

Abstract
The protein islet amyloid polypeptide (IAPP) is a glucose metabolism associated hormone cosecreted with insulin by the β-cells of the pancreas. In humans with type 2 diabetes, IAPP deposits as amyloid fibers. The assembly intermediates of this process are associated with β-cell death. Here, we examine the rat IAPP sequence variant under physiological solution conditions. Rat IAPP is mechanistically informative for fibrillogenesis, as it samples intermediate-like states but does not progress to form amyloid. A central challenge was the development of a bacterial expression system to generate isotopically labeled IAPP without terminal tags, but which does include a eukaryotic post-translational modification. While optical spectroscopy shows IAPP to be natively unfolded, NMR chemical shifts of backbone and β-carbon resonances reveal the sampling of α-helical states across a continuous stretch comprising ∼40% of the protein. In addition, the manifestation of nonrandom coil chemical shifts is confirmed by the relative insensitivity of the amide proton chemical shifts to alterations in temperature. Intriguingly, the residues displaying helical propensity are conserved with the human sequence, suggesting a functional role for this conformational bias. The inability of rat IAPP to self assemble can be ascribed, in part, to several slowly exchanging conformations evident as multiple chemical shift assignments in the immediate vicinity of three proline residues residing outside of this helical region.
Keywords: amylin, amyloid, IAPP, NMR, α-helix, type 2 diabetes, protein folding, intrinsic disorder
Islet amyloid polypeptide (IAPP) is a 37-residue peptide hormone cosecreted with insulin by the endocrine β-cells of the pancreas (Jaikaran and Clark 2001). This protein is a member of the calcitonin gene-related peptide (CGRP) family (Muff et al. 2004). These proteins are hormones, display sequence homology, and are further characterized by C-terminal amidation and a tight disulfide bond separated by five residues. The hormonal actions ascribed to IAPP are diverse (Hay et al. 2004) and include, for example, control of gastric emptying and paracrine/autocrine signaling upon insulin release by the β-cell (Cooper 1994).
In solution, IAPP is widely regarded as a natively unstructured protein (Kayed et al. 1999; Dunker et al. 2001; Jaikaran and Clark 2001; Padrick and Miranker 2001). However, in humans with type 2 diabetes, IAPP undergoes conformational changes to form β-sheets organized into amyloid fibers. The process of IAPP amyloid formation is correlated with pancreatic β-cell dysfunction (Hoppener et al. 2000; Hull et al. 2004) including an increased rate of apoptosis and a reduction in β-cell mass. At a minimum, amyloid cytotoxicity contributes to diabetes pathology by increasing the requirement for insulin replacement therapy. A number of animal models support these conclusions (Westermark et al. 2000; Wang et al. 2001; Butler et al. 2004). For example, the HIP rat is transgenic for human IAPP and spontaneously develops pathology typical of human diabetes (Butler et al. 2004) including elements of β-cell dysfunction and systemic insulin resistance. The latter result is particularly remarkable as it enables the investigators to suggest that IAPP associated β-cell impairment can be causal to systemic manifestations of this disease (Matveyenko and Butler 2006).
Amyloid fibers themselves are highly ordered, allowing for recent structural studies in a number of systems including Aβ from Alzheimer's disease (Petkova et al. 2005) and Sup35 from yeast (Nelson et al. 2005). However, the kinetics of amyloid assembly are nucleation dependent, resulting in poorly populated, transient, and partially structured intermediates (Padrick and Miranker 2002; Uversky and Fink 2004; Chiti and Dobson 2006). Structural studies of these intermediate states are therefore challenging. The principle approaches to this challenge have used mutagenesis to evaluate the rate and capacity of a protein to self assemble. For example, exhaustive mutagenesis of a model peptide (Lopez de la Paz et al. 2005) has enabled amyloid determinants to be characterized in a manner suitable for proteome analysis. Mutagenesis can also be used in a site-directed manner to stabilize an analog of an intermediate state. For folded and globular amyloid precursors, such as β-2 microglobulin, this has enabled mechanistic insights at atomic resolution (Eakin et al. 2006).
In the case of IAPP, the rat sequence variant is a relevant and useful tool for molecular insight. Rat IAPP differs from human at six residues (Fig. 1) and does not form amyloid fibers (Westermark et al. 1990). The principle, but not exclusive, origin of this difference is three proline residues at positions 25, 28, and 29; for example, human IAPP mutated to contain the three proline residues aggregates, but to a greatly reduced extent (Green et al. 2003). Our own investigations show that rat IAPP adopts structures similar to prefibrillar states of human IAPP (Padrick and Miranker 2001). Notably, both rat and human IAPP in physiological buffer demonstrate fluorescence resonance energy transfer between residue Phe15 and Tyr37, indicative of long-range structure. In human IAPP, this interaction becomes more robust upon transition to the fiber state. Recently, we have shown that both rat and human IAPP adopt comparable structures upon binding phopholipid bilayers (Knight et al. 2006), namely, they both cooperatively assemble into oligomeric α-helical states. However, as with IAPP in solution, only the human variant further transforms to the fibrous state. The structures sampled by rat IAPP are therefore important both to an understanding of its normal function as well as serving as a stabilized analog of prefibrillar conformational states.
Figure 1.

Amino acid sequences of human and rat IAPP (Swiss-Prot P10997 and P12969, respectively). The variants differ at six positions, indicated in bold.
NMR spectroscopy is an ideal method to gain structural information of natively disordered states at atomic resolution (Dyson and Wright 2001; McNulty et al. 2006). In this work we produce 13C- and 15N-labeled wild-type IAPP for study by NMR spectroscopy. To date, a number of groups (Mazor et al. 2002; Lopes et al. 2004) have produced recombinant IAPP; however, one construct retains large tags (44 kDa maltose-binding protein) and neither includes the post-translational modification of amidation at residue 37. We note that Tyr37 participates both in prefibrillar structures (Padrick and Miranker 2001), and that inclusion of the amidation is necessary for protein function (Muff et al. 2004). We therefore sought to include this amidation in the expressed product. Preliminary structure assessments were then made based on chemical shifts derived from assignment of 1HN, 1Hα, 13Cα, 13Cβ, 13CO, and 15N resonances determined using established triple-resonance experiments.
Results
Expression and purification
Wild-type IAPP can be expressed and purified from Escherichia coli. Wild-type IAPP has a disulfide bridge between cysteines 2 and 7 and is amidated at the C terminus (Fig. 1). The expression construct was designed to enhance IAPP solubility, to provide a purification tag, and to generate the C-terminal amidation. Our expression method produces rat IAPP initially as a fusion protein in the form leader-IAPP-intein-CBD (Fig. 2). The N-terminal leader sequence [MKIEEG(NANP)3E] contains codons that are highly expressed in E. coli (MKIEEG) and a bulky, hydrophobic group (NANP repeat) that was used to increase solubility and expression of the Aβ peptide (Dobeli et al. 1995). V8 protease cleaves immediately after acidic residues, allowing the N-terminal leader sequence to be removed at the glutamate, leaving only native IAPP.
Figure 2.

Scheme for expression and purification of IAPP. (A) The IAPP construct was expressed from E. coli cells and loaded onto a chitin column. (B) C-terminally amidated IAPP with attached N-terminal leader sequence was washed from the column after intein cleavage. (C) Wild-type IAPP was generated after disulfide oxidation and V8 protease cleavage.
The Mxe intein and chitin-binding domain (CBD) serve as an affinity tag and means to generate C-terminal amidation. Cottingham et al. (2001) demonstrated that ammonium bicarbonate in the intein cleavage buffer results in amide substitution at the cleavage point. After intein cleavage, leader-IAPP-NH2 was dialyzed to remove excess salt and DTT, to oxidize the disulfide bond, and to reduce the urea concentration for subsequent V8 protease digestion. After digestion, the peptide was purified by reverse-phase HPLC to yield wild-type IAPP. Amidation and oxidation result in a 3 Da mass shift compared with the reduced and free-acid form of the peptide. This was confirmed by mass spectrometry using an internal standard (data not shown).
Resonance assignments
The chemical shift assignments of 1HN, 1Hα, 13Cα, 13Cβ, 13CO, and 15N nuclei were determined for IAPP in 100 mM potassium chloride and 50 mM potassium phosphate (pH 5.5) at 5°C using established triple-resonance NMR experiments (Supplemental Table 1). The chemical shift dispersion was typical of a disordered peptide, yet there was sufficient resolution to identify all resonances. The shifts were deposited in the BMRB (Seavey et al. 1991) under accession number 7311.
Rat IAPP samples multiple conformations in solution. In the15N HSQC spectrum, for example, multiple peaks are each assigned to residues Gly24, Val26, and Leu27 (Fig. 3). The presence of multiple peaks indicates that these amides are in slow exchange relative to the chemical shift differences. The multiple shifts of Gly24, Val26, and Leu27 likely result from cis/trans proline isomerization by Pro25, Pro28, and Pro29. The cis/trans interconversion rate is affected by the preceding residue (MacArthur and Thornton 1991) and is generally very slow, with a halftime of ∼20 min at 0°C (Creighton 1993). As prolines preferentially adopt the trans conformation with a relative ratio of about 4:1, trans:cis (Creighton 1993), the peaks with lower intensities (Fig. 3B) are likely the result of cis backbone conformations. In a predominantly unstructured protein, prolines will have a greater effect on the chemical environment of proximal residues. In rat IAPP, the three prolines could adopt up to eight different combinations of cis/trans states, leading to a multitude of peaks. Interestingly, multiple peaks are not observed for Thr30, which is C-terminal to Pro28 and Pro29, but are observed for Val26, which is C-terminal to Pro25. As the second proline of a diproline has a much stronger preference for the trans conformation (MacArthur and Thornton 1991), we surmise Pro29 to be predominantly trans. The proline residues of rat IAPP drastically inhibit, but do not completely abolish aggregation (Green et al. 2003; Abedini and Raleigh 2006). Our chemical shift data indicate that rat IAPP has several stable conformations induced by proline isomerization. Not only are prolines disruptive to secondary structure, these multiple conformations of the peptide backbone would strongly inhibit symmetric self-association of the peptide.
Figure 3.
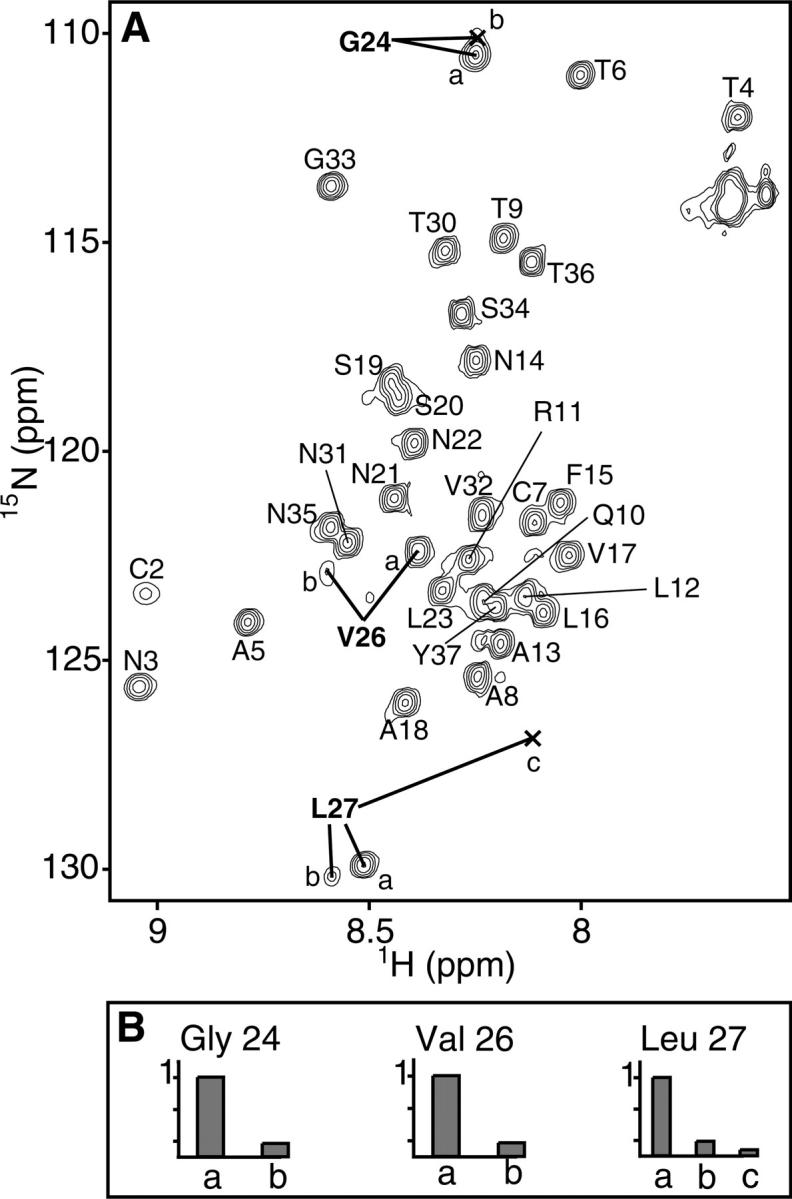
(A) 15N-HSQC spectrum of rat IAPP. Multiple resonances observed for Gly24, Val26, and Leu27 are indicated with bold lines. (B) Relative peak heights for Gly24, Val26, and Leu27 indicated in A normalized to the most intense peak.
Secondary chemical shifts
Secondary chemical shifts of rat IAPP indicate helical propensity from Ala5 to Ser19. The chemical shifts of backbone nuclei are sensitive to the backbone dihedral angles ϕ and ψ (Wishart et al. 1991; Dyson and Wright 2001). In short peptide models of random coil states (Merutka et al. 1995; Wishart et al. 1995; Schwarzinger et al. 2000), these angles have intrinsic bias dependent on residue identity and nearest neighbor effects (Schwarzinger et al. 2001). Deviations from random coil models are indicative of secondary structure propensities, even in denatured proteins. For example, a downfield 1Hα shift indicates β-sheet, whereas an upfield shift indicates α-helix (Dyson and Wright 2001). Secondary shift analysis has been used to determine residual structure in unfolded and denatured states of SH3 domains (Zhang and Forman-Kay 1997) and apomyoglobin (Yao et al. 2001). The secondary chemical shifts for IAPP were calculated by subtracting corrected random coil values from the measured chemical shifts for each nucleus. IAPP has consistent secondary shifts for all nuclei in the N-terminal half of the peptide. These shifts are seen as upfield for 1Hα in residues 5–17, downfield for 13CO in residues 3–19, downfield for 13Cα in residues 5–19, upfield for 1HN in residues 6–22, and upfield for 13Cβ shifts in residues 5–18 (Fig. 4). The consistency of these shifts across consecutive residues is indicative of secondary structure. Importantly, all shifts occur in a field direction associated with α-helix formation (Wishart and Sykes 1994). As 1Hα, 13Cα, and 13CO shifts are the strongest indicators of secondary structure propensity, we assert that residues 5–19 sample α-helical states. The 1HN and 13Cβ shifts, while less robust, are consistent with helicity across this region. The secondary shifts, however, are not indicative of long-lived structure. For example, a Cα nucleus in a fully formed α-helix would have secondary shifts >2.6 δppm (Wishart and Sykes 1994). Finally, the terminal residues (1–4) have no overall secondary structural trend, but have large deviations from random coil. The tight disulfide between cysteines 2 and 7 likely creates a region of local structure.
Figure 4.
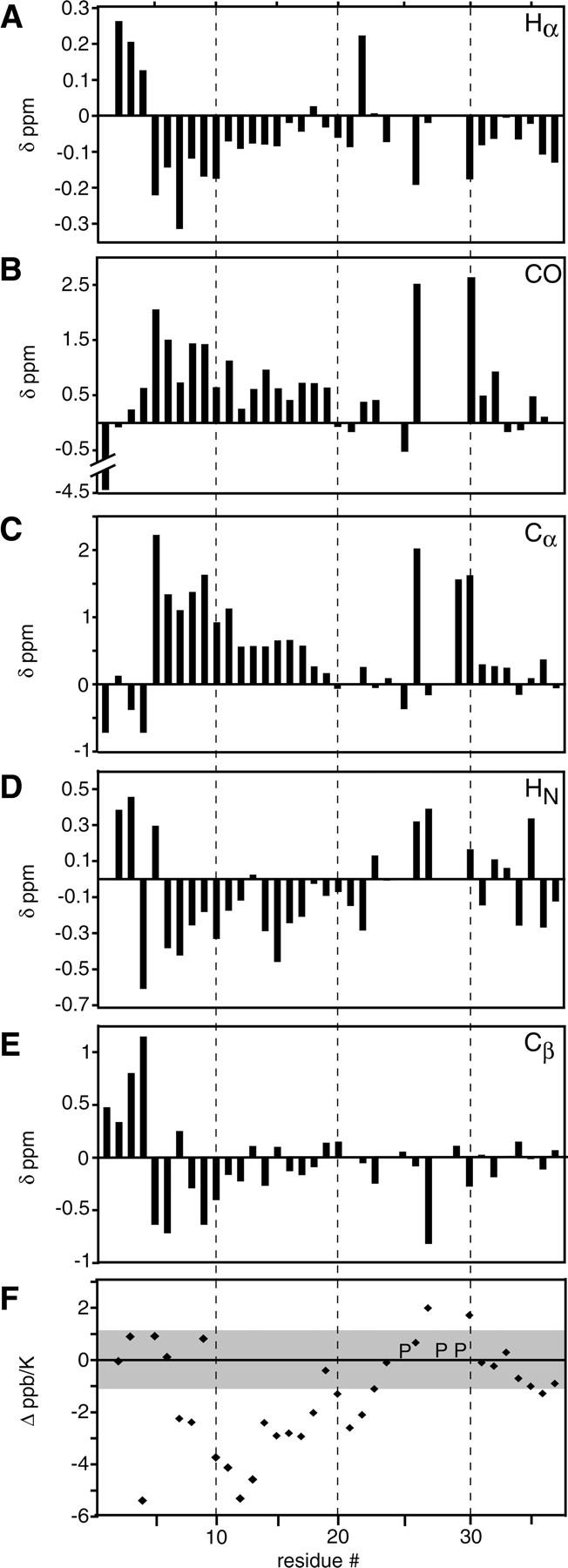
Secondary chemical shifts for Hα (A), CO (B), Cα (C), HN (D), and Cβ (E) of IAPP in solution. Random coil chemical shifts were taken from GGXAGG peptides measured in 1 M urea (Wishart et al. 1995) and corrected for sequence (Schwarzinger et al. 2001) and temperature (Merutka et al. 1995) where applicable. Secondary shifts for the amide nitrogen were also calculated, but produced no trends toward either helix or sheet (data not shown). (F) Deviation of temperature coefficients for rat IAPP from random coil values. The gray box across ±1.1 Δ ppb/K indicates the threshold for increased structure calculated from random coil models. For all chemical shift analyses, the greatest intensity peak was chosen to represent a given residue in the cases of multiple assignments. (P) Positions of proline residues.
Temperature coefficients
Temperature coefficient analysis reveals a stretch of structured residues in the N-terminal half of rat IAPP. The temperature coefficient (ppb/K) is an indicator of the chemical exchange between an intramolecular hydrogen bond and a hydrogen bond with bulk solvent. The chemical shift of an amide proton that participates in an intramolecular hydrogen bond or is buried in a defined structure is less sensitive to temperature than one that is wholly exposed (Cierpicki and Otlewski 2001; Dyson and Wright 2001). As with secondary shifts (above), a consistent stretch of residues with reduced temperature coefficients is indicative of local structure. The temperature coefficients for IAPP were calculated from change in 15N-HSQC chemical shifts for each residue across 5–30°C (Supplemental Table 2). Random coil temperature coefficients from host-guest model peptides (Merutka et al. 1995) were subtracted from measured IAPP coefficients (Δppb/K) (Fig. 4F). This correction has been used to highlight regions of nonrandom structure in, for example, apomyoglobin and SH3 domain (Yao et al. 2001; Zhang et al. 2005). The standard deviation reported in the random coil values ranged from ±0.09 to ±0.56 ppb/K. To establish a conservative threshold of significant deviation from random coil, we arbitrarily assert the following: difference coefficients larger than twice the maximum standard deviation (i.e., 2 × 0.56 ppb/K) are indicative of structure. This can be seen in a stretch of residues, 10–18 (and also 7–8 and 20–22), indicating an ordered region in IAPP.
Discussion
We have produced recombinant, wild-type, 15N-, 13C-labeled rat IAPP and assigned all backbone and Cβ nuclei using triple-resonance NMR experiments. The resonance assignments provided a means for structural assessment of IAPP in solution. In particular, the secondary chemical shifts indicate α-helical propensity from residues 5–19. Furthermore, the temperature coefficients indicate that this same region has residual structure.
Secondary shift and temperature coefficient data support canonical α-helical properties, such as specific hydrogen bonding patterns and helix capping (Pauling et al. 1951; Aurora and Rose 1998). The secondary shifts of IAPP indicate helical propensity from residues 5–19, whereas the temperature coefficients indicate structure from residues 7–22. This disparity is readily explained given that an α-helical amide proton would be affected if hydrogen bonded to a structured i − 4 backbone carbonyl group. The N-terminal end of an α-helix has unsatisfied hydrogen bond donors, which is consistent with the unstructured temperature coefficients of residues 5 and 6. Capping of the N-terminal helix end may be mediated by the side chains of residues in the constricted disulfide region. Similarly, the apparent structure at residues 21 and 22 suggests that these residues might play a role in capping the C-terminal end of the helix at residue 19. Such interactions would likely complement those of serines 19 and 20. Serines have an intrinsically low helical propensity; therefore, the observation that these residues are unstructured in IAPP is consistent with helix termination by C-terminal capping (Aurora and Rose 1998).
Our data are consistent with transient and cooperative sampling of helical structure. CD measurements have indicated IAPP has ∼10% helical content under aqueous solution conditions (Knight et al. 2006). This can arise from two possibilities. First, the peptide is in equilibrium between an α-helical and random coil state. Second, residues 5–19 adopt conformations independent of one another, each of which have 10% α-helical bias. Consistent secondary shift data across a sequential set of residues are generally taken to indicate the former. In addition, temperature coefficients suggest a cooperative conversion as i, i + 4 intra-molecular hydrogen bonding is likely the dominant factor diminishing temperature sensitivity. For a few residues, 10–13, we observe coefficients comparable to that of a folded protein. In a comprehensive study of proteins of known structure, hydrogen bonded protons had temperature coefficients ranging from –5 to 0 ppb/K (Cierpicki and Otlewski 2001). Residues 10–13 have high coefficients ranging from −3.9 to −3.1 ppb/K (Supplemental Table 2). As these residues’ chemical shifts are consistent with partial structure, it is likely that these protons experience additional interactions. One possible source of this is proximal aromatic side chains, which can result in increased temperature coefficients, even in disordered residues (Cierpicki and Otlewski 2001). Residues 10–13 may be perturbed by the neighboring Phe15 and also Tyr37, which has been shown to transiently interact with Phe15 by fluorescence resonance energy transfer measurements (Padrick and Miranker 2001).
Helical sampling by IAPP is consistent with its biological function. A sample of IAPP sequence variants, including human, rat, chicken, salmon, and cat, was analyzed using the AGADIR helical prediction algorithm (Fig. 5). Remarkably, all variants have a strong helical propensity across our experimentally observed region (residues 5–19). Experimentally comparable results have been observed in 25% 1,1,1,3,3,3-hexafluoro-2-propanol, where rat IAPP forms a stable helical domain spanning residues 5–20 (Cort et al. 1994). Similarly, IAPP shares ∼40% identity with calcitonin gene-related peptide (CGRP), a member of the same peptide hormone family (Cooper 1994; Muff et al. 2004). In 50% trifluoroacetic acid, CGRP is reported to form a helical segment spanning residues 6–18 (Breeze et al. 1991). While fluorinated alcohols are not physiological, they can nevertheless serve to illuminate subregions of the peptide with helical propensity. Removal of the helical region decreases CGRP receptor affinity by 50–100 fold (Howitt et al. 2003). As IAPP can also bind to CGRP receptors (Hay et al. 2004), and we observe helical properties of IAPP under physiological conditions, we suggest the conserved helical region is important for IAPP hormonal activity. The helical sampling we observe in aqueous solution may therefore be a reflection of the biologically active state.
Figure 5.

Helical propensities for human, rat, chicken, salmon, and cat IAPP sequence variants predicted with AGADIR at pH 5.5 and 5°C. The arrow from residues 5–19 indicates our experimentally determined helical region. Helical propensities at extracellular pH 7.4 and 37°C were similarly predicted across the same residues (data not shown).
Transient α-helix formation by IAPP may also reflect a possible intermediate of amyloid formation. Indeed, residues 8–14 of IAPP have been predicted to be α-helical and residues 8–20 can be induced to form fibers in isolation (Jaikaran et al. 2001). Intriguingly, IAPP binds lipid bilayers, whereupon it forms oligomers of α-helical states in equilibrium with monomer. For human IAPP, the population of these oligomeric species is correlated both with fiber formation and membrane leakage (Knight et al. 2006). Conditions found in the diabetic islet cell may therefore promote the formation of oligomeric α-helical states. For IAPP on lipid bilayers, it is suggested that the α-helical oligomers preorganize the peptide into parallel assemblies. We speculate that the transient α-helical sampling of IAPP in solution is a structural reflection of similar states stabilized by lipid bilayers. Further assembly into amyloid by IAPP is likely mediated by additional interactions unique to the human IAPP sequence and the type 2 diabetic state. Additionally, helix formation is seen in other natively unstructured amyloid proteins. Aβ peptide from Alzheimer's disease is α-helical on micelle surfaces (Shao et al. 1999) and has been shown to sample α-helical states in aqueous solution prior to transition to amyloid fiber (Kirkitadze et al. 2001). Also, the N terminus of α-synuclein from Parkinson's disease forms helices whose stability affects amyloid formation and toxicity (Kessler et al. 2003). Preassembly of α-helices may therefore be a common factor affecting amyloid formation by natively unstructured peptides.
Materials and methods
Reagents
All oligonucleotides and synthetic peptides were synthesized in-house at the W.M. Keck Foundation Biotechnology Resource Laboratory. Molecular biology reagents were obtained from New England Biolabs (NEB). Buffers, salts, and solvents were obtained from Macalaster Bicknel, American Bioanalytical, J.T. Baker, or Sigma. Isotopic salts and solvent were obtained from Cambridge Isotope Laboratories.
Cloning
The coding sequences of the peptide inserts were designed with optimal bacterial codon usage for expression. Six oligonucleotides, comprising the complimentary forward and reverse sequences, were annealed to create the double-stranded coding sequence for IAPP with SapI and NdeI compatible overhands. The sequence was ligated into the pTXB1 vector (NEB), which had been linearized with SapI and NdeI. The pTXB1:IAPP plasmid was cloned in LE392 E. coli cells (Murray et al. 1977) and purified with a QIAGEN spin miniprep kit and sequenced for verification.
Expression and purification
The construct was transformed into E. coli BL21(DE3) expression cells (Studier and Moffatt 1986) and grown in LB medium with 100 μg/mL ampicillin at 37°C until OD600 reached 0.5–0.6. To express 13C- and/or 15N-labeled protein, cells were switched into Neidhardt minimal medium (Neidhardt et al. 1974; Gehman 2003) containing 2 g/L 13C-glucose and/or 9.5 mM 15N-ammonium chloride. After induction with 1 mM IPTG, protein was expressed overnight (15–17 h) at 15°C. All subsequent steps were carried out at 4°C, except where noted. Cells were collected and resuspended in equilibration buffer (EB) containing 20 mM HEPES, 0.1 mM EDTA, 50 mM NaCl, and 2 M urea (pH 8). Cells were lysed by sonication using a Branson Sonifer at level 5 constant ouput for 5 min on ice with 30-sec pauses every 30 sec. Insoluble cell matter was pelleted by centrifugation at 5000 relative centrifugal force. The soluble cell lysate was run at 0.5 mL/min over a 20-mL chitin (NEB) column pre-equilibrated with EB. The column was washed with 15 column volumes of EB at 1 mL/min followed by three column volumes of cleavage buffer (CB). CB contains EB plus 100 mM DTT and 2 M ammonium bicarbonate. Incubation in CB overnight induced the intein-mediated cleavage and C-terminally amidated IAPP (Cottingham et al. 2001). Soluble leader-IAPP-NH2 was eluted in one column volume and dialyzed in 1000 MWCO Specta/Por 7 tubing at 4°C. The peptide was dialyzed against 2 L of 20 mM HEPES, 0.1 mM EDTA, and 2 M urea (pH 8); then 2 L of 20 mM HEPES, 0.1 mM EDTA, 2 M urea, 6.3 mM cysteamine, and 3.7 mM cystamine (pH 8); and finally, 4 L of 20 mM HEPES, 0.1 mM EDTA, and 0.5 M urea (pH 8). The N-terminal leader sequence was removed by digestion with 50 mg of V8 protease (Sigma) for 2 h at room temperature. Native IAPP was purified by reverse-phase HPLC on a Vydac Protein and Peptide C18 column with a gradient of 5% HPLC-grade acetonitrile (ACN) and 0.5% trifluoroacetic acid (TFA) to 90% ACN and 0.05% TFA. One liter of saturated E. coil culture yielded ∼2 mg of purified protein. Purified peptide was stored lyophilized at –20°C until use.
Mass spectrometry
Purified rat IAPP was sprayed on a Micromass LCT electrospray time of flight spectrometer to verify mass. To verify amidation and oxidation, synthetic human IAPP was used as an internal standard by doping it in equal amounts into rat IAPP or rat IAPP preincubated with 90 mM DTT. Peptides were sprayed from 50% acetonitrile with 0.2% formic acid and masses were calibrated externally using 20 mM CsI.
NMR
Lyophilized peptide was dissolved in water to half the final volume and vortexed; 2× buffer was added to make ∼1.5 mM IAPP in 50 mM potassium phosphate, 100 mM potassium chloride, 10% D2O (pH 5.5), adjusted by HCl, not compensating for deuterium. All spectra were recorded in house on a 500-MHz Varian Unity Inova with a room temperature or cryogenic probe. The proton chemical shifts were referenced to the water peak and the 15N and 13C shifts were referenced indirectly. The temperature was set to 5°C (unless noted) and calibrated with neat methanol. Triple-resonance backbone assignments were made using double- and triple-resonance experiments as described in Supplemental Table 1 (Ikura et al. 1990; Grzesiek and Bax 1992a,b; Kay et al. 1992; Zhang et al. 1994). Mixing times for the NOESY-15N-HSQC and TOCSY-15N-HSQC were 175 and 75 msec, respectively. NMR data were processed in NMRDraw (Delaglio et al. 1995) using standard window functions, zero-filling, and linear prediction. NMR processing and analysis was done in Sparky (T.D. Goddard and D.G. Kneller, University of California, San Francisco).
We chose the random coil values determined by Wishart et al. (1995) measured by GGXGAGG peptides (pH 5) in 1 M urea, as this was closest to our experimental conditions. The random coil shifts (except for 13Cβ) were corrected for sequence using the parameters determined by Schwarzinger et al. (2001). The amide proton shifts were corrected for temperature using random coil temperature coefficients (Merutka et al. 1995). Random coil values were subtracted from the experimental shifts.
A series of 15N-HSQCSs of rat IAPP were measured from 5°C to 30°C. Temperature was calibrated with neat methanol. The chemical shifts of each residue were plotted against temperature and fit by linear regression to extract the temperature coefficients. Random coil temperature coefficients (Merutka et al. 1995) were subtracted from measured coefficients to generate Δ ppb/K values.
AGADIR analysis
AGADIR was implemented at http://www.embl-heidelberg.de/Services/serrano/agadir/agadir-start.html (Munoz and Serrano 1997). Aligned sequences from human, rat, salmon, cat, and chicken IAPP were obtained from Knight et al. (2006). Helicity was predicted under our NMR conditions (pH 5.5 and 278 K) and physiological conditions (pH 7.4 and 310 K) and with the C terminus amidated.
Electronic supplemental material
Supplemental material includes two tables. The first table describes the NMR experiments and parameters used to determine the resonance assignments of the peptide. The second table lists the raw temperature coefficient data for the peptide.
Acknowledgments
We thank Dr. S. Padrick for early discussion and efforts on the expression system and Dr. C. Eakin for critical reading of the manuscript. We also thank the NMR users at Yale University for time and technical support. This work was funded by a grant from the National Institute of Health (DK54899).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Andrew D. Miranker, Department of Molecular Biophysics and Biochemistry, Yale University, 260 Whitney Avenue, New Haven, CT 06520-8114, USA; e-mail: Andrew.miranker@yale.edu; fax: (203) 432-5175.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062486907.
References
- Abedini, A. and Raleigh, D.P. 2006. Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: Is there a critical amyloidogenic domain in human IAPP? J. Mol. Biol. 355: 274–281. [DOI] [PubMed] [Google Scholar]
- Aurora, R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7: 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeze, A.L., Harvey, T.S., Bazzo, R., and Campbell, I.D. 1991. Solution structure of human calcitonin gene-related peptide by 1H NMR and distance geometry with restrained molecular dynamics. Biochemistry 30: 575–582. [DOI] [PubMed] [Google Scholar]
- Butler, A.E., Jang, J., Gurlo, T., Carty, M.D., Soeller, W.C., and Butler, P.C. 2004. Diabetes due to a progressive defect in β-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): A new model for type 2 diabetes. Diabetes 53: 1509–1516. [DOI] [PubMed] [Google Scholar]
- Chiti, F. and Dobson, C.M. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75: 333–366. [DOI] [PubMed] [Google Scholar]
- Cierpicki, T. and Otlewski, J. 2001. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21: 249–261. [DOI] [PubMed] [Google Scholar]
- Cooper, G.J. 1994. Amylin compared with calcitonin gene-related peptide: Structure, biology, and relevance to metabolic disease. Endocr. Rev. 15: 163–201. [DOI] [PubMed] [Google Scholar]
- Cort, J., Liu, Z., Lee, G., Harris, S.M., Prickett, K.S., Gaeta, L.S., and Andersen, N.H. 1994. β-structure in human amylin and two designer β-peptides: CD and NMR spectroscopic comparisons suggest soluble β-oligomers and the absence of significant populations of β-strand dimers. Biochem. Biophys. Res. Commun. 204: 1088–1095. [DOI] [PubMed] [Google Scholar]
- Cottingham, I.R., Millar, A., Emslie, E., Colman, A., Schnieke, A.E., and McKee, C. 2001. A method for the amidation of recombinant peptides expressed as intein fusion proteins in Escherichia coli . Nat. Biotechnol. 19: 974–977. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1993. Proteins: Structures and molecular properties, 2d ed. W.H. Freeman and Company, New York.
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Dobeli, H., Draeger, N., Huber, G., Jakob, P., Schmidt, D., Seilheimer, B., Stuber, D., Wipf, B., and Zulauf, M. 1995. A biotechnological method provides access to aggregation competent monomeric Alzheimer's 1–42 residue amyloid peptide. Biotechnology (N.Y.) 13: 988–993. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Lawson, J.D., Brown, C.J., Williams, R.M., Romero, P., Oh, J.S., Oldfield, C.J., Campen, A.M., Ratliff, C.M., and Hipps, K.W., et al. 2001. Intrinsically disordered protein. J. Mol. Graph. Model. 19: 26–59. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2001. Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol. 339: 258–270. [DOI] [PubMed] [Google Scholar]
- Eakin, C.M., Berman, A.J., and Miranker, A.D. 2006. A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 13: 202–208. [DOI] [PubMed] [Google Scholar]
- Gehman, J.D. 2003. Theoretical macromolecular structure elucidation using solid state nuclear magnetic resonance. In Chemistry, pp. 160–188. Yale University, New Haven, CT.
- Green, J., Goldsbury, C., Mini, T., Sunderji, S., Frey, P., Kistler, J., Cooper, G., and Aebi, U. 2003. Full-length rat amylin forms fibrils following substitution of single residues from human amylin. J. Mol. Biol. 326: 1147–1156. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1992a. Correlating backbone amide and side-chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114: 6291–6293. [Google Scholar]
- Grzesiek, S. and Bax, A. 1992b. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J. Magn. Reson. 99: 201–207. [Google Scholar]
- Hay, D.L., Christopoulos, G., Christopoulos, A., and Sexton, P.M. 2004. Amylin receptors: Molecular composition and pharmacology. Biochem. Soc. Trans. 32: 865–867. [DOI] [PubMed] [Google Scholar]
- Hoppener, J.W., Ahren, B., and Lips, C.J. 2000. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 343: 411–419. [DOI] [PubMed] [Google Scholar]
- Howitt, S.G., Kilk, K., Wang, Y., Smith, D.M., Langel, U., and Poyner, D.R. 2003. The role of the 8-18 helix of CGRP8-37 in mediating high affinity binding to CGRP receptors; coulombic and steric interactions. Br. J. Pharmacol. 138: 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, R.L., Westermark, G.T., Westermark, P., and Kahn, S.E. 2004. Islet amyloid: A critical entity in the pathogenesis of type 2 diabetes. J. Clin. Endocrinol. Metab. 89: 3629–3643. [DOI] [PubMed] [Google Scholar]
- Ikura, M., Kay, L.E., and Bax, A. 1990. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: Heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 29: 4659–4667. [DOI] [PubMed] [Google Scholar]
- Jaikaran, E.T. and Clark, A. 2001. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 1537: 179–203. [DOI] [PubMed] [Google Scholar]
- Jaikaran, E.T., Higham, C.E., Serpell, L.C., Zurdo, J., Gross, M., Clark, A., and Fraser, P.E. 2001. Identification of a novel human islet amyloid polypeptide β-sheet domain and factors influencing fibrillogenesis. J. Mol. Biol 308: 515–525. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc 114: 10663–10665. [Google Scholar]
- Kayed, R., Bernhagen, J., Greenfield, N., Sweimeh, K., Brunner, H., Voelter, W., and Kapurniotu, A. 1999. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol 287: 781–796. [DOI] [PubMed] [Google Scholar]
- Kessler, J.C., Rochet, J.C., and Lansbury Jr., P.T. 2003. The N-terminal repeat domain of α-synuclein inhibits β-sheet and amyloid fibril formation. Biochemistry 42: 672–678. [DOI] [PubMed] [Google Scholar]
- Kirkitadze, M.D., Condron, M.M., and Teplow, D.B. 2001. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 312: 1103–1119. [DOI] [PubMed] [Google Scholar]
- Knight, J.D., Hebda, J.A., and Miranker, A.D. 2006. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry 45: 9496–9508. [DOI] [PubMed] [Google Scholar]
- Lopes, D.H., Colin, C., Degaki, T.L., de Sousa, A.C., Vieira, M.N., Sebollela, A., Martinez, A.M., Bloch Jr., C., Ferreira, S.T., and Sogayar, M.C. 2004. Amyloidogenicity and cytotoxicity of recombinant mature human islet amyloid polypeptide (rhIAPP). J. Biol. Chem. 279: 42803–42810. [DOI] [PubMed] [Google Scholar]
- Lopez de la Paz, M., de Mori, G.M., Serrano, L., and Colombo, G. 2005. Sequence dependence of amyloid fibril formation: Insights from molecular dynamics simulations. J. Mol. Biol. 349: 583–596. [DOI] [PubMed] [Google Scholar]
- MacArthur, M.W. and Thornton, J.M. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218: 397–412. [DOI] [PubMed] [Google Scholar]
- Matveyenko, A.V. and Butler, P.C. 2006. β-Cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes 55: 2106–2114. [DOI] [PubMed] [Google Scholar]
- Mazor, Y., Gilead, S., Benhar, I., and Gazit, E. 2002. Identification and characterization of a novel molecular-recognition and self-assembly domain within the islet amyloid polypeptide. J. Mol. Biol. 322: 1013–1024. [DOI] [PubMed] [Google Scholar]
- McNulty, B.C., Young, G.B., and Pielak, G.J. 2006. Macromolecular crowding in the Escherichia coli periplasm maintains α-synuclein disorder. J. Mol. Biol. 355: 893–897. [DOI] [PubMed] [Google Scholar]
- Merutka, G., Dyson, H.J., and Wright, P.E. 1995. ‘Random coil’ 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR 5: 14–24. [DOI] [PubMed] [Google Scholar]
- Muff, R., Born, W., Lutz, T.A., and Fischer, J.A. 2004. Biological importance of the peptides of the calcitonin family as revealed by disruption and transfer of corresponding genes. Peptides 25: 2027–2038. [DOI] [PubMed] [Google Scholar]
- Munoz, V. and Serrano, L. 1997. Development of the multiple sequence approximation within the AGADIR model of α-helix formation: Comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers 41: 495–509. [DOI] [PubMed] [Google Scholar]
- Murray, N.E., Brammar, W.J., and Murray, K. 1977. Lambdoid phages that simplify the recovery of in vitro recombinants. Mol. Gen. Genet. 150: 53–61. [DOI] [PubMed] [Google Scholar]
- Neidhardt, F.C., Bloch, P.L., and Smith, D.F. 1974. Culture medium for enterobacteria. J. Bacteriol. 119: 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, R., Sawaya, M.R., Balbirnie, M., Madsen, A.O., Riekel, C., Grothe, R., and Eisenberg, D. 2005. Structure of the cross-β spine of amyloid-like fibrils. Nature 435: 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrick, S.B. and Miranker, A.D. 2001. Islet amyloid polypeptide: Identification of long-range contacts and local order on the fibrillogenesis pathway. J. Mol. Biol. 308: 783–794. [DOI] [PubMed] [Google Scholar]
- Padrick, S.B. and Miranker, A.D. 2002. Islet amyloid: Phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry 41: 4694–4703. [DOI] [PubMed] [Google Scholar]
- Pauling, L., Corey, R.B., and Branson, H.R. 1951. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. 37: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova, A.T., Leapman, R.D., Guo, Z., Yau, W.M., Mattson, M.P., and Tycko, R. 2005. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307: 262–265. [DOI] [PubMed] [Google Scholar]
- Schwarzinger, S., Kroon, G.J., Foss, T.R., Wright, P.E., and Dyson, H.J. 2000. Random coil chemical shifts in acidic 8 M urea: Implementation of random coil shift data in NMRView. J. Biomol. NMR 18: 43–48. [DOI] [PubMed] [Google Scholar]
- Schwarzinger, S., Kroon, G.J., Foss, T.R., Chung, J., Wright, P.E., and Dyson, H.J. 2001. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 123: 2970–2978. [DOI] [PubMed] [Google Scholar]
- Seavey, B.R., Farr, E.A., Westler, W.M., and Markley, J.L. 1991. A relational database for sequence-specific protein NMR data. J. Biomol. NMR 1: 217–236. [DOI] [PubMed] [Google Scholar]
- Shao, H., Jao, S., Ma, K., and Zagorski, M.G. 1999. Solution structures of micelle-bound amyloid β-(1-40) and β-(1-42) peptides of Alzheimer's disease. J. Mol. Biol. 285: 755–773. [DOI] [PubMed] [Google Scholar]
- Studier, F.W. and Moffatt, B.A. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189: 113–130. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Fink, A.L. 2004. Conformational constraints for amyloid fibrillation: The importance of being unfolded. Biochim. Biophys. Acta 1698: 131–153. [DOI] [PubMed] [Google Scholar]
- Wang, F., Hull, R.L., Vidal, J., Cnop, M., and Kahn, S.E. 2001. Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes 50: 2514–2520. [DOI] [PubMed] [Google Scholar]
- Westermark, G.T., Gebre-Medhin, S., Steiner, D.F., and Westermark, P. 2000. Islet amyloid development in a mouse strain lacking endogenous islet amyloid polypeptide (IAPP) but expressing human IAPP. Mol. Med. 6: 998–1007. [PMC free article] [PubMed] [Google Scholar]
- Westermark, P., Engstrom, U., Johnson, K.H., Westermark, G.T., and Betsholtz, C. 1990. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. 87: 5036–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994. Chemical shifts as a tool for structure determination. Methods Enzymol. 239: 363–392. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222: 311–333. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Holm, A., Hodges, R.S., and Sykes, B.D. 1995. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5: 67–81. [DOI] [PubMed] [Google Scholar]
- Yao, J., Chung, J., Eliezer, D., Wright, P.E., and Dyson, H.J. 2001. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry 40: 3561–3571. [DOI] [PubMed] [Google Scholar]
- Zhang, O. and Forman-Kay, J.D. 1997. NMR studies of unfolded states of an SH3 domain in aqueous solution and denaturing conditions. Biochemistry 36: 3959–3970. [DOI] [PubMed] [Google Scholar]
- Zhang, O., Kay, L.E., Olivier, J.P., and Forman-Kay, J.D. 1994. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 4: 845–858. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Xu, Y., Zhang, J., Wu, J., and Shi, Y. 2005. Structural and dynamic characterization of the acid-unfolded state of hUBF HMG box 1 provides clues for the early events in protein folding. Biochemistry 44: 8117–8125. [DOI] [PubMed] [Google Scholar]