

Abstract
A general acid–base catalytic mechanism is responsible for the cleavage of the phosphodiester bonds of the RNA by ribonuclease A (RNase A). The main active site is formed by the amino acid residues His12, His119, and Lys41, and the process follows an endonucleolytic pattern that depends on the existence of a noncatalytic phosphate-binding subsite adjacent, on the 3′-side, to the active site; in this region the phosphate group of the substrate establishes electrostatic interactions through the side chains of Lys7 and Arg10. We have obtained, by means of site-directed mutagenesis, RNase A variants with His residues both at positions 7 and 10. These mutations have been introduced with the aim of transforming a noncatalytic binding subsite into a putative new catalytic active site. The RNase activity of these variants was determined by the zymogram technique and steady-state kinetic parameters were obtained by spectrophotometric methods. The variants showed a catalytic efficiency in the same order of magnitude as the wild-type enzyme. However, we have demonstrated in these variants important effects on the substrate's cleavage pattern. The quadruple mutant K7H/R10H/H12K/H119Q shows a clear increase of the exonucleolytic activity; in this case the original native active site has been suppressed, and, as consequence, its activity can only be associated to the new active site. In addition, the mutant K7H/R10H, with two putative active sites, also shows an increase in the exonucleolytic preference with respect to the wild type, a fact that may be correlated with the contribution of the new active site.
Keywords: ribonuclease A, active site, enzyme kinetics, catalytic mechanism, protein engineering, artificial enzymes
Bovine pancreatic ribonuclease A (RNase A) (EC 3.1.27.5) is an endonuclease that catalyzes the breakdown of single-stranded RNA by the cleavage of the 3′,5′-phosphodiester linkages when the base of the nucleotide in the 3′ position of the scissile bond is a pyrimidine. The physical, chemical, and enzymatic properties of RNase A have been extensively characterized (for reviews, see D'Alessio and Riordan 1997; Raines 1998). Kinetic studies, using different oligonucleotides as low molecular mass substrates and homopolynucleotides as homologous to the RNA molecule, together with structural and molecular modeling studies indicated the existence of several substrate binding subsites in the structure of RNase A. This structure gave support to an enzyme-substrate binding model based on the presence of specific sites for the binding of the bases, riboses, and phosphates of the substrate (Fig. 1) (Parés et al. 1991; Boix et al. 1994; Moussaoui et al. 1996). The cleavage of the phosphodiester bond of the RNA by RNase A takes place through an acid–base mechanism in the active site, named p1, formed by the side chains of the amino acids His12, His119, and Lys41. In addition to the active site p1, other two main noncatalytic phosphate-binding subsites were characterized, the p0 site (Lys66), which binds the phosphate group of the nucleotide adjacent upstream to that located in the active site (Fontecilla-Camps et al. 1994; Nogués et al. 1998; Cuchillo et al. 2002), and the p2 site (Lys7 and Arg10), which binds the phosphate group of the corresponding adjacent nucleotide located in the 3′ side (Parés et al. 1980, 1991; de Llorens et al. 1989; Boix et al. 1994; Nogués et al. 1995; Fisher et al. 1998a,b) (Fig. 1). Other phosphate-binding subsites, such as p-1 (Arg85) (Fontecilla-Camps et al. 1994; Fisher et al. 1998a) and those located on the surface of the protein are also involved in the formation of the enzyme–substrate complex. The RNase A cleavage pattern of the homopolynucleotide polycytidylic acid (poly(C)) showed the preference of the enzyme for the longer substrate molecules releasing fragments containing six to eight nucleotide units, indicating a clear endonucleolytic activity (Moussaoui et al. 1996). This preference was also observed in the cleavage pattern of lower molecular mass oligocytidylic acids (Cuchillo et al. 2002), although in this case bonds adjacent to the terminal residues were also broken in a significant proportion. The p2 binding site plays a key role in the endonucleolytic activity of RNase A as has been demonstrated from the analysis of RNase variants obtained either by chemical modification or by site-directed mutagenesis; the variants that lack electrostatic interaction in this site show a clear shift toward the exonucleolytic cleavage of the substrate (Moussaoui et al. 1996; Cuchillo et al. 2002). Based on the knowledge about the role of the p2 binding site, the aim of the present work has been to explore the possibilities offered by protein engineering in the design of new variants of RNase A in which p2 has been transformed into a new catalytic site, instead of just being a phosphate-binding site. This should allow us to better understand the mechanism of action as well as some of the factors involved in the reaction specificity of the enzyme. To this end, RNase A variants with His residues at positions 7 and 10 were obtained. Although we analyzed other variants, in the present work we only describe the mutants that have proved to be more relevant for the studies about the new active site: (1) K7H/R10H, with an unmodified main active site at p1 but with a potential new active site in p2, (2) K7H/R10H/H12K/H119Q, a variant in which a potential new active site in p2 has been introduced at the same time that the main active site histidines of p2 have been replaced by a lysine and a glutamine, respectively, thus maintaining the basic character of the region, and (3) H12K/H119Q, a mutant that corresponds to a negative control for RNase activity.
Figure 1.
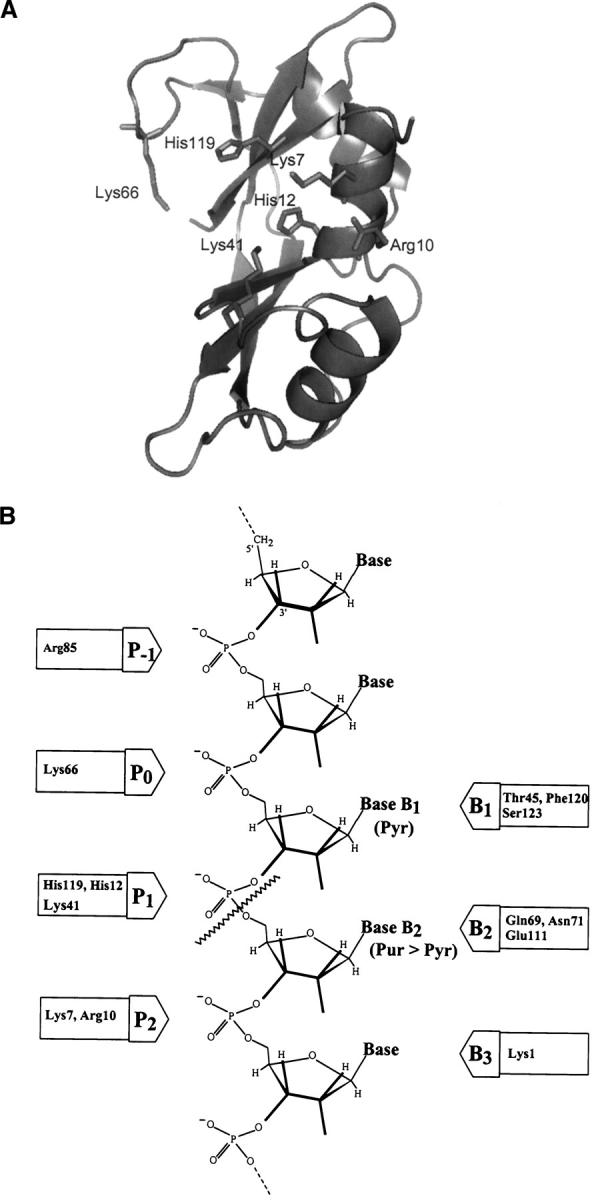
(A) Three-dimensional structure of RNase A. Amino acid residues belonging to the active site p1 (His12, His 119, and Lys41) and to the noncatalytic phosphate-binding subsites p0 (Lys66) and p2 (Lys7 and Arg10) are shown in stick. The picture was obtained using the program PyMOL (DeLano Scientific program, PDB accession code 7RSA). (B) Schematic representation of the substrate-binding sites; p1 corresponds to the active site where the cleavage of the phosphodiester bond takes place (wavy line).
The catalytic activity of the wild-type RNase A and variants was analyzed following different approaches with the aim of characterizing substrate specificity, steady-state kinetic parameters and the preference for an endo- or exonucleolytic activity. The results demonstrate that the presence of His residues at positions 7 and 10 gives rise to a new active site in the p2 region that maintains the specificity for pyrimidines (with C better than U) but with a clear increase in the preference for the scission of phosphodiester bonds near the ends of the substrate molecule. The absence of an efficient phosphate-binding site adjacent in the 3′ side, which contributes to the binding of the RNA chain, would be responsible for the change in the cleavage preference.
Results
Design and preparation of RNase A variants and characterization of the enzyme activity
Previous studies indicated that RNase A obtained either from a commercial source or from recombinant DNA technology displayed the same kinetic parameters both for the hydrolysis reaction of the substrate cytidine 2′,3′-cyclic phosphate (C>p) and for the transphosphorylation reaction of the substrates cytidylyl-3′,5′-adenosine (CpA) and poly(C) (Boix et al. 1994). In the present study, different RNase A variants were obtained by site-directed mutagenesis, with the aim of introducing a potential new catalytic site in RNase A. This new active site was designed from the knowledge about specific regions of the protein involved in the binding of the negatively charged phosphates of the RNA in noncatalytic subsites. These phosphate-binding sites contribute through cooperative binding to the catalytic efficiency of the enzyme (Nogués et al. 1995; Moussaoui et al. 1996). The phosphate-binding site p2 adjacent at the 3′ side of the main active site p1 is formed by amino acids Lys7 and Arg10 (Fig. 1). The role of this site in the RNase A reaction has been extensively characterized: It contributes to the binding efficiency in the formation of the enzyme–substrate complex, is involved in the endonucleolytic activity of the enzyme (Moussaoui et al. 1998; Cuchillo et al. 2002), and plays an indirect role in the catalytic mechanism (Boix et al. 1994). In this context, and from the knowledge of the topology of this part of the protein, we hypothesized that substitution of both amino acids, Lys7 and Arg10, by histidines could create a new active site in this region. At the same time, the main active site at p1, is maintained in the K7H/R10H variant. In the K7H/R10H/H12K/H119Q variant the main active site histidines were substituted by a basic binding region; the amino acid residues Lys and Gln were chosen to replace His12 and His119, respectively, in order to keep the electrostatic binding potential and the overall volume of the side chains in this region. The H12K/H119Q variant was designed as a negative control for RNase activity, as the main active site is suppressed by the lack of histidines and no new potential active site is introduced.
Figure 2 shows the RNase activity present in the intracellular soluble fraction of Escherichia coli cells transformed with the plasmid pET11d as determined by the zymogram technique. Cells transformed with the plasmid lacking the RNase A insert show a high molecular mass band corresponding to the intrinsic E. coli RNase activity (lane 2). Cells transformed with the plasmid containing the RNase gene show, in addition to the endogenous activity, an activity band that corresponds to the electrophoretic mobility of RNase A (lanes 1 and 3); this band is not detected in cells transformed with the plasmid containing the H12K/H119Q-RNase A gene, suggesting that this mutant can be used as a negative control for the main active site.
Figure 2.

Positive and negative controls of recombinant RNases. Activities were determined by the zymogram technique in 15% SDS-PAGE containing poly(C) as substrate. (Lane 1) 300 pg of RNase A. (Lane 2) 5 μl of the intracellular soluble fraction of E. coli cells transformed with the plasmid pET11d lacking the RNase A insert. (Lane 3) 5 μl of the intracellular soluble fraction of E. coli cells transformed with the plasmid pET11d carrying native RNase A as target gene. (Lane 4) 5 μl of the intracellular soluble fraction of E. coli cells transformed with the plasmid pET11d carrying H12K/H119Q-RNase A as target gene.
RNase A variants obtained by site-directed mutagenesis were purified according to the general procedure described by Boix et al. (1999b). However, the steepness of the salt gradient applied to the cation exchange chromatography was adjusted to optimum separation in each case. The molecular mass of the purified RNase A variants was checked by MALDI-TOF mass spectrometry and the N-terminal sequences were checked for confirmation that the right species had been expressed. These techniques were also used to check the absence of contaminating proteins that could interfere in the assays directed to the characterization of the RNase activity.
Figure 3A shows the 15% SDS-PAGE and Coomassie Blue staining of native RNase A (lane 1), the purified H12K/H119Q-RNase A (lane 2), K7H/R10H/H12K/H119Q-RNase A (lane 3), and K7H/R10H-RNase A (lane 4).
Figure 3.

SDS-15% PAGE with Coomassie blue staining (A) and RNase activity staining on gels containing either poly(C) (B) or poly(U) (C) as substrates. (A) (Lane 1) native RNase A. (Lane 2) H12K/H119Q-RNase A. (Lane 3) K7H/R10H/H12K/H119Q-RNase A. (Lane 4) K7H/R10H-RNase A (B) (Lane 1) native RNase A (30 ng). (Lane 2) K7H/R10H/H12K/H119Q-RNase A (30 ng). (Lane 3) K7H/R10H-RNase A (30 ng). (Lane 4) native RNase A (0.2 ng). (Lane 5) H12K/H119Q-RNase A (80 ng). (C) (Lane 1) native RNase A (30 ng). (Lane 2) K7H/R10H/H12K/H119Q-RNase A (30 ng). (Lane 3) K7H/R10H-RNase A (30 ng).
The zymogram technique is a nonquantitative method, although it is very powerful for detecting RNase activities. The comparative activity staining with different homopolynucleotides as substrates was used for the initial characterization of these variants. The presence of the polymeric substrate in the gel and the absence of β-mercaptoethanol in the protein sample may introduce some distortion in the protein electrophoretic mobility. Wild-type RNase A (Fig. 3B, lanes 1,4) shows a minor high molecular mass band that corresponds to a spontaneously formed noncovalent dimeric form. Lanes 2 and 3 of Figure 3B show the activity associated to the variants K7H/R10H/H12K/H119Q and K7H/R10H, respectively. In lane 5 the variant H12K/H119Q was analyzed; in this variant, although the main active site is suppressed by the lack of histidines, a very low residual activity is maintained.
Wild-type RNase A and K7H/R10H/H12K/H119Q and K7H/R10H variants showed a higher activity with poly(C) (Fig. 3B) than with poly(U) (Fig. 3C); no activity was observed with the substrate poly(A) (data not shown). A more detailed kinetic characterization of the activity of each variant was obtained from the determination of the steady-state kinetic parameters (see below).
Kinetic characterization of the K7H/R10H and K7H/R10H/H12K/H119Q variants in relation with the native enzyme
Transphosphorylation reaction
The properties of the enzyme activity associated to the presence of His residues at positions 7 and 10 were analyzed by determining the activity of the native enzyme and the variants by means of spectrophotometric methods. The cleavage of an oligo- or polynucleotide substrate by RNase A takes place in two steps: The first step is a transphosphorylation reaction in which the susceptible 3-5′phosphodiester bonds are cleaved with formation of a 2′3′-cyclic phosphodiester; only when no susceptible 3′-5′ phosphodiester bonds are left, the hydrolysis of the 2′,3′-cyclic phosphodiester bonds to a 3′-phosphate product takes place (second step) (Cuchillo et al. 1993). Consequently, both reactions take place separately, the catalytic efficiency of RNase A for the transphosphorylation reaction being much higher than that for the hydrolysis reaction. Table 1 shows the comparison of the activity of the variants expressed as a percentage with respect to RNase A. The H12K/H119Q variant shows a very small residual activity that has also been observed by the zymogram technique. This activity may be a consequence of the distortion of the substrate structure strain induced by the interaction with the enzyme in a way similar to that occurring in catalytic antibodies (abzymes) by proximity effects (Hilvert 2000). To understand this fact it is important to point out that (1) this variant maintains all the noncatalytic substrate binding subsites that contribute to the correct alignment of the substrate (Parés et al. 1991; Nogués et al. 1998) and (2) in the region of the active site (p1) two electrostatic interactions are still present through the side chain of the new lysine in position 12 and that of the unmodified Lys41. The fact that this residual activity of the H12K/H119Q variant corresponds to only 0.007% of the activity of RNase A and 0.085% of that of the K7H/R10H/H12K/H119Q variant demonstrates the unambiguous specific contribution of the His residues introduced at positions 7 and 10 as responsible for the new active site.
Table 1.
Comparison of the activity of RNase variants related to RNase A

Table 2 shows the steady-state kinetic parameters at pH 7.0 obtained by the spectrophotometric method for the cleavage of the oligocytidylic acid substrate (Cp)4C>p, a pentanucleotide in which C>p indicates a terminal 2′,3′-cyclic phosphate. This substrate is obtained from poly(C) digestion by RNase A and product separation by HPLC (Nogués and Cuchillo 2001; Cuchillo et al. 2002; see below).
Table 2.
Steady state kinetic parameters for the transphophorylation of the pentacytidylic acid (Cp)4C>p and the hydrolysis of C>p by RNase A and variants

The data in Table 2 indicate that with this substrate both variants show a lower catalytic efficiency with respect to the activity of the native enzyme (although within the same order of magnitude). The kcat values obtained for the active variants are well correlated with the relative activity values shown in Table 1; from this correlation we have estimated an approximate kcat value of 1.3 min−1 for the H12K/H119Q variant. As is noted in the Discussion section the kinetic data for the K7H/R10H variant do not allow us to distinguish which is the specific contribution of each active site to the overall reaction. On the other hand, it is important to point out the remarkable catalytic efficiency of the quadruple mutant that only contains the new active site.
Hydrolysis reaction
The cyclic mononucleotide C>p was used as substrate to analyze the hydrolysis reaction that leads to the formation of 3′-CMP. The steady-state kinetic parameters at pH 5.5 obtained with the K7H/R10H variant showed a decrease of the activity as seen by the lower kcat value with respect to the RNase A in a way similar to that obtained for the transphosphorylation reaction. No significant change in the Km value is observed. In the case of the variant K7H/R10H/H12K/H119Q, which only maintains the activity associated to the His residues at positions 7 and 10, no activity for the hydrolysis reaction could be detected, even using up to 1.4 μM final enzyme concentration; the absence of the hydrolysis reaction for this variant was confirmed by reversed-phase HPLC separation of the products; no 3′-CMP was detected (data not shown).
Poly(C) digestion by RNase A and the variants
The analysis of the poly(C) cleavage by RNase A and the variants was carried out by separation of the products of the digestion by reversed-phase HPLC (Nova-Pak C18 column). Product separation and oligonucleotide identification were described by Moussaoui et al. (1996). The initial poly(C) substrate elutes as a homogeneous peak (Fig. 4A). The oligocytidylic acids of increasing size elute with increasing retention time and the resolution between products allows us to quantify the different oligocytidylic acid peaks up to 10 nucleotide units (decacytidylic acid) (Fig. 4B). Taking into account the catalytic mechanism of RNase A, the resulting oligocytidylic acids show the general structure (Cp)nC>p, in which C>p indicates a terminal 2′,3′-cyclic phosphate and n varies according to the size (Cuchillo et al. 2002). Figure 4C shows the products formed (up to 10 nucleotide units) by the activity of RNase A and the variants K7H/R10H and K7H/R10H/H12K/H119Q. For comparison purposes the product formation was determined when a similar amount of undigested poly(C), near 30%, was left. Figure 4B,C shows the characteristic pattern found with RNase A at the intermediate steps of the cleavage with an accumulation of the oligonucleotides containing six to seven residues (Moussaoui et al. 1996). However, both variants show a different cleavage pattern characterized by an increase of the mononucleotide formation, and, in the case of K7H/R10H-RNase A variant, also of the dinucleotide, and a shift in the distribution of intermediate oligonucleotides that indicates an increase in the formation of shorter products (Fig. 4C). In addition, the MALDI-TOF MS analysis of the oligonucleotides containing three, four, and five residues showed that the products originated from the new active site in the K7H/R10H/H12K/H119Q variant have a terminal 2′,3′-cyclic phosphate group, indicating that the same chemical reaction path as that of the native enzyme is followed in the variant. The negative control H12K/H119Q variant was also checked by this technique; the chromatographic profile obtained from poly(C) at the same conditions used for the active variants showed only a residual formation of cleavage products (data not shown).
Figure 4.
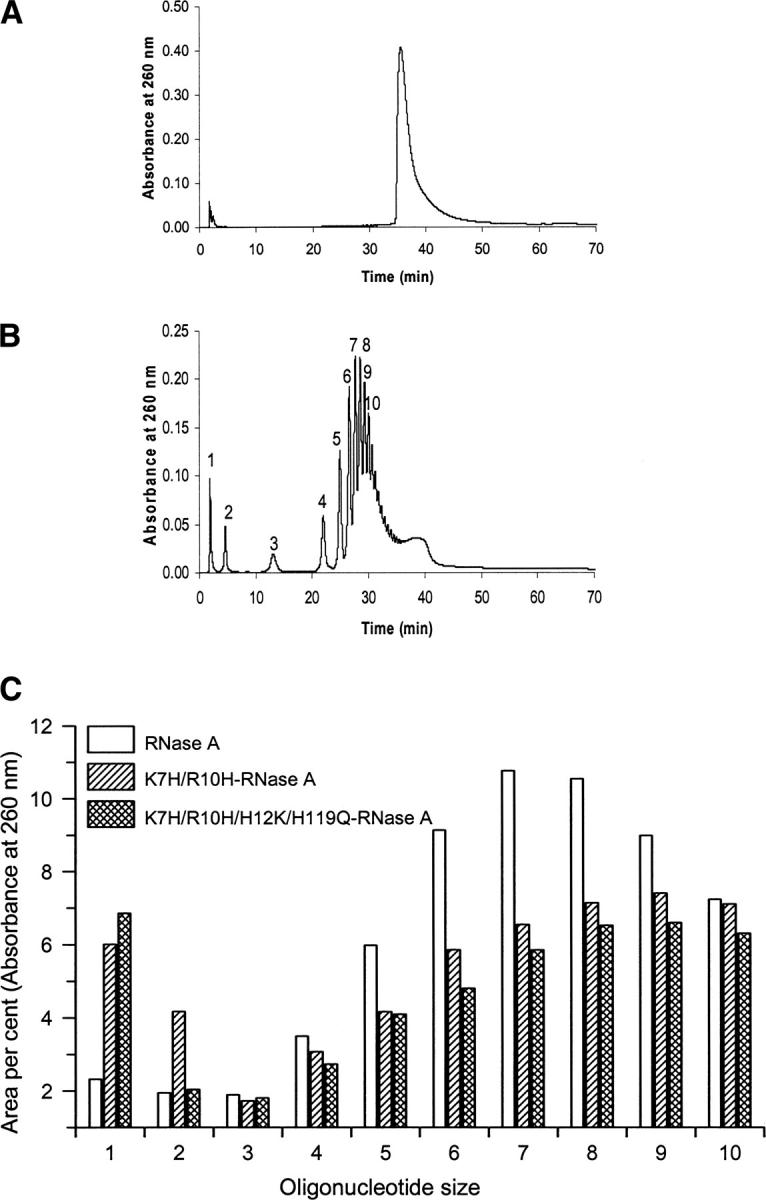
Analysis by reversed-phase HPLC of the poly(C) substrate before addition of enzyme (A) and of the products obtained by digestion with RNase A (B) (the oligonucleotide size of the products is indicated). (C) Comparison of oligocytidilyc acid formation ((Cp)nC>p (n = 0–10)) from the poly(C) cleavage by RNase A and the variants K7H/R10H and K7H/R10H/H112K/H119Q. Area percent for each oligonucleotide was determined from the area of the corresponding peak eluted from a reversed-phase HPLC column (Nova pak C18). Due to the differences in the cleavage pattern and the enzyme activities, the comparison has been established using as reference the moment in which 30% of the poly(C) substrate remained undigested.
Pentanucleotide (Cp)4C>p cleavage pattern by RNase A and the variants
In the analysis of the product formation pattern with high molecular mass substrates, such as poly(C), the interaction with the enzyme involves many binding subsites, including the basic amino acids located at the surface of the protein. However, when smaller substrates, such as a pentanucleotide, are used, the binding of the substrate will be limited to the regions adjacent to the active site and, therefore, the consequences of the modifications introduced in the region of study will be better appreciated.
The characterization of the cleavage preference of RNase A and both variants using the (Cp)4C>p substrate was carried out following the procedure described by Cuchillo et al. (2002), which is based on the size of the products and their ratio. Taking into account the products generated from the specific scissile bond (Fig. 5A), the ratio of formation of the tetranucleotide (Cp)3C>p versus dinucleotide CpC>p as products obtained from the digestion of (Cp)4C>p allows us to distinguish between the preference for an endo- or exonucleolytic activity. Because of the 1:1 stoichiometry CpC>p can be replaced by (Cp)2C>p and (Cp)3C>p by C>p. However, in the latter case the HPLC measurement of C>p concentration is subject to a significantly higher error (Nogués and Cuchillo 2001). Figure 5B shows as an example the separation by reversed-phase HPLC of the products obtained from the cleavage of the pentanucleotide (Cp)4C>p. Each product was quantified from the peak area of each product in absorbance units divided by the corresponding molar extinction coefficient. Figure 6A,B shows the ratio of (Cp)3C>p versus (Cp)C>p formation at different digestion percentages of the initial substrate for the K7H/R10H and K7H/R10H/H12K/H119Q, respectively. Extrapolation of this ratio to 0% digestion of the initial substrate indicates the actual preference of the enzyme in the initial stages of the reaction. High values are indicative of a preference for the exonucleolytic cleavage with respect to the endonucleolytic cleavage and, conversely, low values are indicative of a preference for an endonucleolytic cleavage. Figure 6C shows that, whereas the native enzyme shows a moderate preference for exonucleolytic activity, variants lacking the p2 phosphate-binding site (K7H/R10H/H12K/H119Q and K7Q/R10Q) are much more exonucleolytic. The variant that presents an additional active site (K7H/R10H-RNase A) gives values intermediate between those of the K7H/R10H/H12K/H119Q and the native enzyme. The variant in which the p2 phosphate-binding subsite has been substituted by an active site at the same time that the original active site has been suppressed (K7H/R10H/H12K/H119Q-RNase A) shows a very notorious increase in the ratio of exonuclease/endonuclease activity. The exonuclease activity observed with this variant indicates that for an endonuclease activity a phosphate-binding site in the 3′ side position in relation to an active site must be present. This result is in agreement with the importance of the p2 phosphate-binding site that is located in the 3′ side of the p1 active site in the endonuclease activity of the native RNase A (Cuchillo et al. 2002).
Figure 5.

(A) Possible distribution of products formed by the initial cleavage of (Cp)4C>p. Exonucleolytic cleavage, from either end, yields a mononucleotide plus a tetranucleotide whereas the endonucleolytic cleavage, also from either end, yields a mixture of di- and trinucleotide. The ratio between the tetranucleotide and the dinucleotide formation may be used as an indicator of the preference for the endo- or exonucleolytic cleavage. (B) Separation by reversed-phase HPLC (Nova pak C18 column) of the products obtained from the digestion of (Cp)4C>p by K7H/R10H/H12KH119Q-RNase A in 10 mM HEPES-KOH at pH 7.5. Elution was carried out as described in Material and Methods.
Figure 6.
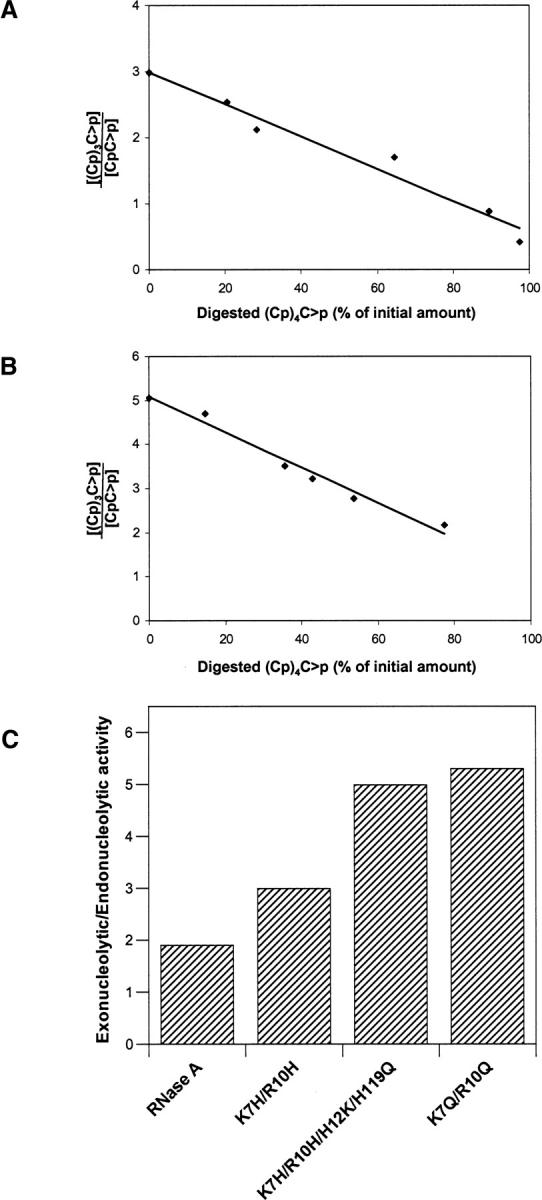
Effect of the presence of a new active site on the exonucleolytic versus endonucleolytic activity of RNase A. Tetranucleotide/dinucleotide ratio for the cleavage of the pentacytidylic acid substrate (Cp)4C>p by K7H/R10H-RNase A (A) and by the K7H/R10H/H12K/H119Q-RNase A (B). With this substrate, tetranucleotide formation indicates exonucleolytic cleavage by the enzyme whereas formation of dinucleotide indicates endonucleolytic cleavage (the trinucleotide molecule could have been used as well). Extrapolation of the ratios to zero time (0% of initial substrate digested) was determined with the program GraFit v.5 (Leatherbarrow 2001) and indicates the preference of the enzymes on the intact substrate. The ratio decreases with time because the tetranucleotide produced at the initial stages of the reaction is later used as a substrate by the enzyme. (C) Exonucleolyticversus endonucleolytic activity of RNase A and variants as determined from the ratios of the products at zero time. The value corresponding to the variant K7Q/R10Q was taken from Cuchillo et al. (2002).
Discussion
The breaking of a 3′,5′-phosphodiester bond in RNA by RNase A involves a general acid–base mechanism in which the imidazole group of His12 acts as a base and the imidazole group of His119 acts as an acid. There is also ample evidence that the positive ɛ-NH2 group of the Lys41 side chain stabilizes the pentacovalent phosphate intermediate that it is produced during the reaction. These three residues (His12, His119, and Lys41) belong to what is known as subsite p1, one of several phosphate-binding subsites that are present in the enzyme molecule. Among the other subsites the best characterized are known as p0, p1, and p2 (Fisher et al. 1998b; Cuchillo et al. 2002) (Fig. 1B). In addition, other phosphate-binding subsites have been hypothesized (p-1, p-2, p3, etc.) (McPherson et al. 1986; de Llorens et al. 1989; Fisher et al. 1998a) that could be involved in the formation of the enzyme–substrate complex and that could expand up to 9 or 10 nucleotides of the substrate molecule.
The p1 phosphate-binding subsite is special in that, apart from interacting with a phosphate group, it promotes the cleavage of the substrate's phosphodiester bond (Fig. 1B). The enzyme shows a mandatory specificity for a pyrimidine in B1, whereas the base bound in B2 can be either a purine or a pyrimidine (with a preference for the former). We thought that transforming the p2 phosphate-binding site into a catalytic site, similar to p1, at the same time that the original active site was suppressed could yield an enzyme with an altered specificity. In addition, we could get more information about the details of the catalytic mechanism.
The well-studied p2 binding subsite is formed by residues Lys7 and Arg10, which establish electrostatic interactions with the phosphate group located at the 3′ side of the RNA chain with respect to that bound to the active site (p1) (Fig. 1). We have selected this region with the aim of changing this noncatalytic phosphate-binding subsite into an active catalytic site by replacing both basic residues by His residues. Site-directed mutagenesis has contributed to characterize the role of RNase A specific amino acids on its mechanism, its high catalytic efficiency, and its specificity (Raines 2004). The design of the new active site has also taken into account as models the catalytic cleavage of RNA by imidazole buffer and by β-cyclodextrin derivatives carrying two imidazole groups (Breslow 1991; Breslow et al. 1991); in both cases the catalytic mechanism is analogous to that described for RNase A, and intermediate products ending in a 2′,3′-cyclic phosphate group are also produced.
The predicted three-dimensional structure of the variants characterized in this study (K7H/R10H- and K7H/R10H/H12K/H119Q-RNase A) did not show significant deviations from the overall structure of RNase A (Fig. 7). The RNase activity assays of both variants determined by the zymogram technique did not allow us to detect any activity with the poly(A) substrate; this result indicates that no changes in the specificity for the pyrimidine have appeared as a consequence of the new active site in the p2 region. Kinetic studies using dinucleotides as substrate indicate a preference for purine bases in region B2 of RNase A (Witzel and Barnard 1962) (Fig. 1B). From this preference and taking into account the enzyme–substrate binding model, initially we hypothesized a change in the cleavage specificity preference of RNase A from pyrimidines to purines. However, our results indicate that the pyrimidine specificity was maintained, probably as a consequence of the better alignment of the substrate, which is favored by the low size of the pyrimidine bases, or, alternatively, the pyrimidine specificity could be maintained by the original B1 site due to the proximity between the p1 and p2 binding sites.
Figure 7.
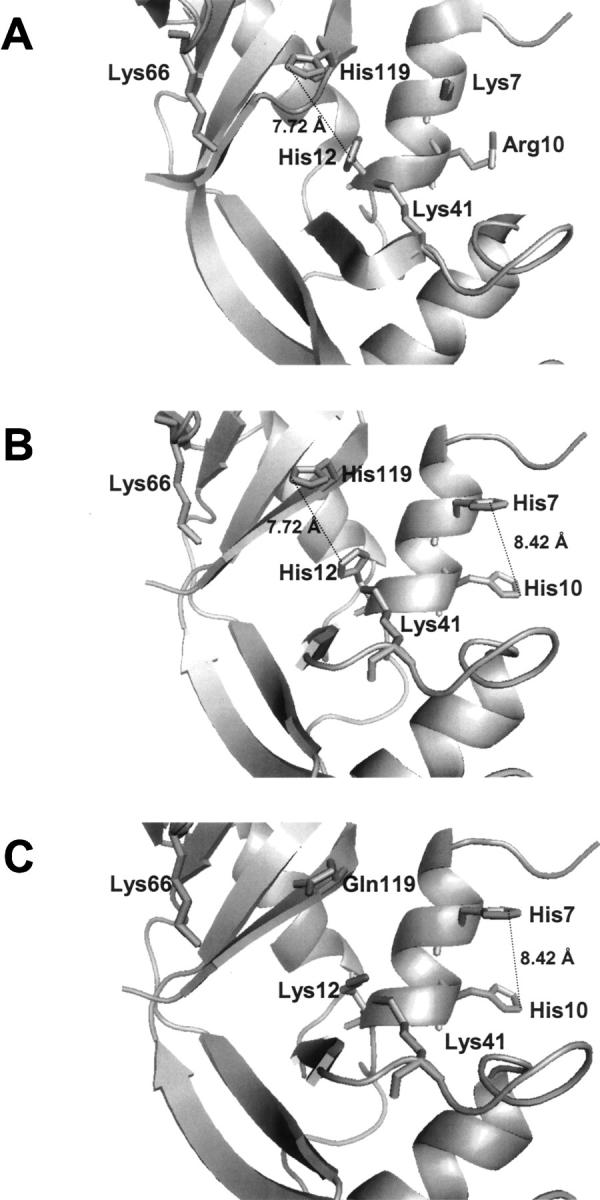
Three-dimensional structure of the region around the RNase A active site. (A) RNase A. (B,C) Molecular modeling of the variants K7H/R10H-RNase A and K7H/R10H/H12K/H119Q-RNase A, respectively. Molecular modeling was carried out with the program Deep View/Swiss PDB Viewer and the picture was drawn using PyMOL, DeLano Scientific program (PDB accession code 7RSA).
The steady-state kinetic parameters for the transphosphorylation reaction using the pentanucleotide (Cp)4C>p as substrate show a decrease in the k cat values of the active variants with respect to the wild-type-RNase A (Table 2). However, the catalytic efficiency (kcat/Km) for the two variants is of the same order of magnitude (106 M−1·s−1) as the wild-type enzyme. It is important to emphasize that the activity of the K7H/R10H/H12K/H119Q-RNase A variant is only due to the contribution of the new artificial active site. The accepted catalytic mechanism for RNase A includes, in addition to the roles of His12 and His119 in the acid–base mechanism, an essential role for Lys41 in the stabilization of the transition state; the predicted three-dimensional structure of this variant does not allow a clear identification of a putative amino acid residue that could play a role analogous to that of the wild-type Lys41. However, taking into account the structural proximity in this variant of Lys12 to the p2 active site, a possible contribution of its side chain to the catalytic mechanism should not be discarded (Fig. 7C). For the variant K7H/R10H-RNase A the kinetic studies using spectrophotometric methods do not allow us to dissect the individual contribution of the two possible active sites (p1 and p2) to the catalysis. For this variant the decrease in the k cat value may be a consequence of the role of Lys7 in the catalytic mechanism of wild-type RNase A in which a stabilization of the transition state through an intervening water network has been postulated (Brünger et al. 1985; Boix et al. 1994).
The MALDI-TOF MS analysis of the products of the cleavage of the substrate (Cp)4C>p by both variants showed that in all cases a terminal 2′,3′-cyclic phosphate group was formed following the same transphosphorylation reaction as in native RNase A. The absence of the hydrolysis reaction for the K7H/R10H/H12K/H119Q-RNase A variant as determined with the C>p substrate may be explained as a consequence of the need for an induced-fit mechanism in the p2 active site that could not be brought about by a very small substrate such as the water molecule. In the case of the K7H/R10H variant all the observed hydrolytic activity should, therefore, be ascribed to the original active site located in p1.
The pattern of product formation using a substrate such as (Cp)4C>p can be very illuminating about the preference of the enzyme for an endo- or exonucleolytic cleavage. The exonucleolytic cleavage of the pentanucleotide substrate yields a mononucleotide plus a tetranucleotide, and in the case of the endonucleolytic activity, a dinucleotide and a trinucleotide are produced (Fig. 5A). Given that in the RNase mechanism a 2′,3′-cyclic phosphate intermediate is formed, it is not possible with our method to distinguish between the two bonds that can, theoretically, be broken in both exo- and endonucleolytic cleavages. Taking into account these specific scissile sites and the corresponding formed products, we have chosen the ratio between the tetranucleotide and the dinucleotide formation to determine the preference for the exo- or endonucleolytic activity for RNase A and the variants (Fig. 5A). Studies with wild-type RNase A indicated that the electrostatic interactions of the substrate in the p2 site are extremely important for the endonucleolytic activity; abolition of this binding site by site-directed mutagenesis (K7Q/R10Q-RNase A variant) results in a strong preference for the exonucleolytic activity (Fig. 6; Cuchillo et al. 2002). Studies of substrate cleavage with the eosinophil cationic protein (ECP) (also known as RNase 3) have also emphasized the relevance of this binding site for its endonucleolytic activity; this protein, a member of the RNase A protein family, shows a clear preference for the exonucleolytic cleavage of the substrate (Boix et al. 1999b) at the same time that the X-ray three-dimensional structure indicates the absence of an equivalent substrate-binding region (Boix et al. 1999a). In the case of the K7H/R10H/H12K/H119Q-RNase A variant a clear preference for the exonucleolytic versus endonucleolytic activity is seen (Fig. 6); the predicted three-dimensional structure of this variant does not show any binding region capable of establishing an electrostatic interaction with the substrate through the phosphate group located at the 3′ side with respect to the phosphate that binds at the active site; this structural conformation may explain the similarity between the activity of this variant and the cleavage preference found with either K7Q/R10Q-RNase A or ECP (Boix et al. 1999b; Cuchillo et al. 2002). The behavior of the K7H/R10H-RNase A variant in what refers to the preference for an endo- or exonucleolytic activity is consistent with a small increase for the exo- versus endonucleolytic activity with respect to the RNase A cleavage; the analysis of the cleavage pattern does not allow us to determine the specific contribution to the total reaction rate of each catalytic site in this variant (p1 and p2). However, the results, which do not show an increase in the mononucleotide product formation (data not shown), indicate that a simultaneous activity in both sites is not very likely.
In summary, this work demonstrates that from structural and functional information about RNase A it has been possible to introduce a new active site in this enzyme. The new active site, which shows a remarkably high catalytic efficiency, modifies the substrate cleavage specificity with respect to the wild-type enzyme to a higher preference for the exo- with respect to the endonucleolytic process. Further studies will be addressed to obtain structural and mechanistic information related to the active site and the enzyme–substrate interactions in these variants.
Materials and methods
Site-directed mutagenesis, expression, and purification of RNase A variants
K7H/R10H, K7H/R10H/H12K/H119Q, and H12K/H119Q-RNase A mutants were obtained from an RNase A synthetic gene (Boix et al. 1994) using the PCR techniques. The RNase A gene was previously cloned in pET11d vector (Novagen) and the modifications were performed using the Quick change site-directed mutagenesis kit (Novagen). All constructs were confirmed by DNA sequencing. Protein expression in the E. coli BL21 (DE3) strain (Novagen) as well as the initial purification steps and folding of the proteins from inclusion bodies were carried out as previously described (Boix et al. 1999b). A cation-exchange chromatography (Resource S column, Amersham Pharmacia Biotech) using an FPLC system (Amersham Biosciences) was carried out in the subsequent purification step. The proteins were injected dissolved in 15 mM HEPES/KOH at pH 7.0, and the elution was performed by applying a linear gradient from 0 to 150 mM sodium acetate. Differences in the elution pattern were the result of the different net charge of each mutant. An additional process by reversed-phase chromatography (Vydac C18 column, Scharlau) using an HPLC system (Waters Corp.) was carried out for further purification. The proteins were injected dissolved in water containing 0.1% trifluoroacetic acid (TFA), and the elution was carried out with a linear gradient between 0% and 100% acetonitrile containing 0.1% TFA. The homogeneity of the purified protein was checked with 15% SDS-PAGE and Coomassie Blue staining and N-terminal sequencing. The molecular mass of the purified RNase A variants was checked by MALDI-TOF mass spectrometry. Native RNase A was purified by cation exchange chromatography from a commercial preparation (Sigma) (Alonso et al. 1986).
Activity-staining gels (Zymogram)
The RNase activity assays were performed by zymogram on 15% SDS-PAGE gels containing 2.5 mg/mL of either poly(C), poly(U), or poly(A) (Sigma) as substrate according to the method described by Bravo et al. (1994). For each variant the protein concentration was adjusted depending on its catalytic activity. The gels were stained with 0.2% Toluidine Blue.
Analysis of the digestion products of poly(C)
The high molecular mass substrate poly(C) was dissolved at a concentration of 2.5 mg/mL in 10 mM HEPES-KOH at pH 7.5. Fifty microliters of the poly(C) solution were digested with 10 μl of enzyme solution at 25°C. Enzyme concentrations were adjusted according to the activity of each variant. At different digestion times the products of the reaction were separated by reversed-phase HPLC (Nova Pak C18 column, Waters) according to the previously described procedure (Moussaoui et al. 1996; Nogués and Cuchillo 2001). Briefly, 40 μl of the reaction mixture were injected to the column equilibrated with solvent A (10% (w/v) ammonium acetate and 1% (v/v) acetonitrile) and the elution was carried out by an initial 10-min wash and 50-min gradient from 100% solvent A to 10% solvent A plus 90% solvent B (10% (w/v) ammonium acetate and 11% (v/v) acetonitrile). Product elution was detected from the absorbance at 260 nm, and peak identification was performed according to Moussaoui et al. (1996). The mononucleotide fraction is eluted at the initial conditions and oligonucleotides of increasing size elute with increasing retention times. The elution profile allows us to distinguish between consecutive sizes up to 9 or 10 nucleotides. The area percentage in each case was determined from the area of the corresponding isolated peak. Due to the different enzyme activities, comparisons were established using as reference the same undigested poly(C) fraction.
Preparation of the pentacytidylic acid (Cp)4C>p from poly(C) digestion
The oligocytidylic acid (Cp)4C>p used as substrate was obtained by RNase A digestion of a poly(C) solution (Moussaoui et al. 1996; Nogués and Cuchillo 2001). Five hundred microliters of a 10 mg/mL poly(C) solution in 10 mM HEPES-KOH at pH 7.5 were digested with 50 μl of 0.7 μM RNase A at 25°C for 5 min. The reaction products were separated by reversed-phase column chromatography as described in the procedure for the separation of the digestion products of poly(C). The peak corresponding to the pentanucleotide (Cp)4C>p was collected and lyophilized.
Analysis of the oligocytidylyc acid (Cp)4C>p cleavage pattern by RNase A and variants
Fifty microliters of a (Cp)4C>p solution in 10 mM HEPES-KOH at pH 7.5 were digested with 10 μl of the enzyme solution at 25°C. Enzyme concentrations were adjusted to the activity of each variant. The products of the reaction were separated by column chromatography as described in the procedure for the separation of the digestion products of poly(C). The amount of each oligonucleotide product was calculated by first integrating the areas (in OD units) and dividing these values by the corresponding absorbance coefficients at 260 nm (ɛ260): 7845 M−1cm−1 for C>p, 15,175 M−1cm−1 for CpC>p, 20,745 M−1cm−1 for (Cp)2C>p, 24,282 M−1cm−1 for (Cp)3C>p, and 28,683 M−1cm−1 for (Cp)4C>p (Cuchillo et al. 2002). The program GraFit v.5 (Leatherbarrow 2001) was used to fit the plots.
Steady-state kinetic parameters using (Cp)4C>p as substrate
The oligonucleotide pentacytidylic acid (Cp)4C>p was used as substrate for the transphosphorylation reaction of RNase A and variants and the activity was determined by the spectrophotometric method (Spectrophotometer Cary 100 Bio, Varian). The activities were determined in 0.2 M HEPES-KOH buffer at pH 7.0. The cleavage of the substrate was followed by measuring the increase in absorbance at 280 nm. The substrate concentration range was from 10 to 150 μM and the measurements were carried out at 25°C using 2-mm path-length cells. The activity was measured by following the initial reaction velocities using the difference molar absorbance coefficient in relation to the substrate digestion end point: Δɛ280 = 3600 M−1cm−1. The program GraFit v.5 (Leatherbarrow 2001) was used for the determination of the kinetic parameters Km and V max from the initial velocity values. The final enzyme concentration was 0.3 nM for RNase A and 1 nM for both active variants.
Analysis of the hydrolysis reaction of cytididine 2′,3′cyclic phosphate (C>p)
The kinetics of the hydrolysis reaction of the substrate cytidine 2′,3′-cyclic phosphate (C>p) (Sigma) by RNase A and variants were analyzed by the spectrophotometric method based on the absorbance increase of the reaction mixture at 296 nm due to the formation of 3′-CMP (Δɛ296 = 516.4 M−1cm−1) (Moussaoui et al. 1998). The activities were determined in 0.2 M sodium acetate buffer at pH 5.5. The substrate concentration range was from 0.05 mM to 3 mM and the measurements were carried out at 25°C using 1-cm path-length cells. The program GraFit v.5 (Leatherbarrow 2001) was used for the determination of the kinetic parameters Km and V max from the initial velocity values. The final enzyme concentration was 0.1 μM for RNase A and the K7H/R10H variant. As no activity was detected, the enzyme concentration for the K7H/R10H/H12K/H119Q variant was increased up to 1.4 μM. In addition, the formation of 3′-CMP from the hydrolysis of C>p was also determined by reversed-phase HPLC according to the procedure described by Cuchillo et al. (2002).
Matrix assisted laser desorption ionization time of flying mass spectrometry (MALDI-TOF MS)
Mass determination by MALDI-TOF MS of the tri-, tetra-, and pentanucleotide products obtained from the HPLC separation of cleavage products by the variant K7H/R10H/H12K/H119Q was performed with a Brucker Biflex mass spectrometer. The protocol described for the analysis of the products formed by the RNase A digestion of poly(C) was followed (Cuchillo et al. 2002).
Acknowledgments
This work was supported by Grants 2001SGR-00196 from the Direcció General de Recerca of the Generalitat de Catalunya, BMC2003-08485-C02-01 from the Dirección General de Investigación of the Ministerio de Educación y Ciencia (Spain) and from the funds FEDER of the European Union.
Footnotes
Reprint requests to: M. Victòria Nogués, Departament de Bioquímica i Biologia Molecular, Facultat de Ciències, Universitat Autònoma de Barcelona, 08193-Bellaterra, Spain; e-mail: victoria.nogues@uab.es; fax: 34-93-5811264.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062251707.
Abbreviations: RNase A, bovine pancreatic ribonuclease; poly(C), polycytidylic acid; poly(U), polyuridylic acid; poly(A), polyadenylic acid; C>p, cytidine 2′,3′-cyclic phosphate; (Cp)nC>p, an oligocytidylic acid of n + 1 residues ending in a 2′,3′-cyclic phosphate; CpA, cytidylyl-3′,5′-adenosine; MALDI-TOF MS, matrix assisted laser desorption ionization-time of flight mass spectrometry.
References
- Alonso, J., Nogués, M.V., and Cuchillo, C.M. 1986. Modification of bovine pancreatic ribonuclease A with 6-chloropurine riboside. Arch. Biochem. Biophys. 246: 681–689. [DOI] [PubMed] [Google Scholar]
- Boix, E., Nogués, M.V., Schein, C.H., Benner, S.A., and Cuchillo, C.M. 1994. Reverse transphosphorylation by ribonuclease A needs an intact p2-binding site. Point mutations at Lys-7 and Arg-10 alter the catalytic properties of the enzyme. J. Biol. Chem. 269: 2529–2534. [PubMed] [Google Scholar]
- Boix, E., Leonidas, D.D., Nikolovski, Z., Nogués, M.V., Cuchillo, C.M., and Acharya, K.R. 1999a. Crystal structure of eosinophil cationic protein at 2.4 Å resolution. Biochemistry 38: 16794–16801. [DOI] [PubMed] [Google Scholar]
- Boix, E., Nikolovski, Z., Moiseyev, G.P., Rosenberg, H.F., Cuchillo, C.M., and Nogués, M.V. 1999b. Kinetic and product distribution analysis of human eosinophil cationic protein indicates a site arrangement that favors exonuclease-type activity. J. Biol. Chem. 274: 15605–15614. [DOI] [PubMed] [Google Scholar]
- Bravo, J., Fernández, E., Ribó, M., de Llorens, R., and Cuchillo, C.M. 1994. A versatile negative-staining ribonuclease zymogram. Anal. Biochem. 219: 82–86. [DOI] [PubMed] [Google Scholar]
- Breslow, R. 1991. How do imidazole groups catalyze the cleavage of RNA in enzyme models and in enzymes? Evidence from “negative catalysis. Acc. Chem. Res. 24: 317–324. [Google Scholar]
- Breslow, R., Anslyn, E., and Huang, D.L. 1991. Ribonuclease mimics. Tetrahedron 47: 2365–2376. [Google Scholar]
- Brünger, A.T., Brooks, C.L., and Karplus, M. 1985. Active site dynamics of ribonuclease. Proc. Natl. Acad. Sci. 82: 8458–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchillo, C.M., Parés, X., Guasch, A., Barman, T., Travers, F., and Nogués, M.V. 1993. The role of 2′,3′-cyclic phosphodiesters in the bovine pancreatic ribonuclease A catalysed cleavage of RNA: Intermediates or products? FEBS Lett. 333: 207–210. [DOI] [PubMed] [Google Scholar]
- Cuchillo, C.M., Moussaoui, M., Barman, T., Travers, F., and Nogués, M.V. 2002. The exo- or endonucleolytic preference of bovine pancreatic ribonuclease A depends on its subsites structure and on the substrate size. Protein Sci. 11: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio, G. and Riordan, J.F. 1997. Ribonucleases. Structures and functions. Academic Press, New York.
- de Llorens, R., Arús, C., Parés, X., and Cuchillo, C.M. 1989. Chemical and computer graphics studies on the topography of the ribonuclease A active site cleft. A model of enzyme-pentanucleotide substrate complex. Protein Eng. 2: 417–429. [DOI] [PubMed] [Google Scholar]
- Fisher, B.M., Grilley, J.E., and Raines, R.T. 1998a. A new remote subsite in ribonuclease A. J. Biol. Chem. 273: 34134–34138. [DOI] [PubMed] [Google Scholar]
- Fisher, B.M., Ha, J.-H., and Raines, R.T. 1998b. Coulombic forces in protein–RNA interactions: Binding and cleavage by ribonuclease A and variants at Lys7, Arg10, and Lys66. Biochemistry 37: 12121–12132. [DOI] [PubMed] [Google Scholar]
- Fontecilla-Camps, J.C., de Llorens, R., le Du, M.H., and Cuchillo, C.M. 1994. Crystal structure of ribonuclease A·d(ApTpApApG) complex direct evidence for extended substrate recognition. J. Biol. Chem. 269: 21526–21531. [DOI] [PubMed] [Google Scholar]
- Hilvert, D. 2000. Critical analysis of antibody catalysis. Annu. Rev. Biochem. 69: 751–793. [DOI] [PubMed] [Google Scholar]
- Leatherbarrow, R.J. 2001. GraFit Version 5, Erithacus Software.
- McPherson, A., Brayer, G.D., Cascio, D., and Williams, R. 1986. The mechanism of binding of a polynucleotide chain to pancreatic ribonuclease. Science 232: 765–768. [DOI] [PubMed] [Google Scholar]
- Moussaoui, M., Guasch, A., Boix, E., Cuchillo, C.M., and Nogués, M.V. 1996. The role of non-catalytic binding subsites in the endonuclease activity of bovine pancreatic ribonuclease A. J. Biol. Chem. 271: 4687–4692. [DOI] [PubMed] [Google Scholar]
- Moussaoui, M., Nogués, M.V., Guasch, A., Barman, T., Travers, T., and Cuchillo, C.M. 1998. The subsite structure of bovine pancreatic ribonuclease A accounts for the abnormal kinetic behavior with cytidine 2′,3′-cyclic phosphate. J. Biol. Chem. 273: 25565–25572. [DOI] [PubMed] [Google Scholar]
- Nogués, M.V. and Cuchillo, C.M. 2001. Analysis by HPLC of distributive activities and the synthetic (back) reaction of pancreatic-type ribonucleases. In Methods in molecular biology: Nucleases methods and protocols (ed. C.H. Schein). Vol. 160, pp. 15–24. Humana Press Inc., Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Nogués, M.V., Vilanova, M., and Cuchillo, C.M. 1995. Bovine pancreatic ribonuclease A as model of an enzyme with multiple substrate binding sites. Biochim. Biophys. Acta 1253: 16–24. [DOI] [PubMed] [Google Scholar]
- Nogués, M.V., Moussaoui, M., Boix, E., Vilanova, M., Ribó, M., and Cuchillo, C.M. 1998. The contribution of noncatalytic phosphate-binding subsites to the mechanism of bovine pancreatic ribonuclease A. Cell. Mol. Life Sci. 54: 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parés, X., Llorens, R., Arús, C., and Cuchillo, C.M. 1980. The reaction of bovine pancreatic ribonuclease A with 6-chloropurine riboside 5′monophosphate. Evidence on the existence of a phosphate-binding sub-site. Eur. J. Biochem. 105: 571–579. [DOI] [PubMed] [Google Scholar]
- Parés, X., Nogués, M.V., de Llorens, R., and Cuchillo, C.M. 1991. Structure and function of ribonuclease A binding subsites. In Essays in biochemistry (ed. K.F. Tipton). Vol. 26, pp. 89–103. Portland Press Ltd., London. [PubMed] [Google Scholar]
- Raines, R.T. 1998. Ribonuclease A. Chem. Rev. 98: 1045–1065. [DOI] [PubMed] [Google Scholar]
- Raines, R.T. 2004. Active site of ribonuclease A. In Nucleic acids and molecular biology: Artificial nucleases (ed. M.A. Zenkova). Vol. 13, pp. 19–32. Springer-Verlag, Berlin. [Google Scholar]
- Witzel, H. and Barnard, E.A. 1962. Mechanism and binding sites in the ribonuclease reaction. II. Kinetic studies on the first step of the reaction. Biochem. Biophys. Res. Commun. 7: 295–299. [DOI] [PubMed] [Google Scholar]