

Abstract
Docking ligands into an ensemble of NMR conformers is essential to structure-based drug discovery if only NMR structures are available for the target. However, sequentially docking ligands into each NMR conformer through standard single-receptor-structure docking, referred to as sequential docking, is computationally expensive for large-scale database screening because of the large number of NMR conformers involved. Recently, we developed an efficient ensemble docking algorithm to consider protein structural variations in ligand binding. The algorithm simultaneously docks ligands into an ensemble of protein structures and achieves comparable performance to sequential docking without significant increase in computational time over single-structure docking. Here, we applied this algorithm to docking with NMR structures. The HIV-1 protease was used for validation in terms of docking accuracy and virtual screening. Ensemble docking of the NMR structures identified 91% of the known inhibitors under the criterion of RMSD < 2.0 Å for the best-scored conformation, higher than the average success rate of single docking of individual crystal structures (66%). In the virtual screening test, on average, ensemble docking of the NMR structures obtained higher enrichments than single-structure docking of the crystal structures. In contrast, docking of either the NMR minimized average structure or a single NMR conformer performed less satisfactorily on both binding mode prediction and virtual screening, indicating that a single NMR structure may not be suitable for docking calculations. The success of ensemble docking of the NMR structures suggests an efficient alternative method for standard single docking of crystal structures and for considering protein flexibility.
Keywords: molecular docking, ligand–protein interaction, protein flexibility, HIV-1 protease, NMR structures
Molecular docking plays an important role in structure-based drug design (Halperin et al. 2002; Shoichet et al. 2002; Brooijmans and Kuntz 2003). Given two molecules, referred to as the receptor and the ligand, molecular docking attempts to predict the binding mode by evaluating the energy scores of different bound conformations with a scoring function. A pre-condition for molecular docking is the availability of the receptor structure. Two types of experimental techniques are commonly used for protein structure determination, X-ray crystallography and NMR spectroscopy. X-ray structures normally have higher precision than NMR structures. On the other hand, NMR structures are determined in solution and therefore are often thought to be more biologically relevant than X-ray structures (Garbuzynskiy et al. 2005).
To date, most molecular docking studies use high-resolution crystal structures for protein targets. However, for some proteins, their X-ray structures may not be achievable because they cannot be crystallized, whereas the NMR structures of these proteins may be acquired. In these cases, docking ligands into NMR structures is inevitable. Furthermore, NMR spectroscopy is ideal for observing protein dynamic motions; the solved structures reflect protein structural variations. It is therefore necessary to study docking ligands into NMR structures.
It is challenging to use NMR structures for molecular docking because a typical NMR structure usually contains tens of conformers. A simple method is to select a representative structure from one of the NMR structures, or simply to use the NMR minimized average structure as a representative of the protein target for docking. However, it is difficult to determine which NMR conformer is the representative structure. Our calculated results also revealed that the NMR minimized average structure or a single NMR conformer usually performs poorly for prediction on binding modes and affinities/scores (see Results and Discussion). An alternative method, referred to as sequential docking, is to sequentially dock ligands into each NMR conformer; scores for individual docking are then merged into one list by re-ranking. The run time of sequential docking is thus proportional to the number of protein conformers. Consequently, sequential docking is computationally too expensive for large database screening because of the large number of NMR conformers involved.
Kuntz and colleagues developed an elegant method to use NMR structures in molecular docking (Knegtel et al. 1997). In their pioneer work, a composite energy grid was constructed by combining the energy grids generated from every NMR structure with a weighting scheme; the grid was then used for standard ligand docking (Meng et al. 1992). Despite the success of the composite-grid algorithm, the method may lead to loss of geometric accuracy because of its averaging nature (Knegtel et al. 1997; Broughton 2000).
Recently, we developed a novel algorithm for molecular docking that uses multiple protein structures to account for protein structural variations in ligand binding (Huang and Zou 2006a). This algorithm, referred to as ensemble docking, is able to simultaneously dock ligands into multiple protein structures by optimizing both the ligand orientation and the receptor conformation. Our ensemble docking algorithm obtains a good balance between computational efficiency and docking accuracy, with a run time comparable to that of standard single docking and a performance comparable to that of sequential docking. In this study, we used this fast docking algorithm to dock against NMR structures, which normally consist of tens of conformers.
Here, we chose HIV-1 protease (HIVp) as a test case for docking evaluations. HIVp is an important drug target for treatment of AIDS. Several HIVp inhibitors are FDA-approved anti-HIV drugs (Babine and Bender 1997). Protein flexibility plays a significant role in ligand binding to HIVp (Erickson et al. 2004; Meagher and Carlson 2004). Numerous X-ray and NMR structures of HIVp are available in the Protein Data Bank (Berman et al. 2000), which allows us to assess ensemble docking against NMR structures. The assessment is twofold. First, can the ensemble docking algorithm dock against NMR structures instead of a single X-ray crystal structure without a significant increase in run time? Second, how is the efficacy of NMR structures on incorporating protein flexibility in ligand binding?
Results and Discussion
Docking accuracy
One common goal of molecular docking is to predict ligand binding modes, referred to as docking accuracy. Here, the docking accuracy was evaluated in terms of the root mean square deviation (RMSD) between the docked position and the experimentally determined position for the ligand. In this study, a docking prediction was considered successful if the RMSD value is <2.0 Å for the best-scored conformation (Cole et al. 2005). This is the default criterion unless specified again. As described in Materials and Methods, ligand flexibility was treated by pre-generating multiple ligand conformers with the OMEGA software (OpenEye Scientific Inc.) and then docking each ligand conformer into the protein target as a rigid molecule. The energy scores of the conformers were then merged for each ligand. The conformer with the lowest score represented the docked ligand.
A total of 11 HIVp ligands, which were extracted from their bound crystal structures (see Table 1 and Fig. 1), were used to evaluate our ensemble docking algorithm. Figure 2A shows the RMSD results of ensemble docking against the ensemble of the HIVp NMR conformers (see the last column). For comparison, the panel also lists the RMSD results of standard single-receptor docking against the individual X-ray crystal structures (including the apo structure, 3PHV), the NMR minimized average structure (i.e., 1BVG), and a randomly selected NMR conformer of 1BVE (i.e., the first model of the NMR structures, referred to as 1NMR). In addition, the panel shows the results of sequential docking against the X-ray crystal structures (denoted as XSEQ), in which the energy scores were pooled from single docking of all 12 X-ray structures (i.e., 11 bound structures and one apo structure) and re-ranked.
Table 1.
Tested HIV-1 protease structures used for binding mode prediction and virtual screening

Figure 1.

Chemical structures of the inhibitors of HIV-1 protease used in the present test studies. The ligand code (defined in Table 1) and its corresponding PDB entry were labeled for each inhibitor. The chemical structures were drawn using the MarvinSketch tool of ChemAxon Ltd. (http://www.chemaxon.com/marvin/).
Figure 2.

The gray-scaled table of the RMSD values (Å) for the predictions of ligand binding modes using different docking methods. The rows refer to different HIVp inhibitors. The columns refer to single rigid-receptor docking against different HIVp structures, sequential docking against the X-ray crystal structures (XSEQ), and ensemble docking against the NMR structures (ENMR), respectively. The cells with darker borders represent native docking. SR stands for success rate of binding mode prediction. The RMSD values were calculated based on the best-scored ligand conformation/orientation (A) and the top five ligand conformations/orientations (B).
It can be seen from Figure 2A that ensemble docking of the NMR structures identified the near-native modes of 10 ligands and yielded a success rate of 91%, only slightly lower than that of sequential docking against the crystal structures (100%). Notice that none of the 11 ligands extracted from the crystal structures is present in the NMR structures. Overall, single docking using the crystal structures yielded lower success rates (66% on average), ranging from 9% for the apo structure to 91% for 1HVR and 1PRO. Single docking against 1BVG and 1NMR gave low success rates (27% and 18%), suggesting that the NMR energy-minimized average structure or a randomly selected NMR conformer may not be efficient for docking calculations.
It is also noted from Figure 2A that some X-ray structures such as 1HVR and 1PRO still gave success rates as high as 91% on binding mode prediction. One may then conclude that these crystal structures would be good targets for structure-based drug design. However, in practice (e.g., virtual screening, for which sequential docking is too time consuming), it is usually impossible to know which crystal structure would yield a high success rate before docking calculations are performed. In the present test case, the docking accuracy of ensemble docking using the NMR structures is encouraging, compared to the docking accuracy of standard single docking with individual X-ray structures. When no crystal structure is available, ensemble docking using the NMR structures is particularly useful.
Figure 2A also shows that single docking correctly identified all the near-native binding modes when the 11 ligands were re-docked to their co-crystallized protein structures (referred to as native docking; see the diagonal cells with darker borders in the panel). The success of native docking served as a validation of the docking algorithm and the scoring function used in this study. When the ligands were cross-docked into the HIVp crystal structures originally bound with other ligands via single-structure docking (referred to as cross docking), the success rates decreased significantly, as shown in the figure panel. The decrease is because of the protein flexibility and the resulting protein conformational changes upon ligand binding (“induced fit”). Different ligands may induce different protein conformational changes. Therefore, it is difficult to cope with all the induced conformational changes using standard single-structure docking, leading to failure of some cross-docking runs. As reviewed by Carlson (2002) and Teodoro and Kavraki (2003), one way to account for protein flexibility is to use multiple protein structures (Knegtel et al. 1997; Broughton 2000; Carlson et al. 2000; Claussen et al. 2001; Cavasotto and Abagyan 2004; Ferrari et al. 2004; Meagher and Carlson 2004; Wei et al. 2004; Zavodszky et al. 2004; Cavasotto et al. 2005; Huang and Zou 2006a). Our ensemble docking results substantiate this view. As shown in Figure 2A, ensemble docking using the NMR structures yielded a high success rate of 91%, implying the effectiveness of incorporating multiple protein structures to consider protein flexibility. Notice that none of the 11 ligands is present in the solved NMR structures. It is therefore impressive that ensemble docking using the NMR structures successfully identified the near-native binding modes of 10 out of the 11 ligands.
To further evaluate our docking algorithm, we also examined the docking accuracy when considering several top conformations/orientations in addition to the best-scored one. This is because it is common to save multiple top conformations/orientations for each ligand for further evaluation in hierarchical database screenings (Lamb et al. 2001; Wang et al. 2005). Here, we defined that a prediction is successful if any of the top five conformations satisfies the criterion of RMSD < 2.0 Å. If the RMSD values of all the top five conformations/orientations were >2.0 Å, the best-scored conformation/orientation was used for further analysis. Figure 2B shows the RMSD values of the 11 ligands for different docking runs. It can be seen from this panel that although the success rates of some docking runs increased slightly with a looser RMSD criterion, the relative performances of different docking methods remained the same. Specifically, although the average success rate of single docking with the crystal structures was slightly higher with the new criterion (71% vs. 66%), the success rate of ensemble docking with the NMR structures also improved (100% vs. 91%), matching that of sequential docking against the crystal structures (100%). The success rate of single docking with the NMR average minimized structure and the single NMR conformer remained low (27% each). These results again imply the efficiency of ensemble docking using NMR structures.
To examine whether the NMR structures indeed reflect the ligand-induced conformational changes, we took q82 of 1HVH as an example. This is the inhibitor with which many cross-docking tests failed on mode prediction, as shown in Figure 2. Figure 3 plots two typical HIVp structures, 1AJV and 2UPJ, which yielded large RMSD values for q82 (above 10 Å). In contrast, re-docking q82 into its co-crystallized protein structure (1HVH) was successful (RMSD = 0.2 Å). One of the NMR conformers also led to the success of ensemble docking of q82 (RMSD = 1.0 Å), as shown in Figure 2B. These two HIVp structures were also plotted in Figure 3. All the protein structures were superimposed by matching four backbone carbon atoms in the binding site (Ferrin et al. 1988). Figure 3 also displays the binding mode of q82 observed in the crystal structure (1HVH). It can be seen from the figure that there exists a large backbone movement at the loop ranging from K45 to K55. The HIVp structures of 1AJV and 2UPJ exhibit severe atomic clashes between the loop and the inhibitor q82, with the closest distance <1.0 Å. Consequently, single docking against 1AJV and 2UPJ resulted in poor predictions of the binding mode of q82. In contrast, in the co-crystallized HIVp structure of q82 (1HVH), the loop (K45–K55) moves downward for ∼2 Å in order to accommodate q82. Figure 3 shows that the NMR conformer correctly reflects this ligand-induced fit effect; it superimposes very well with the native structure (1HVH) around this loop region.
Figure 3.
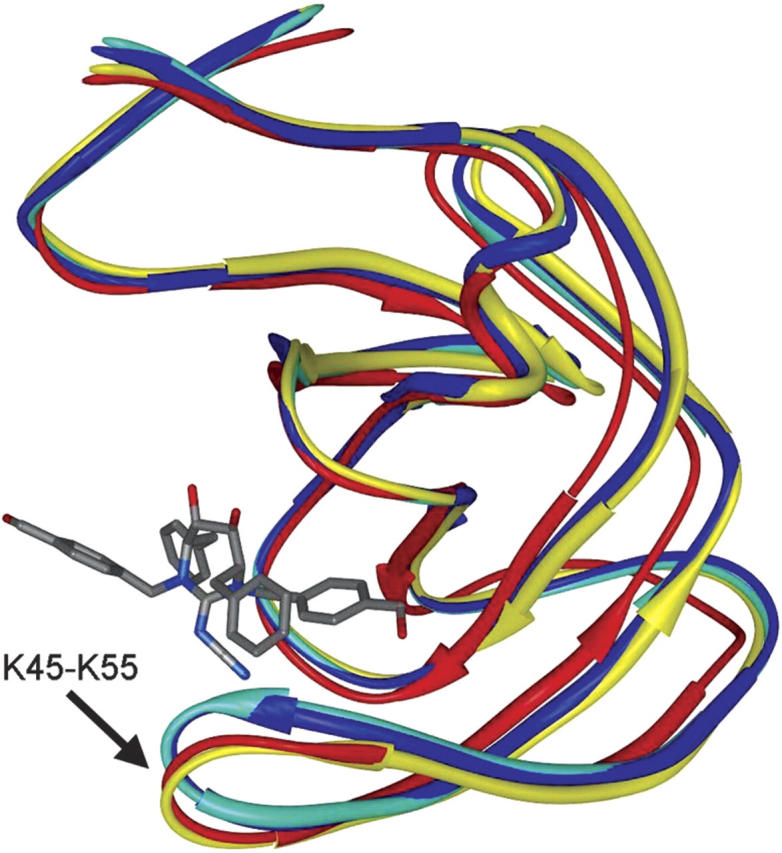
Comparison of four typical HIVp structures. The superimposed protein structures are shown in strand mode and colored in red (one of the NMR conformers), yellow (1HVH), cyan (1AJV), and blue (2UPJ). The co-crystallized ligand of 1HVH, q82, is displayed in stick mode. The loop segment, K45-K55, exhibits a movement as large as ∼2 Å. For sake of clarity, only one monomer of the dimeric HIVp is shown in this figure (see text for detail). The graphic image was produced using the MidasPlus program (University of California, San Francisco).
Computational efficiency
In addition to good docking accuracy, our ensemble docking algorithm is also computationally efficient with a run time comparable to that of standard single rigid-receptor docking, although it simultaneously addressed multiple protein structures, as demonstrated in our previous study (Huang and Zou 2006a). In the present test, ensemble docking using the NMR structures took an average of 3.4 sec for each ligand conformer, comparable to the run time for single docking of individual HIVp structures (∼3.0 sec). The run time for sequential docking against the 12 crystal structures was 35.4 sec, proportional to the number of protein structures in consideration.
Virtual database screening
In addition to predicting binding modes, another practical goal of molecular docking is to screen potential inhibitors from a large compound database, namely, identify ligands that bind to a target protein and distinguish them from other ligands that do not bind through energy ranking. Single rigid-structure docking may miss certain true inhibitors because of neglect of the induced-fit effect. Therefore, we next evaluated the performance of ensemble docking of the NMR structures on database screening.
As described in Materials and Methods, 1000 compounds were randomly selected from the Available Chemical Directory (ACD, distributed by Molecular Design Ltd., San Leandro, CA), and served as a database of decoy molecules. The 11 ligands extracted from the X-ray structures (see Fig. 1) were chosen as the active compounds. Thus, the screened database consists of 11 HIVp inhibitors and 1000 decoys. Similar to above, every ligand was allowed to be flexible in the docking calculations; up to 200 conformers generated via OMEGA were considered for each ligand. For each ligand, the conformer with the lowest energy score was kept. The ligands in the database were then ranked according to their energy scores.
A widely used index for success in virtual screening is the enrichment factor, defined as the fraction or accumulated rate of known inhibitors identified in a certain top percentage of the ranked database. At a fixed percentage of the ranked database, the higher the enrichment factor, the better the performance of docking in identifying known binders. Figure 4 shows the enrichment results of ensemble docking of the NMR structures. For comparison, the figure also shows the enrichments of single rigid-receptor and sequential docking with the 12 X-ray crystal structures, and enrichments of single docking of the NMR minimized average structure and the single NMR conformer (1NMR).
Figure 4.

Comparison of different docking algorithms on the enrichment test against HIV-1 protease. The dashed lines stand for single rigid-receptor docking against individual X-ray crystal structures except the apo structure (3PHV, dark yellow solid line). The magenta solid line shows the average enrichment of all single docking runs with the X-ray crystal structures (XAVE). The red solid line represents ensemble docking with the NMR structures (ENMR). The blue and green solid lines represent single-receptor docking with the NMR minimized average structure (1BVG) and a randomly selected NMR conformer of 1BVE (1NMR), respectively. The black solid line stands for sequential docking against all the X-ray crystal structures (XSEQ).
As shown in Figure 4, ensemble docking with the NMR structures matched single docking of the crystal structures in the top 1.2% of the ranked database, but performed significantly better than most single docking after this percentage. For example, in the top 2.7% of the ranked database, ensemble docking obtained a high enrichment factor of 91%, but the average enrichment of single docking with the crystal structures was 71%. Sequential docking of crystal structures still performed the best. Overall, ensemble docking of NMR structures performed better than single-structure docking. The NMR minimized average structure, and the single NMR conformer again performed less satisfactorily (Fig. 4).
Note that for a small top percentage of the ranked database, the average enrichment of single docking against the crystal structures was comparable to the enrichment of ensemble docking against the NMR structures. The reason can be understood as follows. The co-crystallized HIVp inhibitors were included in the compound database for 11 out of the 12 tested crystal structures. Therefore, these inhibitors were easily identified by their bound HIVp structures. On the other hand, the inhibitor co-bound to the NMR structures was not included in this study to mimic realistic docking calculations. Therefore, identification of the non-native inhibitors of the NMR structures was more difficult. Moreover, some of the inhibitors were similar to each other (see Fig. 1), and would likely induce similar conformational changes to the protein. These inhibitors could therefore be easier to identify by each other's co-bound HIVp structures, leading to higher enrichments for these particular HIVp structures. A contrasting example is that the apo structure (3PHV), which contains no induced-fit information, yielded a much lower enrichment (see Fig. 4).
It is also noted from Figure 4 that single docking against certain crystal structures (e.g., 1HVR, the red dashed line) performed better than ensemble docking against the NMR structures for any percentage of the ranked database. The reason is twofold. First, the co-crystallized ligands of these protein structures (e.g., xk2 of 1HVR) were similar to several other inhibitors in study (see Fig. 1). These similar inhibitors would likely induce similar protein conformational changes. Therefore, their co-bound HIVp structures would not be very different and may likely identify similar inhibitors in database screening, resulting in high enrichments. Second, the crystal structures bound with larger inhibitors tend to have larger binding pockets that have sufficient space to hold most inhibitors well. In contrast, single docking of the crystal structures that have very different co-crystallized ligands would yield low enrichments (e.g., 7UPJ; magenta dashed line in Fig. 4).
Considering that the NMR structures contain no structural information of the known inhibitors, the results of ensemble docking with the NMR structures are encouraging. Our virtual screening test suggests that the use of NMR structures seems to be a good way to consider protein flexibility.
Conclusions
In this study, we applied our novel ensemble docking algorithm to docking ligands into the NMR structures of HIV-1 protease. The results were evaluated in terms of binding mode prediction and virtual database screening. Although the NMR structures contain no information about the tested ligands, ensemble docking of the NMR structures yielded a success rate of 91% in predicting the binding modes, higher than the average success rate of single rigid-receptor docking of X-ray crystal structures (66%), under the criterion of RMSD < 2.0 Å for the best-scored ligand conformation. The results also showed that the NMR minimized average structure and a randomly selected NMR conformer may not be efficient for structure-based drug design because of their low success rates. In contrast, the ensemble of NMR structures may incorporate protein conformational changes in ligand binding. A similar trend was also observed in the virtual screening test. In addition to good docking accuracy, the ensemble docking algorithm was also computationally efficient, with a run time comparable to that of standard single-structure docking. Our results suggest that docking against NMR structures provides an efficient alternative method for standard docking against individual X-ray crystal structures.
Today, it remains unknown what structures give the best representation of protein flexibility, the crystal structures or the NMR structures (for reviews, see Carlson 2002; Teodoro and Kavraki 2003). In this study, ensemble docking of the NMR structures matched sequential docking of the crystal structures on binding mode prediction, and sequential docking performed better on virtual screening. The NMR structures seem to be a good alternative to a significant number of the crystal structures for docking studies, especially when multiple crystal structures are unavailable.
Materials and methods
Docking algorithm
We have recently developed a novel ensemble docking algorithm that can simultaneously dock against an ensemble of protein structures, for example, multiple crystal structures of a protein bound with different ligands (Huang and Zou 2006a). The key idea of the algorithm is to consider the conformational state of the protein as an additional variable. A brief description of the algorithm is as follows.
Without losing generality, we explain the ensemble docking algorithm by considering the ligand as a rigid body. This is because ligand flexibility can be accounted for by pre-generating multiple ligand conformers and then docking each ligand conformer, treated as a rigid body, into the protein target. Normally, a rigid ligand has six degrees of freedom, three translational (x, y, and z) and three rotational (θ, ϕ, and ψ). Here, x, y, and z represent the coordinates of the center of mass of the ligand, and θ, ϕ, and ψ denote the three Euler angles. A traditional scoring procedure is therefore described as:
In the ensemble docking algorithm, we introduce the seventh variable, m, which stands for the m-th structure of the protein ensemble. m is an integer and ranges from 1 to M, where M is the total number of protein conformations in the ensemble. The energy function is then not only a function of (x, y, z, θ, ϕ, and ψ), which characterizes a ligand orientation, but also a function of m, which indicates the protein conformation that the ligand is docked to:
Introduction of the integer variable m enables the step for selection of an optimal protein conformation to be effectively incorporated into the energy optimization procedure through Equation 2. Consequently, the ensemble docking algorithm is efficient on docking against multiple protein structures; the corresponding computational time has been demonstrated to be comparable to that of standard single-structure docking (Huang and Zou 2006a).
A simplified FORTRAN version of the DOCK4.0 program (Ewing et al. 2001) was developed to implement our ensemble docking algorithm. Specifically, we adopted the exhaustive matching algorithm of Ewing and Kuntz (1997) to generate possible ligand orientations. The “distance tolerance” parameter, representing the maximum difference between all intraligand and intrareceptor distances in a match (Ewing et al. 2001), was set to 0.5 Å. The ligand orientation that matched the most sphere points was first selected for further scoring evaluation. The maximum number of ligand orientations for scoring, “maximum orientations,” was set to 500. Equations 1 and 2 were used to calculate the energy score of each ligand orientation for single and ensemble docking, respectively. The specific form of the pairwise energy function is described in the next subsection. To enable rapid energy score evaluation during docking, the score potentials were precalculated on a three-dimensional grid using the GRID algorithm of DOCK (Meng et al. 1992). The grid spacing was set to 0.3 Å, and trilinear interpolation was used for energy score calculations. The binding energy of each ligand orientation was minimized with the SIMPLEX method (Nelder and Mead 1965). The maximum number of SIMPLEX iterations for each cycle of minimization, “maximum iterations,” was set to 200. The optimized ligand orientations were then ranked from low to high according to their scores. Next, the ligand orientations were clustered. For two orientations with RMSDs less than 1.0 Å, only the one with the lower score was kept. The ligand conformation search algorithm of DOCK4.0 was not used. Instead, we accounted for ligand flexibility by generating multiple conformations for each ligand molecule with the OMEGA software (OpenEye, Inc.), and then docking each (rigid) ligand conformation into the target site. The default OMEGA parameters were used. The maximum number of output conformers per ligand, MAXCONFS, was set to one of the recommended values (200). The scores of different conformations of the same ligand were merged, and the conformation with the lowest energy score would be selected to represent the ligand binding mode. The details of our ensemble docking algorithm were given in our previous study (Huang and Zou 2006a).
Scoring function
In this study, the energy score of ligand binding is calculated as
where EL represents the intramolecular energy of the ligand, which was obtained from the output file of the OMEGA software. EP–L denotes the intermolecular energy between the protein and the ligand, which was calculated with the iterative knowledge-based scoring function recently developed by our group (referred to as ITScore) (Huang and Zou 2006b,c). Specifically, the ITScore was obtained by summing up all protein (P)–ligand (L) atom pair interaction energies as
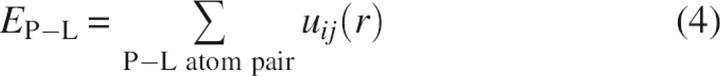 |
where uij(r) is the pair potential between the protein atom of type i and the ligand atom of type j at the atom pair distance r. A total of 26 atom types were used, based on the definitions provided by the SYBYL software (Tripos, Inc.). Hydrogens were not explicitly considered. The pair potentials uij(r) were derived via an efficient iterative method, using 786 protein–ligand complexes extracted from the Protein Data Bank (PDB) (Berman et al. 2000). The main idea of the iterative method is to adjust the pair potentials by iteration until they correctly discriminate native binding modes from decoy structures. The ITScore was validated by testing against ∼200 other diverse protein–ligand complexes in terms of native structure identification and binding affinity prediction. Its efficiency and generality were further demonstrated through virtual database screening against four target proteins. Details of the theory of ITScore and related test studies were described in our previous studies (Huang and Zou 2006b,c).
The pairwise feature of ITScore enables rapid calculation of this scoring function, and also allows us to further decrease computational time by precomputing the protein contributions to the binding energy score and storing the calculated contributions on a three-dimensional grid for future docking use (Meng et al. 1992).
Protein and ligand preparation
HIV-1 protease (HIVp) was chosen to test our algorithm for the following reasons: First, it is an essential enzyme for the life cycle of the HIV virus and is an important target for the development of therapeutic agents against AIDS. Second, HIVp is flexible and the “induced-fit” effect is prominent upon ligand binding. Third, multiple atomic structures of HIVp, in free or ligand-bound form, are available. We found only one NMR structure in the Protein Data Bank (PDB entry 1BVE) (Berman et al. 2000), which contains 28 conformers and no structural water. The 1BVE structures were downloaded from the PDB. Hydrogens and the bound ligand were removed from each structure in 1BVE. The 28 conformers of the NMR structures were then used as an ensemble of HIVp structures for ensemble docking.
To evaluate the efficiency of ensemble docking, the results were compared with the performance of standard single-structure docking against the HIVp crystal structures extracted from the Protein Data Bank. It is noticeable that for many of the HIVp complexes, there exists one conserved (structural) water molecule placed between the flaps of HIVp and the inhibitors (e.g., HOH301 of 7HVP). This structural water molecule, important for ligand binding, is usually kept as part of the protein during docking calculations (Knegtel et al. 1997). However, the structural water is absent in some HIVp complexes; the corresponding position is either occupied by or overlapped with inhibitor atoms. Because the NMR structures of 1BVE do not have the structural water, only the crystal structures in the Protein Data Bank that contain no structural water were used as single docking targets for consistency, yielding a total of 12 complex structures. One of them is bound with the same inhibitor as that of 1BVE, which was removed to mimic realistic docking calculations. For a control study, the apo crystal structure, 3PHV, was added for single docking calculations. Notice that the PDB code 3PHV is a monomeric structure. Transformations were performed with 3PHV according to its crystallographic symmetry to generate a dimeric apo structure of HIVp (still denoted as 3PHV). Therefore, a total of 12 crystal structures of HIVp were used as single docking targets. Their PDB entries are listed in Table 1. Their co-crystallized ligands (shown in Fig. 1) were used for docking evaluations and also served as active compounds in the virtual screening test. In addition, the NMR minimized average protein structure (PDB entry 1BVG) and the first NMR conformer of 1BVE (referred to as 1NMR) were also used to evaluate the performance of a single representation of NMR structures.
All of the HIVp structures listed in Table 1 were superimposed by using the algorithm of Ferro and Hermans (1977). Four backbone carbon atoms near the binding site were selected for matching use. The inhibitors were then separated from the superimposed protein structures. The atom types for HIVp and the ligands were assigned using the SYBYL software. The superimposed protein structures were used for sequent docking calculations (including scoring and optimization).
Similar to our previous study (Huang and Zou 2006a), a reference protein structure was constructed based on the 28 NMR conformers to generate the initial ligand orientations for ensemble docking. The principle for constructing the reference protein is that the binding pocket should be as large as possible to provide sufficient sampling space for the initial ligand orientations with the least change of the overall shape of the binding site (Huang and Zou 2006a). Generation of initial ligand orientations for single docking is based on individual protein structures.
It should be addressed that the reference protein would not affect the final results of ensemble docking because the reference protein is solely used to guide the generation of initial ligand orientations. The real protein structures rather than the reference protein structure will be used for later binding score calculations and orientation optimization (Equation 2). Therefore, the single docking results would remain unchanged even if the reference protein structure was used to generate initial ligand orientations in single-structure docking. One additional note is that there can be multiple ways to construct the reference protein. One alternative method is to use the average structure of multiple protein conformations as the reference protein structure, for which no binding pocket information is needed. In that case, sufficient sampling of initial ligand orientations could be achieved by allowing some atomic clashes between the reference protein and the initial ligand orientations. Either method for constructing the reference protein is expected to not change the results of ensemble docking.
Database preparation
For a small-scale virtual screening test, 1000 compounds were randomly selected from the Available Chemical Directory (ACD, distributed by Molecular Design Ltd., San Leandro, CA). The selected compounds contain 10 ∼ 50 heavy atoms and consist of no atoms other than C, H, O, N, S, P, F, Cl, Br, and I. They also span the range of physical properties of the active compounds (e.g., molecular weight, number of hydrogen bond donors or acceptors). These 1000 molecules served as a set of inactive compounds for virtual database screening. The coordinates of the molecules were generated using the CONCORD program (Rusinko et al. 1989).
Acknowledgments
We thank the UCSF Computer Graphics Laboratory for use of the Midas molecular graphics software. Support to X.Z. from OpenEye Scientific Software Inc., MDL Information Systems, Inc., and Tripos, Inc. is gratefully acknowledged. This work is supported by NIH grant DK61529 and AHA grant 0265293Z (Heartland Affiliate) to X.Z.
Footnotes
Reprint requests to: Xiaoqin Zou, Dalton Cardiovascular Research Center & Department of Biochemistry, University of Missouri, 134 Research Park, Columbia, MO 65211, USA; e-mail: zoux@missouri.edu; fax: (573) 884-4232.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062501507.
References
- Babine, R.E. and Bender, S.L. 1997. Molecular recognition of protein–ligand complexes: Applications to drug design. Chem. Rev. 97: 1359–1472. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooijmans, N. and Kuntz, I.D. 2003. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 32: 335–373. [DOI] [PubMed] [Google Scholar]
- Broughton, H.B. 2000. A method for including protein flexibility in protein-ligand docking: Improving tools for database mining and virtual screening. J. Mol. Graph. Model. 18: 247–257. [DOI] [PubMed] [Google Scholar]
- Carlson, H.A. 2002. Protein flexibility is an important component of structure-based drug discovery. Curr. Pharm. Des. 8: 1571–1578. [DOI] [PubMed] [Google Scholar]
- Carlson, H.A., Masukawa, K.M., Rubins, K., Bushman, F.D., Jorgensen, W.L., Lins, R.D., Briggs, J.M., and McCammon, J.A. 2000. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 43: 2100–2114. [DOI] [PubMed] [Google Scholar]
- Cavasotto, C.N. and Abagyan, R.A. 2004. Protein flexibility in ligand docking and virtual screening to protein kinases. J. Mol. Biol. 337: 209–225. [DOI] [PubMed] [Google Scholar]
- Cavasotto, C.N., Kovacs, J.A., and Abagyan, R.A. 2005. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 127: 9632–9640. [DOI] [PubMed] [Google Scholar]
- Claussen, H., Buning, C., Rarey, M., and Lengauer, T. 2001. FlexE: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 308: 377–395. [DOI] [PubMed] [Google Scholar]
- Cole, J.C., Murray, C.W., Nissink, J.W.M., Taylor, R.D., and Taylor, R. 2005. Comparing protein-ligand docking programs is difficult. Proteins 60: 325–332. [DOI] [PubMed] [Google Scholar]
- Erickson, J.A., Jalaie, M., Robertson, D.H., Lewis, R.A., and Vieth, M. 2004. Lessons in molecular recognition: The effects of ligand and protein flexibility on molecular docking accuracy. J. Med. Chem. 47: 45–55. [DOI] [PubMed] [Google Scholar]
- Ewing, T.J.A. and Kuntz, I.D. 1997. Critical evaluation of search algorithms for automated molecular docking and database screening. J. Comput. Chem. 18: 1175–1189. [Google Scholar]
- Ewing, T.J.A., Makino, S., Skillman, A.G., and Kuntz, I.D. 2001. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 15: 411–428. [DOI] [PubMed] [Google Scholar]
- Ferrari, A.M., Wei, B.Q., Costantino, L., and Shoichet, B.K. 2004. Soft docking and multiple receptor conformations in virtual screening. J. Med. Chem. 47: 5076–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrin, T.E., Huang, C.C., Jarvis, L.E., and Langridge, R. 1988. The MIDAS display system. J. Mol. Graph. 6: 13–27. [Google Scholar]
- Ferro, D.R. and Hermans, J. 1977. A different best rigid-body molecular fit routine. Acta Crystallogr. A 33: 345–347. [Google Scholar]
- Garbuzynskiy, S.O., Melnik, B.S., Lobanov, M.Y., Finkelstein, A.V., and Galzitskaya, O.V. 2005. Comparison of X-ray and NMR structures: Is there a systematic difference in residue contacts between X-ray and NMR resolved protein structures? Proteins 60: 139–147. [DOI] [PubMed] [Google Scholar]
- Halperin, I., Ma, B., Wolfson, H., and Nussinov, R. 2002. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 47: 409–443. [DOI] [PubMed] [Google Scholar]
- Huang, S.Y. and Zou, X. 2006a. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins DOI 10.1002/prot.21214. [DOI] [PubMed]
- Huang, S.Y. and Zou, X. 2006b. An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials. J. Comput. Chem. 27: 1866–1875. [DOI] [PubMed] [Google Scholar]
- Huang, S.Y. and Zou, X. 2006c. An iterative knowledge-based scoring function to predict protein-ligand interactions: II. Validation of the scoring function. J. Comput. Chem. 27: 1876–1882. [DOI] [PubMed] [Google Scholar]
- Knegtel, R.M., Kuntz, I.D., and Oshiro, C.M. 1997. Molecular docking to ensembles of protein structures. J. Mol. Biol. 266: 424–440. [DOI] [PubMed] [Google Scholar]
- Lamb, M.L., Burdick, K.W., Toba, S., Young, M.M., Skillman, K.G., Zou, X., Arnold, J.R., and Kuntz, I.D. 2001. Design, docking, and evaluation of multiple libraries against multiple targets. Proteins 42: 296–318. [DOI] [PubMed] [Google Scholar]
- Meagher, K.L. and Carlson, H.A. 2004. Incorporating protein flexibility in structure-based drug discovery: Using HIV-1 protease as a test case. J. Am. Chem. Soc. 126: 13276–13281. [DOI] [PubMed] [Google Scholar]
- Meng, E.C., Shoichet, B.K., and Kuntz, I.D. 1992. Automated docking with grid-based energy approach to macromolecule-ligand interactions. J. Comput. Chem. 13: 505–524. [Google Scholar]
- Nelder, J.A. and Mead, R. 1965. A simplex method for function minimization. Computer J. 7: 308–313. [Google Scholar]
- Rusinko III, A., Sheridan, R.P., Nilakantan, R., Nilakantan, K.S., Bauman, N., and Venkataraghavan, R. 1989. Using CONCORD to construct a large database of three-dimensional coordinates from connection tables. J. Chem. Inf. Comput. Sci. 29: 251–255. [Google Scholar]
- Shoichet, B.K., McGovern, S.L., Wei, B., and Irwin, J.J. 2002. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 6: 439–446. [DOI] [PubMed] [Google Scholar]
- Teodoro, M.L. and Kavraki, L.E. 2003. Conformational flexibility models for the receptor in structure based drug design. Curr. Pharm. Des. 9: 1635–1648. [DOI] [PubMed] [Google Scholar]
- Wang, J., Kang, X., Kuntz, I.D., and Kollman, P.A. 2005. Hierarchical database screenings for HIV-1 reverse transcriptase using a pharmacophore model, rigid docking, solvation docking, and MMPB/SA. J. Med. Chem. 48: 2432–2444. [DOI] [PubMed] [Google Scholar]
- Wei, B.Q., Weaver, L.H., Ferrari, A.M., Matthews, B.W., and Shoichet, B.K. 2004. Testing a flexible-receptor docking algorithm in a model binding site. J. Mol. Biol. 337: 1161–1182. [DOI] [PubMed] [Google Scholar]
- Zavodszky, M.I., Lei, M., Thorpe, M.F., Day, A.R., and Kuhn, L.A. 2004. Modeling correlated main-chain motions in proteins for flexible molecular recognition. Proteins 57: 243–261. [DOI] [PubMed] [Google Scholar]