

Abstract
The translocated intimin receptor (TIR) of enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC) is required for EPEC and EHEC infections, which cause widespread illness across the globe. TIR is translocated via a type-III secretion system into the intestinal epithelial cell membrane, where it serves as an anchor for E. coli attachment via its binding partner intimin. While many aspects of EPEC and EHEC infection are now well understood, the importance of the intermolecular contacts made between intimin and TIR have not been thoroughly investigated. Herein we report site-directed mutagenesis studies on the intimin-binding domain of EPEC TIR, and how these mutations affect TIR-intimin association, as analyzed by isothermal titration calorimetry and circular dichroism. These results show how two factors govern TIR's binding to intimin: A three-residue TIR hot spot is identified that largely mediates the interaction, and mutants that alter the β-hairpin structure of TIR severely diminish binding affinity. In addition, peptides incorporating key TIR residues identified by mutagenesis are incapable of binding intimin. These results indicate that hot spot residues and structural orientation/preorganization are required for EPEC, and likely EHEC, TIR-intimin binding.
Keywords: EPEC, EHEC, intimin, translocated intimin receptor (TIR)
Intimin and the translocated intimin receptor (TIR) of enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC) play an integral role in attachment to intestinal epithelial cells (Marches et al. 2000; Vallance and Finlay 2000). These two proteins are encoded in the locus of enterocyte effacement and are expressed (TIR) or up-regulated (intimin) when EPEC/EHEC cells come into close contact with a host cell (McDaniel et al. 1995; Knutton et al. 1997). TIR, upon expression, is localized to the E. coli cell membrane and then translocated through the type-III secretion needle complex (Kenny et al. 1997; Gauthier and Finlay 2003) into the host cell. TIR then becomes immobilized in the intestinal epithelium with its intimin binding domain (IBD) displayed on the intestinal cell surface. Intimin, a transmembrane protein whose extracellular domain (ECD) is displayed on the E. coli surface, binds TIR-IBD, intimately attaching the E. coli cell to the intestinal epithelium (Kenny et al. 1997). This attachment ultimately leads to actin recruitment within the epithelial cell and the formation of attaching and effacing lesions (Knutton et al. 1987). Lesion formation is the precursor of intestinal cell membrane disruption and diarrhea in infected individuals (Knutton et al. 1987, 1997). In severe cases, infection leads to hemolytic uremic syndrome, a disease that kills 12% of those infected (Gerber et al. 2002; Garg et al. 2003).
The structure of intimin-ECD has been solved by NMR (Batchelor et al. 2000) and X-ray crystallography (Luo et al. 2000). In addition, the cocrystal structure of intimin-ECD bound to TIR-IBD has also been determined (Fig. 1; Luo et al. 2000). These structures have had a great impact on our understanding of these two proteins, including the elucidation of their binding interface. However, very limited additional work has been conducted to determine which specific residues of these two proteins are most responsible for their interaction. To our knowledge, the only studies along these lines have been reported by the Leong group, who carried out random mutagenesis studies on the ECD of intimin (Liu et al. 1999, 2002). However, because very few mutants were generated at the binding surface, the vast majority of this protein–protein interface has, until now, remained unstudied.
Figure 1.

(A) Depiction of intimin-ECD (surface) and TIR-IBD (wire) in their cocrystallized form. (B) Close-up representation of the intimin-ECD/TIR-IBD interface showing the β-hairpin of TIR-IBD examined in this study.
In this article, we report the systematic mutation of all of the nonalanine residues from the β-hairpin of EPEC TIR-IBD that lie in close proximity to intimin, thereby providing the most complete understanding of the TIR-intimin binding interface to date. The ability of each of these point mutants to bind to intimin-ECD has been characterized by isothermal titration calorimetry (ITC) and their secondary structures examined by circular dichroism (CD). From this study of 11 point mutants, we have identified a three-residue TIR-IBD binding hot spot. Several other mutants implicate key structural requirements for TIR-intimin association. Finally, we demonstrate that short peptides derived from the intimin-binding region of TIR-IBD do not have the ability to bind intimin-ECD. These peptides support our hypothesis that sequential, unstructured mimics alone are not sufficient to bind intimin-ECD, and that additional structural requirements are necessary to modulate this protein–protein interaction. Due to the 85% sequence identity between EPEC and EHEC TIR within the β-hairpin region (DeVinney et al. 1999), these results could apply to EHEC TIR as well.
Results
Alanine point mutants were generated for all nonalanine residues in the β-hairpin of TIR-IBD whose side chains were within 5 Å of intimin, as determined based on an inspection of the Luo et al. (2000) X-ray crystal structure (Protein Data Bank [PDB] code 1F02). These mutants were then screened for their ability to bind intimin-ECD by ITC. The results of this thermodynamic analysis of 11 mutants are shown in Tables 1 and 2 (results are the average ± SD of at least three independent titrations for each TIR-IBD mutant). Figure 2A depicts the location of each of these residues in the β-hairpin of TIR. The side chains are color-coded according to their respective ΔΔG values for intimin-ECD binding relative to wild-type TIR-IBD.
Table 1.
Analysis of native and mutant TIR-IBD proteins by ITC

Table 2.
The effect of mutating a given residue is compared to wild-type TIR-IBD/intimin-ECD binding

Figure 2.
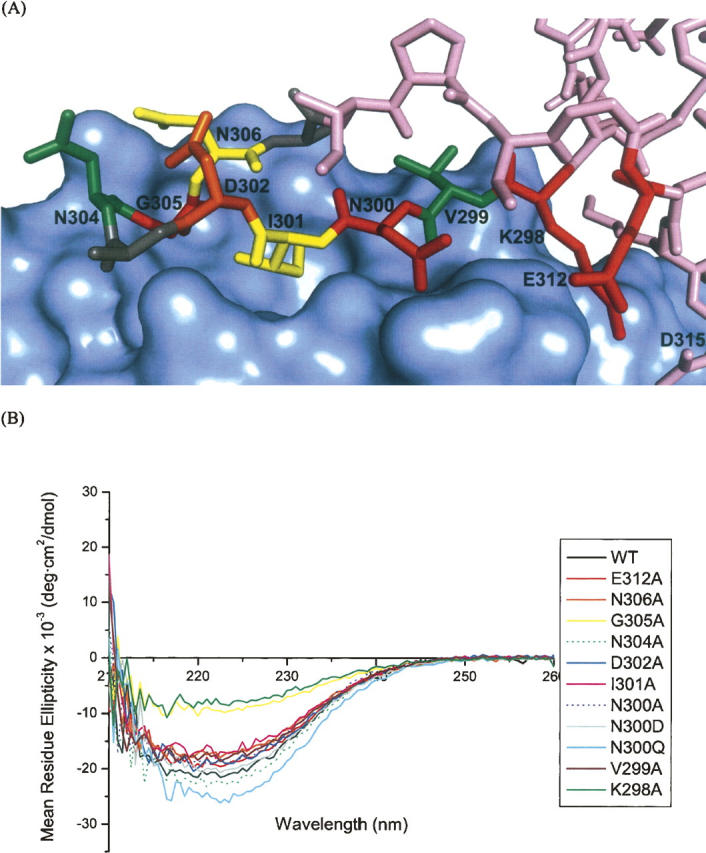
(A) TIR-IBD residues studied by alanine scanning mutagenesis are shown in colors corresponding to their effect on binding free energy change, ΔΔG (green indicates −0.50 to +0.50 kcal/mol; yellow, +0.50 to +1.0 kcal/mol; orange, +1.0 to +1.75 kcal/mol; red, +1.75 to +3.0 kcal/mol). Alanine residues that are part of the β-hairpin but were not mutated in this study, are colored gray. (B) Circular dichroism results from each of the mutants studied. The data are normalized for concentration and are buffer subtracted. While the majority of the mutations show the same global fold as native TIR-IBD, K298A and G305A both show a greater contribution to their overall fold from random coil.
Two of the alanine point mutants, V299A and N304A, had no significant effect on TIR-IBD/intimin-ECD binding (ΔΔG = −0.5 to +0.5 kcal/mol). Mutation of I301 or N306 to alanine had a modest effect (ΔΔG = +0.5 to +1.0 kcal/mol), while D302A caused a more substantial change in affinity (ΔΔG = 1.37 kcal/mol). Four alanine mutants (G305, E312, K298, and N300) had a large impact on the binding interaction, with ΔΔG values between 1.75 and 3.00 kcal/mol.
The secondary structure of each of the mutants was examined using CD spectroscopy to ensure that the changes in binding affinity observed by ITC were due to side-chain interactions themselves, and not changes in the structure of the protein. As can been seen in Figure 2B, most of the mutants appear to have similar CD spectra to that of wild-type TIR-IBD, with the exception of G305A and K298A. These two mutants, which were designed to probe the structural importance of TIR-IBD's β-hairpin, altered the overall structure of TIR-IBD as expected. The loss of binding affinity upon mutation of G305 or K298 likely reflects the significance of both the β-hairpin's conformation, and its positioning relative to the rest of TIR-IBD.
Mutation of asparagine-300 to alanine was found to have the most significant impact on TIR-IBD's ability to bind to intimin-ECD without altering the global fold (ΔΔG = 2.81 kcal/mol). In an effort to further probe this interaction, two additional mutations were made. N300D and N300Q both showed a significant decrease in binding free energy (ΔΔG = 1.32 and 2.09 kcal/mol, respectively) compared with wild-type TIR-IBD, further supporting the pivotal role of asparagine-300 in this interaction (Tables 1, 2).
In order to further examine the importance of conformational constraint on TIR-IBD's ability to bind intimin-ECD, we synthesized and tested three short peptides. These peptides (INV, AINV, and INVK) were designed to represent potential minimal binding domains of TIR, incorporating key residues identified by mutagenesis while minimizing total length in an effort to reduce the entropic penalty for binding. None showed any measurable affinity for intimin-ECD, as determined by ITC (data not shown).
Discussion
The binding interface of TIR and intimin is relatively small, burying 1335 Å2 of solvent-exposed surface area (Luo et al. 2000; Headd et al. 2007). It is therefore not surprising that most of the residues of TIR located near the interface have some impact on intimin binding. However, this study implicates three key residues, asparagine-300, isoleucine-301, and glutamic acid-312, as the binding hot spot (Clackson and Wells 1995; Jones and Thornton 1997) of this protein–protein interaction interface. Additionally, more conservative mutations of N300 allowed for the observation that the ability of this asparagine to form two intermolecular bonds, and their placement, affects binding to intimin-ECD.
Mutagenesis also revealed which residues contribute to the binding interaction to a lesser extent, and some that do not appear to participate in binding at all. Two residues, asparagine-304 and valine-299, were found to have no significant effect on TIR-IBD's ability to bind intimin-ECD. N304 points directly toward bulk solvent, and away from intimin, in the cocrystal structure of these two proteins. Accordingly, it was not surprising that mutation of this residue did not have a pronounced effect on binding. Valine-299 was slightly more surprising, because its side chain lies directly on the surface of intimin. However, because it does not fit into a pocket or other surface feature, replacing this hydrophobic residue with another hydrophobic residue, albeit one with a smaller side chain, did not reduce binding.
Two mutants, N306A and I301A, have only a modestly decreased binding free energy (ΔΔG = +0.5 to +1.0 kcal/mol) relative to wild-type TIR-IBD. Asparagine-306 is within hydrogen-bonding distance of lysine-919 of intimin, and the absence of this H-bonding capability could account for the 0.61 kcal/mol loss in ΔG observed in N306A. Many investigators have estimated the energetic value of a hydrogen bond in protein association, and it is now generally accepted that a range of 0.6–1.0 kcal/mol (ΔG) is a reasonable approximation (Myers and Pace 1996; Klostermeier and Millar 2002). I301A has a ΔΔG of 0.76 kcal/mol. Isoleucine-301 buries its side chain in a pocket on the surface of intimin (Fig. 2A), potentially serving to “set the register” for the binding interaction via this hydrophobic burial (Chothia and Janin 1975; Tsai et al. 1997). The low temperature factors for this residue (Supplemental Fig. S2) also suggest that I301 is critical for TIR-intimin interaction. Why, then, does mutating the residue to alanine only result in a ΔΔG of 0.76 kcal/mol? It is possible that the alanine methyl group is able to provide some of the registration function of the isoleucine side chain in the native structure. Additionally, we can speculate that the less favorable binding entropy for I301A binding to intimin is largely due to differences in solvent release, although this is partially compensated by the removal of the entropic penalty required for freezing out I301 side-chain rotation (Harpaz et al. 1994; Stites 1997; Lee et al. 2000).
Mutation of D302 to alanine produces a ΔΔG of binding of 1.37 kcal/mol. This was initially somewhat puzzling, because D302 does not appear to have any direct interactions with intimin. The side-chain oxygen does appear to make a cross-strand hydrogen bond to the backbone amide of TIR N306. However, mutation of this residue to alanine did not alter the CD spectrum of TIR-IBD. Aspartic acid cross-strand hydrogen bonds are well-documented, stabilizing factors in type-II β-turns (Hutchinson and Thornton 1994); thus, mutation of this residue likely caused small changes in the orientation or conformational dynamics of the β-hairpin (accounting for the change in binding free energy), without altering global fold of the protein.
Four amino acids have a greater than 1.5 kcal/mol effect on ΔG when mutated to alanine. The first, lysine-298, participates in both an intramolecular salt bridge with TIR aspartic acid-315, and also buries its methylene groups on the surface of intimin (Fig. 2A). Mutation of K298 to alanine resulted in a ΔΔG of 2.86 kcal/mol. The salt bridge made by K298 to D315 is located exactly at the boarder of the β-hairpin and α-helix of TIR-IBD. This bridge may serve to maintain the angle and positioning of the β-hairpin of TIR-IBD with respect to the rest of the protein. In fact, when this bond is broken (by mutation of lysine to alanine), random coil features become a more predominant component of the secondary structure of the protein (Fig. 2B). Additionally, the K298A mutation causes a very large decrease in TΔΔS (−13.28 kcal/mol), consistent with a greater binding-induced loss of conformational freedom for the K298A mutant. Accordingly, alteration of the structure or alignment of the β-hairpin of TIR is likely responsible for the majority of this loss in binding affinity.
The second mutation that had a very pronounced effect on TIR-IBD/intimin-ECD binding was glycine-305. While it is unusual to mutate a glycine residue when conducting alanine scanning mutagenesis, we were interested in determining whether converting G305 to alanine would disrupt the type-II β-hairpin, thereby serving as an indicator of its structural importance. Indeed, mutation to alanine at position 305 caused a loss of binding affinity (ΔΔG) of 1.82 kcal/mol and also altered the overall fold of the protein (Fig. 2B). This structural mutation also caused a very large entropic penalty for binding (TΔΔS = −16.50 kcal/mol). Taken together, the K298A and the G305A mutants indicate that both the fold and orientation of the β-hairpin of TIR-IBD are essential for submicromolar binding to intimin-ECD.
While K298A and G305A have significant effects on binding by altering the structure of TIR-IBD, two other residues also exhibit large effects on binding without disrupting the fold of the protein. Glutamic acid-312 appears to make a direct salt bridge to lysine-927 of intimin and also forms part of a pocket on the surface of TIR that is occupied by intimin K927. Thus, it is unsurprising that the E312A mutant has a destabilizing effect of 2.11 kcal/mol. It is also noteworthy that E312 is the key residue differentiating EPEC TIR-IBD from EHEC TIR-IBD (DeVinney et al. 1999; Luo et al. 2000). In EHEC TIR-IBD, glutamic acid is replaced by valine at position 312. It therefore makes sense that our E312A mutant roughly matches the affinity of EHEC TIR-IBD for EPEC intimin (DeVinney et al. 1999).
Mutation of asparagine-300 to alanine had the most destructive effect on TIR-IBD binding affinity (ΔΔG = 2.81 kcal/mol), without affecting the overall fold of the protein. A detailed examination of the cocrystal structure revealed that the side chain of asparagine-300 likely participates in two direct contacts with intimin-ECD (Fig. 3). The side-chain amino group appears to form a hydrogen bond with glutamic acid-930, and the side-chain oxygen H-bonds to the side-chain amino group of asparagine-932. This hydrogen bond to N932 may also orient the side chain of N932 in such a way that its oxygen can accept a hydrogen bond from the backbone amide nitrogen of isoleucine-301 of TIR-IBD.
Figure 3.

Key bonds between N300 of TIR-IBD and intimin-ECD are shown with black dashed lines. Proposed hydrogen bonds between the backbone of TIR-IBD and intimin-ECD are shown as gray dashed lines. Mutagenesis, CD, and peptidomimetic screening results indicate that while N300, I301, and E312 are integral residues for TIR-IBD/intimin-ECD association, structural preorganization of the β-hairpin residues of TIR-IBD is required for binding.
To further investigate N300, two additional mutations were made. First, asparagine-300 was mutated to glutamine in an effort to determine the importance of the precise placement of this side chain. If unimportant, this would argue against asparagine-300 being involved in a more highly coordinated network of hydrogen bonds between intimin and TIR. N300Q had a substantial effect on binding (ΔΔG = 2.09 kcal/mol), slightly less than the loss in affinity displayed by the N300A mutant, but large nevertheless. Second, asparagine-300 was changed to an aspartic acid. We anticipated that this would have a smaller effect on ΔG than the N300A mutation, since only one direct H-bond would be lost. This was the case, with a ΔΔG of 1.32 kcal/mol compared with wild-type TIR-IBD. Interestingly, this change in binding free energy more closely resembles what would be expected if two H-bonds were broken, providing evidence that the H-bond between N300 and intimin N932 may serve to orient N932 in such a way that its side-chain oxygen can H-bond with I301 (Fig. 3).
The importance of asparagine-300 is further reinforced when the temperature factors from the cocrystal structure of intimin-ECD and TIR-IBD are examined (Supplemental Fig. S2). In comparison with the rest of TIR-IBD, N300 atoms have very low B-values (Luo et al. 2000). It appears that asparagine-300 is part of a more complex group of interactions between intimin-ECD and TIR-IBD, and that it may serve as a central anchoring point for TIR-IBD. This idea is further supported by a recent review of protein–protein interactions (Headd et al. 2007), which found that a common theme in key contact areas is the presence of two to three backbone–backbone H-bonds, as may be the case in this region of TIR-IBD and intimin-ECD (Fig. 3).
These mutagenesis results can be summed up in an overall hypothesis as follows: The key hydrogen-bonding interactions provided by asparagine-300, the lock and key type fit of isoleucine-301, and the salt bridge of glutamic acid-312 of EPEC TIR serve as a hot spot for intimin-TIR binding. This is also consistent with the results of Liu et al. (1999), who observed that asparagine-932 of EHEC intimin (which would interact with N300 of EPEC/EHEC TIR) is important for TIR-IBD binding. The complete lack of affinity demonstrated by synthetic peptides INV, AINV, and INVK show that the hot spot sequence alone is not enough to confer intimin binding ability, consistent with our results for the secondary structure-altering mutants K298A and G305A. These residues are important for preorganizing TIR-IBD into a binding-competent conformation. Glycine-305 and lysine-298 allow for the proper type-II β-turn, and its positioning, respectively. Due to the very high sequence identity between the β-hairpin of EPEC and EHEC TIR (85%), it seems likely that these same interactions (with the exception of E312 explained earlier) govern the binding of both proteins to their cognate receptors. This, for the first time, provides a detailed explanation of how this critical protein–protein interaction event is controlled in enteropathogenic and enterohemorrhagic E. coli. We also anticipate that this information will prove useful in the design of peptidomimetics able to interfere with intimin-TIR binding.
Materials and Methods
Molecular biology
Intimin-ECD (amino acids 658–939) and TIR-IBD (272–363) constructs were kindly provided by Professors Natalie Strynadka and Brett Finlay (University of British Columbia) in pET-28a(+) vectors prepared as described previously (Luo et al. 2000). These constructs were transformed into E. coli BL21(DE3)pLysS and BL21(DE3) cell lines, respectively. Cells were grown at 37°C in Lauria-Bertani media with 25 μg/mL kanamycin until an optical density (595 nm) of 0.5–0.8 was reached. They were then shifted to room temperature, and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. Following a 24-h induction period, cells were pelleted at 3000 rpm in a Beckman GS-6R swinging bucket centrifuge (Beckman Coulter). Cell pellets were then resuspended in buffer (20 mM HEPES, 500 mM NaCl, pH 7.5) and sonicated for 15 min. The suspension was then pelleted at 15,000 rpm for 1 h, filtered through a 0.45-μm filter, and applied to a Pharmacia Hi-trap nickel chelating column using a peristaltic pump at a flow rate of 0.5 mL/min. The column was washed with 20 mM HEPES, 500 mM NaCl, and 50 mM imidazole (pH 7.5), to remove any nonspecifically bound protein. Bound protein was eluted in 20 mM HEPES, 500 mM NaCl, and 300 mM imidazole, pH 7.5, and examined for purity by 12% SDS-PAGE. Before ITC and CD experiments, all protein samples were dialyzed for 24 h with one buffer change against 200 volumes of dialysis buffer (50 mM HEPES, 150 mM NaCl, pH 7.2). Following dialysis, concentrations were determined by UV-vis spectrophotometry using the calculated molar extinction coefficients of 37,080 and 1280 (M−1cm−1) for intimin-ECD and TIR-IBD, respectively (Ross et al. 2007).
TIR-IBD point mutants were prepared using a Stratagene QuikChange mutagenesis kit. Each residue of the TIR-IBD β-hairpin was analyzed to determine if its side chain was within interacting distance (limit: 5 Å) of intimin. This was determined using the previously published cocrystal structure of intimin-ECD and TIR-IBD (Luo et al. 2000) as visualized with PyMOL (v. 0.99; DeLano Scientific). Primers for the selected mutants were designed according to kit recommendations, and sequences for each are available in the Supplemental material (Supplemental Table S1). The sequences of individual mutants were confirmed at the University of Rochester Functional Genomics Center (Rochester, NY). Mutants with the desired sequences were transformed into BL21(DE3) E. coli cells, and protein was prepared, dialyzed (50 mM HEPES, 150 mM NaCl, pH 7.2), and analyzed for purity using 12% SDS-PAGE in the same manner as wild-type TIR-IBD. Concentrations were determined using the same molar extinction coefficient as used for native TIR-IBD. The buffer used for all experiments discussed herein was the same as used for protein dialysis.
Isothermal titration calorimetry
ITC experiments were carried out using a MicroCal VP-ITC isothermal titration calorimeter. All experiments were carried out at 25°C (unless otherwise stated) with dialysis buffer (described above) in the reference cell. All titrations were reference subtracted to control for heats of dilution, which in all cases were negligible. All experiments were performed with 10 μcal/sec reference power, 290 rpm stir rate, and 60 5-μL injections with 300 sec spacing between injections. Between all experiments, the calorimeter sample cell and injection needle were each cleaned with at least 500 mL distilled, deionized water, followed by a final cleaning with dialysis buffer. All protein samples were degassed with stirring at 25°C for three 5-min intervals. Data were fit using the MicroCal Origin 7.0 software on the ITC instrument. Binding curves were analyzed using a one-site binding model in Origin (v 7.0; Origin Labs). Representative isotherms generated for each TIR-IBD mutant are available in the Supplemental material (Supplemental Fig. S1). Reported binding constants are average values obtained from at least three independent measurements.
Circular dichroism
CD experiments were conducted using an AVIV 202 spectrometer at 25°C (AVIV Biomedical). Scans were conducted from 260–200 nm in 0.5-nm increments with 5-sec sampling time per 0.5 nm, using a 1-mm path length cell (Hellma). Spectra were normalized, truncated at 210 nm (where strong buffer signal started to obscure the protein signal), and plotted using Excel (Microsoft Office 2002; Microsoft).
Peptide synthesis
Unless otherwise stated all chemicals and reagents used in peptide synthesis were purchased from Sigma-Aldrich and used as obtained from the manufacturer. The INV, AINV, and INVK peptides were synthesized on para-methyl-benzhydrylamine resin (0.4 mmol/g loading) (Advanced ChemTech). Resin was initially swollen in methylene chloride (DCM; Mallinckrodt Baker) and treated with 10% N,N′-diisopropylethylamine (DIPEA; Advanced ChemTech) in DCM. Resin was then washed with a sequence of 3× DCM, 3× N,N-dimethylformamide (DMF; Mallinckrodt Baker), 3× methanol (BDH/VWR), 3× DCM, 1× DMF, nutating for 5 min per wash step. Couplings were conducted using 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU; 3.0 equivalents with respect to resin; GenScript), N-α-(9-fluorenylmethyloxycarbonyl; Fmoc)–protected amino acids (3.0 eq; Advanced ChemTech), and DIPEA (5.0 eq) in DMF for 30 min (3 min HBTU, Fmoc-amino acid, and DIPEA premixing in DMF). Amino acids were double-coupled and then washed as above. The Fmoc protecting group was removed with two cycles of 20% piperidine/DMF (30 min each), followed by washing as above. This cycle of double-couple, double-deprotect was repeated until the synthesis was completed. After each coupling and deprotection, a small (∼2 mg) aliquot of resin was heated in ninhydrin stain to determine if free amines were present. In all cases, the desired presence or lack of free amines were observed.
Following the end of chain elongation, global deprotection was undertaken to first remove the Fmoc group, followed by washing and removal of the trityl side-chain protecting group (2% trifluoroacetic acid [TFA], 2% triethylsilane [TES; Oakwood Products], DCM, 2 × 30 min). After a final wash sequence, the peptides were liberated from the resin with 5% TES in TFA (2 mL/100 mg resin) for 10 min at 0°C, followed by addition of trifluoromethanesulfonic acid (TFMSA; 200 μL/100 mg resin). The reaction vessels were allowed to cool, and then the resin and solvent mixture were stirred at room temperature for 2 h. Resin and solvent were then filtered through glass wool, and peptides were precipitated with cold ether (∼5 volumes; EMD Biosciences). Acid/ether solutions were left at −20°C overnight, and then precipitates were centrifuged at 2000 rpm for 30 min at 4°C. The acid/ether solution was removed, and pellets were washed with cold ether and centrifuged as before. Pellets were washed in this fashion three times (total) and then allowed to dry for 6 h before being placed under high vacuum overnight. Peptides were solubilized in protein dialysis buffer, to a final concentration of 5 mM. They were then screened for their ability to bind 100 μM intimin-ECD by ITC.
Electronic supplemental material
Supporting information is available online, including primer sequences, ITC data, and an additional depiction of the intimin-TIR binding interface.
Acknowledgments
We thank Jessica L. Goodman (Yale University) and Professor Joseph E. Wedekind (University of Rochester) for helpful discussions regarding mutagenesis studies. We also thank Professor Natalie Strynadka (University of British Columbia) for providing us with the intimin-ECD and TIR-IBD constructs used in our initial work.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Benjamin Miller, University of Rochester, Department of Dermatology, 601 Elmwood Avenue, Box 697, Rochester, NY 14642, USA; e-mail: Benjamin_Miller@urmc.rochester.edu; fax: (585) 273-1346.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073128607.
References
- Batchelor M., Prasannan, S., Daniell, S., Reece, S., Connerton, I., Bloomberg, G., Dougan, G., Frankel, G., and Matthews, S. 2000. Structural basis for recognition of the translocated intimin receptor (Tir) by intimin from enteropathogenic Escherichia coli . EMBO J. 19: 2452–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C. and Janin, J. 1975. Principles of protein–protein recognition. Nature 256: 705–708. [DOI] [PubMed] [Google Scholar]
- Clackson T. and Wells, J.A. 1995. A hot spot of binding energy in a hormone-receptor interface. Science 267: 383–386. [DOI] [PubMed] [Google Scholar]
- DeVinney R., Stein, M., Reinscheid, D., Abe, A., Ruschkowski, S., and Finlay, B.B. 1999. Enterohemorrhagic Escherichia coli 0157:H7 produces Tir, which is translocated to the host cell membrane but is not tyrosine phosphorylated. Infect. Immun. 67: 2389–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A.X., Suri, R.S., Barrowman, N., Rehman, F., Matsell, D., Rosas-Arellano, M.P., Salvadori, M., Haynes, R.B., and Clark, W.F. 2003. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome. JAMA 290: 1360–1370. [DOI] [PubMed] [Google Scholar]
- Gauthier A. and Finlay, B.B. 2003. Translocated intimin receptor and its chaperone interact with ATPase of the type III secretion apparatus of enteropathogenic Escherichia coli . J. Bacteriol. 185: 6747–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber A., Karch, H., Allerberger, F., Verweyen, H.M., and Zimmerhackl, L.B. 2002. Clinical course and the role of Shiga toxin-producing Escherichia coli infection in the hemolytic–uremic syndrome in pediatric patients, 1997-2000, in Germany and Austria: A prospective study. J. Infect. Dis. 186: 493–500. [DOI] [PubMed] [Google Scholar]
- Harpaz Y., Gerstein, M., and Chothia, C. 1994. Volume changes on protein folding. Structure 2: 641–649. [DOI] [PubMed] [Google Scholar]
- Headd J.J., Ban, Y.E.A., Brown, P., Edelsbrunner, H., Vaidya, M., and Rudolph, J. 2007. Protein–protein interfaces: Properties, preferences, and projections. J. Proteome Res. 6: 2576–2586. [DOI] [PubMed] [Google Scholar]
- Hutchinson E.G. and Thornton, J.M. 1994. A revised set of potentials for β-turn formation in proteins. Protein Sci. 3: 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. and Thornton, J.M. 1997. Analysis of protein–protein interaction sites using surface patches. J. Mol. Biol. 272: 121–132. [DOI] [PubMed] [Google Scholar]
- Kenny B., DeVinney, R., Stein, M., Reinscheild, D.J., Frey, E.A., and Finlay, B.B. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91: 511–520. [DOI] [PubMed] [Google Scholar]
- Klostermeier D. and Millar, D.P. 2002. Energetics of hydrogen bond networks in RNA: Hydrogen bonds surrounding G+1 and U42 are the major determinants for the tertiary structure stability of the hairpin ribozyme. Biochemistry 41: 14095–14102. [DOI] [PubMed] [Google Scholar]
- Knutton S., Lloyd, D.R., and McNeish, A.S. 1987. Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect. Immun. 55: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutton S., Adu-Bobie, J., Bain, C., Phillips, A.D., Dougan, G., and Frankel, G. 1997. Down regulation of intimin expression during attaching and effacing enteropathogenic Escherichia coli adhesion. Infect. Immun. 65: 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.L., Kinnear, S.A., and Wand, A.J. 2000. Redistribution and loss of side chain entropy upon formation of a calmodulin-peptide complex. Nat. Struct. Biol. 7: 72–77. [DOI] [PubMed] [Google Scholar]
- Liu H., Magoun, L., Luperchio, S., Schauer, D.B., and Leong, J.M. 1999. The Tir-binding region of enterohemorrhagic Escherichia coli intimin is sufficient to trigger actin condensation after bacterial-induced host cell signaling. Mol. Microbiol. 34: 67–81. [DOI] [PubMed] [Google Scholar]
- Liu H., Radhakrishnan, P., Magoun, L., Prabu, M., Campellone, K.G., Savage, P., He, F., Schiffer, C.A., and Leong, J.M. 2002. Point mutants of EHEC intimin that diminish Tir recognition and actin pedestal formation highlight a putative Tir binding pocket. Mol. Microbiol. 45: 1557–1573. [DOI] [PubMed] [Google Scholar]
- Luo Y., Frey, E.A., Pfuetzner, R.A., Creagh, A.L., Knoechel, D.G., Haynes, C.A., Finlay, B.B., and Strynadka, N.C.J. 2000. Crystal structure of enteropathogenic Escherichia coli intimin-receptor complex. Nature 405: 1073–1077. [DOI] [PubMed] [Google Scholar]
- Marches O., Nougayrede, J., Boullier, S., Mainil, J., Charlier, G., Raymond, I., Pohl, P., Boury, M., De Rycke, J., Milon, A., et al. 2000. Role of Tir and intimin in the virulence of rabbit enteropathogeic Escherichia coli serotype 0103:H2. Infect. Immun. 68: 2171–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel T.K., Jarvis, K.G., Donnenberg, M.S., and Kaper, J.B. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. 92: 1664–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers J.K. and Pace, N.C. 1996. Hydrogen bonding stabilizes globular proteins. Biophys. J. 71: 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross N.T., Mace, C.R., and Miller, B.L. 2007. Biophysical analysis of the EPEC translocated intimin receptor-binding domain. Biochem. Biophys. Res. Commun. 362: 1073–1078. [DOI] [PubMed] [Google Scholar]
- Stites W.E. 1997. Protein-protein interactions: Interface structure, binding thermodynamics, and mutational analysis. Chem. Rev. 97: 1233–1250. [DOI] [PubMed] [Google Scholar]
- Tsai C., Lin, S.L., Wolfson, H.J., and Nussinov, R. 1997. Studies of protein–protein interfaces: A statistical analysis of the hydrophobic effect. Protein Sci. 6: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance B.A. and Finlay, B.B. 2000. Exploitation of host cells by enteropathogenic Escherichia coli . Proc. Natl. Acad. Sci. 97: 8799–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]