomolar inhibitors of cyclin-dependent protein kinases (CDKs), while the non-CDKs RSK1, Erk1 (p42MAPK), Erk2 (p44MAPK), LCK, CSK, and PKA are inhibited in the low micromolar range (Bain et al. 2003). Purvalanol A, 2-(1R-isopropyl-2-hydroxyethylamino)-6-(3-chloroanilino)-9-isopropyl-purine, leads to an arrest of G1 and G2 phase in cell cycle progression in vitro. The chemical structure of Purvalanol B differs from Purvalanol A only in the addition of a carboxylate on the m-chloro-phenyl ring. Purvalanol A binds in the RSK1 adenosine pocket with two H-bonds to the hinge region (Fig. 4B). A pair of hydrogen bonds is present between the N7 imidazole nitrogen and the backbone NH of Leu144, and between the N6 amino group and the backbone carbonyl of Leu144. Purvalnol A maintains van der Waals interactions with residues Leu141, Asp142, Phe143, Leu144, Arg145, Gly147 in the hinge region; Leu68 and Val76 in the P-loop; Ala92 and Leu194 in β-strands 3 and 7. Comparison of RSK1-bound Purvalnol A with the structure of the CDK2–Purvalnol B complex (Gray et al. 1998) shows that the orientation of the m-chloro-phenyl ring plane is different between RSK and CDK2 complex structures. The orientation of this ring in RSK1 is close to the orientation in Src–Purvalnol A complex structure (Breitenlechner et al. 2005). In the complex of Purvalnol B and CDK2, the orientation of the chlorine is ambiguous. The Cl atom on the m-chloro-phenyl ring makes a polar interaction to the side chain of Asp86 in about two thirds of the molecules in the CDK2–Purvalnol B crystals. This conformation stabilizes the binding of the m-chloro-phenyl ring. In the other conformation, the m-chloro-phenyl ring is flipped ∼160°. The chlorine of Purvalanol A adopts a single position in RSK1.
Discussion
In order to accelerate the crystal structure determination, we used a combination of highly parallel cloning, protein expression, purification, and crystallization approaches to obtain crystals of RSK1 NTKD. Interestingly, expression in E. coli was extremely poor. Whereas significant differences in expression level and amount of soluble protein generated from different ORF length variants with the same promoter and expression tag were observed in insect cells, some length variants did not produce soluble protein, demonstrating the importance of expression testing of multiple clones of various lengths due to the unpredictable outcomes. Of the 24 variants tested, only four showed reasonable protein expression and two of which contained both the NTKD and the hydrophobic motif. However, only the variant containing the NTKD yielded crystals. It is hard to rationalize why only the polypeptide containing residues 33–353 was amenable to crystallization. The disordered residues at the N terminus (33–55) would suggest that polypeptides starting at residue 55 would have served as better candidates for crystallization, however, no detectable expression was observed for protein constructs starting at residue 44. It is conceivable that the construct ending with residue 387 at the C terminus would have hindered crystallization based on the disorder noticeable beyond residues 340. For longer constructs that contain the hydrophobic motif, the lack of phosphorylation of the key residues may have contributed to crystallization failure.
Comparison of the RSK1 structure with active PKA structure suggests that the low kinase activity of RSK1 is due to several structural constraints. First, critical amino acid side chains in the ATP-binding site (Lys94 and Asp205) as well as the β-phosphate of AMP–PCP in RSK1 adopt an unfavorable conformation for phosphotransfer to occur. The different conformations of the ATP-binding pockets in RSK1 and PKA are due to differences in nearby secondary structure elements. Within the N-terminal lobe of RSK1, residues 97–114, equivalent to the αB-helix and the majority of the αC-helix of PKA, are disordered. In active PKA structure, an invariant glutamate residue (Glu91) in αC-helix interacts with Lys72. The corresponding interaction (Glu112/Lys94) is not possible in RSK1 because of the disorder of αC-helix.
RSK1 is activated by the phosphorylation of regulatory residues Ser221 of the activation loop and Ser380 of the hydrophobic motif that are structurally and functionally conserved within the AGC kinase family. Active kinase structures indicate that the role of Ser380 phosphorylation might be to promote the engagement of the hydrophobic motif with the N-lobe of the kinase domain, resulting in a disorder-to-order transition of the αC-helix. The crystal structures of RSK1 show that residues corresponding to the regions of the αB- and αC-helices are disordered. This disorder probably results from loss of interactions with the hydrophobic motif of RSK1. The ordered αC-helix by interaction with pSer221 in the activation loop may restructure and order the activation loop, generating an active conformation. In RSK1 structure, the majority of the activation loop (residues 212–224) is disordered, and contacts between Arg108 and pSer221 cannot be formed. These observations are consistent with observed inactivity of the RSK1 enzyme.
The structure of RSK1 Staurosporine complex explains the tight binding of this inhibitor and suggests features to be exploited in the design of specific inhibitors of RSK1. Specific inhibitors may be designed that both utilize novel interactions within the ATP-binding site cleft and exploit the sequence differences between kinases in the residues surrounding it (Zhao et al. 2002). Although Staurosporine is a potent inhibitor of most protein kinases, it has some selectivity among protein kinases. The IC50 of Staurosporine against RSK1 is 0.3–1 nM. On the other hand, CK1 shows unusually low affinity for Staurosporine (IC50 = 164 μM) (Meggio et al. 1995). The sequence differences may contribute to the specificity among kinases.
Inhibiting RSK1 is an attractive approach for developing anticancer agents. Inhibitor SL0101 was identified as a RSK-specific inhibitor and shown to inhibit proliferation of the human breast cancer cell line MCF-7 (Smith et al. 2005). Recently, a potent RSK1 inhibitor, BI-D1870 (Sapkota et al. 2007), which inhibits RSK1 with an IC50 of 10 nM, was reported. However, this inhibitor does not significantly inhibit 10 other AGC kinase members and >40 other protein kinases tested at 100-fold higher concentrations. Knowledge of structure of RSK1 with different bound ligands will undoubtedly be critical in the development of potent and selective RSK1 inhibitors.
Materials and Methods
Cloning and expression
Twenty-four various lengths of the RSK1 ORF were generated by PCR from a full-length cDNA clone. The PCR products were cloned into custom destination vectors designed for baculovirus recombination. The vectors are derived from the baculovirus transfer vector pVL1392 (Pharmingen), which was customized by adding His6 for N-fusion and then Gateway adapted (Kornienko et al. 2005). Generation of recombinant baculovirus using the flashBAC (NextGen Sciences) system was performed using standard procedures. RSK1 variants were expressed in Sf-21 at an MOI = 0.01. The cells were harvested after 72 h of growth at 27°C. Of the 24 variants screened, four expressed in reasonable amounts. Attempts to express the protein in E. coli were unsuccessful.
Protein purification and crystallization
Baculovirus-infected Sf-21 cells of four RSK1 variants were resuspended in Lysis buffer (50 mM HEPES, pH 7.5, 250 mM NaCl, 5% glycerol, 0.2% Chaps, 0.1% β-octyl glucoside, 2 mM DTT, protease inhibitor cocktails) and disrupted by sonication on ice. The insoluble material was removed by centrifugation. The supernatant was loaded on to a 5 mL HisTrap column (Amersham Biosciences), the column was eluted using a linear imidazole gradient from 25 to 500 mM imidazole. The fractions containing the RSK1 were pooled and buffer was exchanged into buffer A (50 mM HEPES, pH 7.5, 50 mM NaCl, 5% glycerol, 5 mM DTT). The material was loaded on to a 5 mL Heparin column (Amersham Biosciences) and eluted using a linear NaCl gradient from 50 to 1000 mM NaCl. To remove the His-tag, TEV protease was added to the pooled fractions. The cleaved protein was loaded on to a 10 mL Heparin column and eluted using a linear NaCl gradient. The RSK1 was pooled and concentrated and loaded on to a superdex200 column (Amersham Biosciences) that had been equilibrated in buffer B (50 mM HEPES, pH 7.5, 200 mM Li2SO4, 5% glycerol, 5 mM DTT). The purified protein was concentrated to 10 mg/mL and AMP–PCP/MgCl2 was added to a final concentration of 5 mM. Crystals were grown by vapor diffusion at 20°C by mixing protein solution (10 mg/mL RSK1, 200 mM Li2SO4, 5% glycerol, 5 mM DTT, 5 mM AMP–PCP–MgCl2) with a crystallization solution of 12%–16% (w/v) PEGMME2000, 150 mM DL-malic acid, pH 7.0. Crystals were soaked for 7 d in buffer containing Staurosporine and Purvalnol A before data collection.
Data collection and structure determination
Crystals were transferred to a cryoprotection buffer comprising 35% (w/v) glycerol, 16% (w/v) PEGMME2000, 150 mM DL-malic acid, pH 7.0, and then frozen by plunging into liquid nitrogen.
X-ray diffraction data were collected at Industrial Macromolecular Crystallographer Association's beamline 17-ID at the Advanced Photon Source. Data were processed using HKL2000 (Otwinowski and Minor 1997). The structure was determined by molecular replacement using the program Molrep (CCP4) (Bailey 1994) using RSK1 homology model based on MSK1 structure as a search model. The structure was refined using CNX (Brünger et al. 1998). Model building was performed with Xtalview (McRee 1999). Figures 2, 3, and 4 were prepared using PyMOL (DeLano Scientific).
Coordinates
Coordinates have been deposited with the Protein Data Bank with the following accession codes: 2Z7Q (AMP–PCP), 2Z7R (Staurosporine), and 2Z7S (Purvalnol A).
Acknowledgments
We thank Sujata Sharma, Dawn L. Hall, and Paul L. Darke for invaluable advice with cloning and purification of RSK1 protein; Sachie Arai and Takeshi Sagara for building the RSK1 homology model based on MSK1 structure; and Lawrence C. Kuo and Yoshikazu Iwasawa for their support throughout the project. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Reprint requests to: Mari Ikuta, Department of Structural Biology, Merck Research Laboratories, 770 Sumneytown Pike, West Point, PA 19486, USA; e-mail: mari_ikuta@merck.com; fax: (215) 652-6452; or Sanjeev K. Munshi, Department of Structural Biology, Merck Research Laboratories, 770 Sumneytown Pike, West Point, PA 19486, USA; e-mail: sanjeev_k_munshi@merck.com; fax: (215) 652-6452.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073123707.
References
- Atwell S., Adams, J.M., Badger, J., Buchanan, M.D., Feil, I.K., Froning, K.J., Gao, X., Hendle, J., Keegan, K., Leon, B.C., et al. 2004. A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J. Biol. Chem. 279: 55827–55832. [DOI] [PubMed] [Google Scholar]
- Bailey S. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Bain J., McLauchlan, H., Elliott, M., and Cohen, P. 2003. The specificities of protein kinase inhibitors: An update. Biochem. J. 371: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorbaek C., Zhao, Y., and Moller, D.E. 1995. Divergent functional roles for p90rsk kinase domains. J. Biol. Chem. 270: 18848–18852. [DOI] [PubMed] [Google Scholar]
- Breitenlechner C.B., Kairies, N.A., Honold, K., Scheiblich, S., Koll, H., Greiter, E., Koch, S., Schafer, W., Huber, R., and Engh, R.A. 2005. Crystal structures of active Src kinase domain complexes. J. Mol. Biol. 353: 222–231. [DOI] [PubMed] [Google Scholar]
- Brünger A., Adams, P.D., and Clore, G.M. 1998. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- Clark D.E., Errington, T.M., Smith, J.A., Frierson Jr, H.F., Weber, M.J., and Lannigan, D.A. 2005. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 65: 3108–3116. [DOI] [PubMed] [Google Scholar]
- Dalby K.N., Morrice, N., Caudwell, F.B., Avruch, J., and Cohen, P. 1998. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 273: 1496–1505. [DOI] [PubMed] [Google Scholar]
- De Cesare D., Jacquot, S., Hanauer, A., and Sassone-Corsi, P. 1998. Rsk-2 activity is necessary for epidermal growth factorinduced phosphorylation of CREB protein and transcription of c-fos gene. Proc. Natl. Acad. Sci. 95: 12202–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer P.M. 2004. The design of drug candidate molecules as selective inhibitors of therapeutically relevant protein kinases. Curr. Med. Chem. 11: 1563–1583. [DOI] [PubMed] [Google Scholar]
- Fisher T.L. and Blenis, J. 1996. Evidence for two catalytically active kinase domains in pp90rsk. Mol. Cell. Biol. 16: 1212–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furet P., Caravatti, G., Lydon, N., Priestle, J.P., Sowadski, J.M., Trinks, U., and Traxler, P. 1995. Modelling study of protein kinase inhibitors: Binding mode of Staurosporine and origin of the selectivity of CGP 52411. J. Comput. Aided Mol. Des. 9: 465–472. [DOI] [PubMed] [Google Scholar]
- Gavin A.C. and Nebreda, A.R. 1999. A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr. Biol. 9: 281–284. [DOI] [PubMed] [Google Scholar]
- Ghoda L., Lin, X., and Greene, W.C. 1997. The 90-kDa ribosomal S6 kinase (pp90rsk) phosphorylates the N-terminal regulatory domain of IκBα and stimulates its degradation in vitro . J. Biol. Chem. 272: 21281–21288. [DOI] [PubMed] [Google Scholar]
- Gray N.S., Wodicka, L., Thunnissen, A.M., Norman, T.C., Kwon, S., Espinoza, F.H., Morgan, D.O., Barnes, G., LeClerc, S., Meijer, L., et al. 1998. Exploiting chemical libraries, structures, and genomics in the search of kinase inhibitors. Science 281: 533–538. [DOI] [PubMed] [Google Scholar]
- Huang X., Begley, M., Morgenstern, K.A., Gu, Y., Rose, P., Zhao, H., and Zhu, X. 2003. Crystal structure of an inactive Akt2 kinase domain. Structure 11: 21–30. [DOI] [PubMed] [Google Scholar]
- Huse M. and Kuriyan, J. 2002. The conformational plasticity of protein kinases. Cell 109: 275–282. [DOI] [PubMed] [Google Scholar]
- Jensen C.J., Buch, M.B., Krag, T.O., Hemmings, B.A., Gammeltoft, S., and Frodin, M. 1999. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositidedependent protein kinase-1. J. Biol. Chem. 274: 27168–27176. [DOI] [PubMed] [Google Scholar]
- Joel P.B., Traish, A.M., and Lannigan, D.A. 1998. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J. Biol. Chem. 273: 13317–13323. [DOI] [PubMed] [Google Scholar]
- Jones S.W., Erikson, E., Blenis, J., Maller, J.L., and Erikson, R.L. 1988. A Xenopus ribosomal S6 kinase has two apparent kinase domains that are each similar to distinct protein kinases. Proc. Natl. Acad. Sci. 85: 3377–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornienko M., Montalvo, A., Carpenter, B.E., Lenard, M., Abeywickrema, P., Hall, D.L., Darke, P.L., and Kuo, L.C. 2005. Protein expression plasmids produced rapidly: Streamlining cloning protocols and robotic handling. Assay Drug Dev. Technol. 3: 661–674. [DOI] [PubMed] [Google Scholar]
- Lawrie A.M., Noble, M.E., Tunnah, P., Brown, N.R., Johnson, L.N., and Endicott, J.A. 1997. Protein kinase inhibition by Staurosporine revealed in details of the molecular interaction with CDK2. Nat. Struct. Biol. 4: 796–801. [DOI] [PubMed] [Google Scholar]
- Leighton I.A., Dalby, K.N., Caudwell, F.B., Cohen, P.T., and Cohen, P. 1995. Comparison of the specificities of p70 S6 kinase and MAPKAP kinase-1 identifies a relatively specific substrate for p70 S6 kinase: The N-terminal kinase domain of MAPKAP kinase-1 is essential for peptide phosphorylation. FEBS Lett. 375: 289–293. [DOI] [PubMed] [Google Scholar]
- McRee D.E. 1999. XtalView/Xfit-A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125: 156–165. [DOI] [PubMed] [Google Scholar]
- Meggio F., Donella Deana, A., Ruzzene, M., Brunati, A.M., Cesaro, L., Guerra, B., Meyer, T., Mett, H., Fabbro, D., Furet, P., et al. 1995. Different susceptibility of protein kinases to Staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur. J. Biochem. 234: 317–322. [DOI] [PubMed] [Google Scholar]
- Nowakowski J., Cronin, C.N., McRee, D.E., Knuth, M.W., Nelson, C.G., Pavletich, N.P., Rogers, J., Sang, B., Scheibe, D.N., Swanson, R.V., et al. 2002. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure 10: 1659–1667. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Palmer P., Gavin, A.C., and Nebreda, A.R. 1998. A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) Phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J. 17: 5037–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteet-Smith C.E., Smith, J.A., Lannigan, D.A., Freed, T.A., and Sturgill, T.W. 1999. Generation of constitutively active p90 ribosomal S6 kinase in vivo. Implications for the mitogen-activated protein kinase-activated protein kinase family. J. Biol. Chem. 274: 22135–22138. [DOI] [PubMed] [Google Scholar]
- Prade L., Engh, R.A., Girod, A., Kinzel, V., Huber, R., and Bossemeyer, D. 1997. Staurosporine-induced conformational changes of cAMP-dependent protein kinase catalytic subunit explain inhibitory potential. Structure 5: 1627–1637. [DOI] [PubMed] [Google Scholar]
- Richards S.A., Fu, J., Romanelli, A., Shimamura, A., and Blenis, J. 1999. Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Curr. Biol. 9: 810–820. [DOI] [PubMed] [Google Scholar]
- Roux P.P., Ballif, B.A., Anjum, R., Gygi, S.P., and Blenis, J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. 101: 13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota G.P., Cummings, L., Newell, F.S., Armstrong, C., Bain, J., Frodin, M., Grauert, M., Hoffmann, M., Schnapp, G., Steegmaier, M., et al. 2007. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten G.J., Vertegaal, A.C., Whiteside, S.T., Israel, A., Toebes, M., Dorsman, J.C., van der Eb, A.J., and Zantema, A. 1997. IκBα is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 16: 3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C., Schalk-Hihi, C., Struble, G., Ma, H., Petrounia, I., Brandt, B., Deckman, I.C., Patch, R.J., Player, M.R., Spurlino, J.C., et al. 2007. Crystal structure of the tyrosine kinase domain of colony-stimulating factor-1 recpetor (cFMS) in complex with two inhibitors. J. Biol. Chem. 282: 4094–4101. [DOI] [PubMed] [Google Scholar]
- Schwab M.S., Roberts, B.T., Gross, S.D., Tunquist, B.J., Taieb, F.E., Lewellyn, A.L., and Maller, J.L. 2001. Bub1 is activated by the protein kinase p90(Rsk) during Xenopus oocyte maturation. Curr. Biol. 11: 141–150. [DOI] [PubMed] [Google Scholar]
- Shaeffer H.J. and Webber, M.J. 1999. Mitogen-activated protein kinases: Specific messages from ubiquitous messengers. Mol. Cell. Biol. 19: 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.A., Poteet-Smith, C.E., Malarkey, K., and Sturgill, T.W. 1999. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo . J. Biol. Chem. 274: 2893–2898. [DOI] [PubMed] [Google Scholar]
- Smith K.J., Carter, P.S., Bridges, A., Horrocks, P., Lewis, C., Pettman, G., Clarke, A., Brown, M., Hughes, J., Wilkinson, M., et al. 2004. The structure of MSK1 reveals a novel autoinhibitory conformation for a dual kinase protein. Structure 12: 1067–1077. [DOI] [PubMed] [Google Scholar]
- Smith J.A., Poteet-Smith, C.E., Xu, Y., Errington, T.M., Hecht, S.M., and Lannigan, D.A. 2005. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 65: 1027–1034. [PubMed] [Google Scholar]
- Sutherland C., Leighton, I.A., and Cohen, P. 1993. Inactivation of glycogen synthase kinase-3h by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 296: 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaoki T., Nomoto, H., Takahashi, I., Kato, Y., Morimoto, M., and Tomita, F. 1986. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent kinase. Biochem. Biophys. Res. Commun. 135: 397–402. [DOI] [PubMed] [Google Scholar]
- Xing J., Ginty, D.D., and Greenberg, M.E. 1996. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273: 959–963. [DOI] [PubMed] [Google Scholar]
- Yang J., Cron, P., Good, V.M., Thompson, V., Hemmings, B.A., and Barford, D. 2002. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP–PNP. Nat. Struct. Biol. 9: 940–944. [DOI] [PubMed] [Google Scholar]
- Zhao B., Bower, M.J., McDevitt, P.J., Zhao, H., Davis, S.T., Johanson, K.O., Green, S.M., Concha, N.O., and Zhou, B.B. 2002. Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem. 277: 46609–46615. [DOI] [PubMed] [Google Scholar]
- Zheng J., Knighton, D.R., Ten Eyck, L.F., Karlsson, R., Xuong, N., Taylor, S.S., and Sowadski, J.M. 1993. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 32: 2154–2161. [DOI] [PubMed] [Google Scholar]