

Abstract
Six transmembrane segments, S1–S6, cluster around the central pore-forming region in voltage-gated K+ channels. To investigate the structural characteristics of the S2 segment in the Shaker K+ channel, we replaced each residue in S2 singly with tryptophan (or with alanine for the native tryptophan). All but one of the 23 Trp mutants expressed voltage-dependent K+ currents in Xenopus oocytes. The effects of the mutations were classified as being of low or high impact on channel gating properties. The periodicity evident in the effects of these mutations supports an α-helical structure for the S2 segment. The high- and low-impact residues cluster onto opposite faces of a helical wheel projection of the S2 segment. The low-impact face is also tolerant of single mutations to asparagine. All results are consistent with the idea that the low-impact face projects toward membrane lipids and that changes in S2 packing occur upon channel opening. We conclude that the S2 segment is a transmembrane α helix and that the high-impact face packs against other transmembrane segments in the functional channel.
Keywords: tryptophan scanning, secondary structure
introduction
With the determination of the high-resolution structure of the KcsA K+ channel from Streptomyces lividans (Doyle et al., 1998), structure–function studies of ion channel proteins can henceforth include structure. This advance has revealed in unprecedented detail the molecular makeup of a K+-selective permeation pathway. Moreover, homology considerations argue that the pores of all K+ channels are built along the basic outlines observed in KcsA, a member of the family of K+ channels constructed from two membrane-spanning sequences. Until structures of other K+ channels become available, however, indirect means must be used to attack questions of molecular architecture of protein domains not directly forming the pore. In contrast to KcsA, Kv-type channels are built from six membrane-spanning sequences, the first four of which, S1–S4, are associated with the voltage-dependent conformational changes that culminate in pore opening.
Functional K+ channels are formed by the assembly of four identical or similar subunits (MacKinnon, 1991). The canonical topology-model for the individual Kv channel subunit, shown in Fig. 1 A, is supported by ample biochemical and electrophysiological evidence (Hoshi et al., 1990; MacKinnon and Yellen, 1990; Zagotta et al., 1990; Isacoff et al., 1991; Yellen et al., 1991; Santacruz-Toloza et al., 1994; Sigworth, 1994; Larsson et al., 1996). Comparison to KcsA firmly establishes the pore-forming sequences S5 and S6 as α helical, but no experimental results directly bearing on the secondary structures of the S1–S4 sequences have yet been advanced; likewise, S1 and S4 are definitively known to span the membrane, but this cannot be asserted for S2 or S3, since the S2–S3 “loop” has not yet been nailed down as cytoplasmic. Because of the proposed importance of S2 in cooperating with the charge-bearing S4 sequence in the early steps of gating (Papazian et al., 1995; Seoh et al., 1996; Cha and Bezanilla, 1997; Tiwari-Woodruff et al., 1997), we chose S2 as the focus of a tryptophan-perturbation mutagenesis study designed to achieve two goals: (a) to ascertain the secondary structural character of this putative transmembrane sequence, and (b) to locate the lipid-facing residues of S2. The experimental premise is that bulky Trp substitutions will wreak functional havoc if made at positions that interact intimately with other parts of the channel protein, while they will often be accommodated at lipid-exposed positions. We find that S2 accommodates point mutations with a periodicity that strongly implicates an α-helical structure and identifies a lipid-exposed face of the helix.
Figure 1.
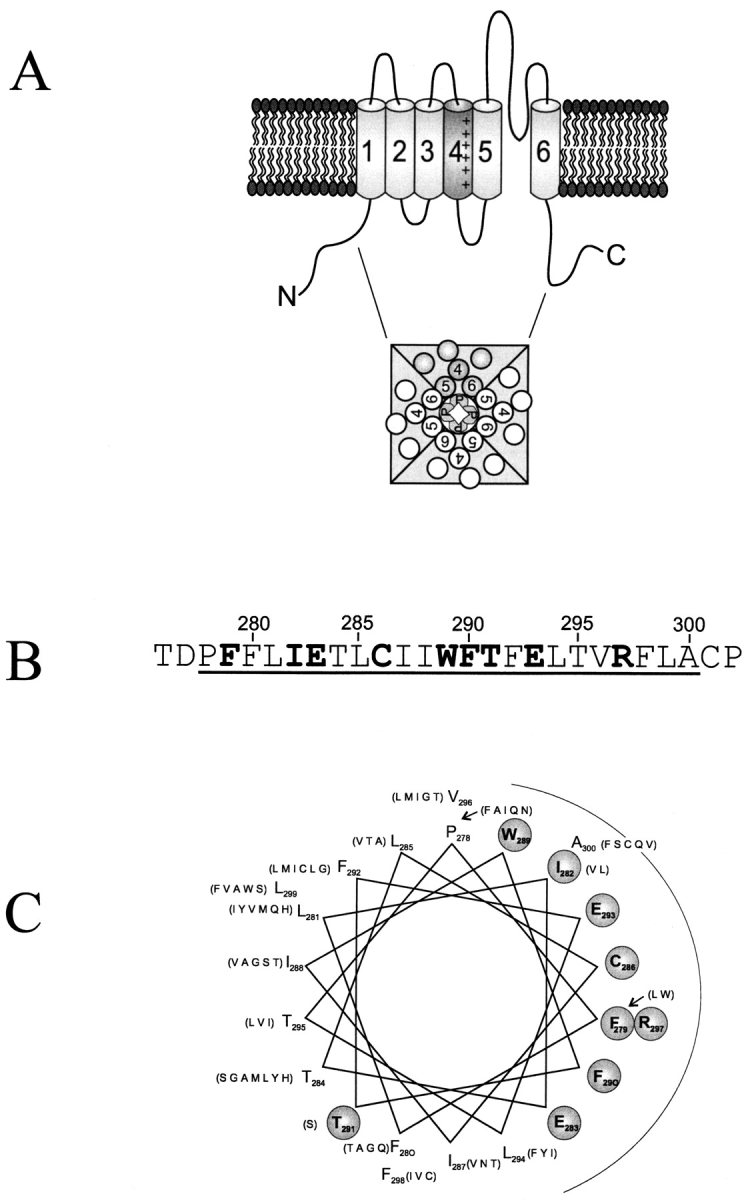
Sequence characteristics of the S2 segment. (A) Topological model for the Shaker K+ channel subunit. Putative transmembrane segments are labeled S1–S6. (B) Amino acid sequence of the Shaker S2 segment showing in boldface those residues conserved within 137 K+ channel S2 sequences identified using a BLAST search (Altschul et al., 1997) of the nonredundant database, using Shaker residues 275–303, which includes all of S2, as query. (C) An α-helical projection of the S2 segment. Conserved residues are circled. Residues in parentheses list other amino acids found at equivalent positions of S2 sequences in other K+ channels. Following Donnelly et al. (1993), we identify conserved residues by two criteria. First, the most common residue is found in over 65% of instances; second, no more than four alternative substitutes are observed, and these must all be of similar chemical character. These criteria lead to a natural distinction between conserved and variable positions.
materials and methods
Recombinant DNA Methods
The channel used here is inactivation-removed (Δ6-46) Shaker B (Schwarz et al., 1988; Hoshi et al., 1990) carrying two point mutations: L338R in the S3–S4 loop to create a MluI site, and F425G in the external vestibule. This Shaker variant, which we denote “wild-type,” is functionally distinguishable from traditional inactivation-removed Shaker B in three respects. Our construct binds charybdotoxin 2,000-fold more strongly than Shaker B (Goldstein and Miller, 1992), its voltage activation curve is right-shifted ∼15 mV, and C-type inactivation is ∼10-fold slower. Mutations were generated on this background in pBluescript KS(+) using PCR-based mutagenesis incorporating 3′ XbaI and 5′ MluI or BamHI restriction sites. PCR products were purified by agarose gel electrophoresis, digested, and reintroduced into the Shaker cDNA. All constructs were confirmed by sequencing through the cloning cassette. cRNA was transcribed in vitro from a FspI-linearized plasmid using T7 RNA polymerase (Promega Corp.) or the T7 mMessage mMachine (Ambion Inc.).
Electrophysiology
Defolliculated Xenopus oocytes were injected with 1–5 ng cRNA (enough to produce ∼5 μA of current 1–5 d after injection) and stored at 17°C in ND96 solution containing (mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.6, and also containing gentamicin (10 mg/liter). Oocytes were examined 2–5 d after injection using two-electrode voltage clamp (Warner Instruments, New Haven, CT) in ND96 solution (containing 0.3 mM CaCl2) to check expression levels and K+ selectivity. Electrodes were filled with 3 M KCl, 5 mM EGTA, and 10 mM HEPES or 10 mM Tris, pH 7.6. Tail currents were recorded in KD98 solution containing (mM): 98 KCl, 0.3 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.6. Standard pulse protocols used a holding potential of −90 mV, a test pulse between −60 and +50 mV in 5- or 10-mV increments (50-ms duration), followed by a tail pulse to −70 mV (30-ms duration). Values of test and tail voltages and pulse duration were modified according to the kinetic properties of individual mutants.
Data Analysis
Voltage-activation curves were calculated using standard tail-current analysis (Liman et al., 1992), with tail amplitudes measured 2–3 ms into the pulse. The activation curve was fit using a Boltzmann function to produce the usual parameters Vo (half-activation voltage), z (slope factor), and ΔG o, the free energy of channel opening at zero voltage (a nearly model-free measure of the intrinsic stability of the open conformation with respect to the closed), according to ΔG o = zFVo.
Activation kinetics were compared among the various mutants at Vo by the time required for half-opening, t o, and the time constant for deactivation, τd, determined by fitting the falling exponential component of the tail.
results
The S2 segment in Shaker spans residues 278–300 (Fig. 1 B). In integral membrane proteins of known structure, lipid-exposed residues in transmembrane helices tend to be more hydrophobic and less well conserved than residues facing the interior of the protein (Rees et al., 1989; Taylor et al., 1994; Wallin et al., 1997). Comparison of S2 sequences in K+ channels highlights the variable residues (Fig. 1 B), which when mapped onto a helical projection lie predominantly on one face (Fig. 1 C). The present experiments seek to test this sequence-based suggestion for an α-helical disposition of S2. We proceed from the idea that a bulky, hydrophobic tryptophan substitution is more likely to disrupt channel structure (and hence ablate function) when placed at positions packed against other parts of the protein than when introduced at lipid-exposed areas. We therefore mutated each S2 residue individually to tryptophan (or the single tryptophan to alanine), anticipating that positions functionally forgiving of mutation would be interspersed, possibly in helical periodicity, with positions marked by failure to express K+ current. Though not rigorously justified, this expectation is plausible since such substitution-toleration patterns have been observed with several membrane proteins, originally with randomly selected residues (Hinkle et al., 1990), and more recently with specifically inserted tryptophans (Choe et al., 1995; Sharp et al., 1995; Collins et al., 1997).
Our results contradict this expectation. Of the 23 substitutions made, only one (R297W) failed to express current in Xenopus oocytes. Representative currents in response to depolarizing voltage pulses are shown in Fig. 2 for the wild-type channel and two mutants. These particular mutant channels display altered voltage dependencies; the activation curve for I287W is right-shifted 27 mV, while that for E293W is left-shifted 22 mV. These two mutants are altered in other ways; the slope of the activation curve is decreased in I287W, and both activation and deactivation kinetics of E293W are slowed substantially. Similar recordings were collected for the remaining Trp-substituted channels. The effects of the substitutions were analyzed using several empirical parameters: ΔG o (the zero-voltage free energy of opening), t o (the activation half-time measured at the half-point on the activation curve), and τd (the deactivation time constant at −70 mV). These parameters are shown in Table I for all mutant channels. Fig. 3 A plots the open-state stabilization energy ΔΔG o; i.e., the difference in ΔG o between mutant and wild type. In all, 13 mutant channels have electrophysiological properties similar to wild type; we define these residues as “low- impact” or “tolerant,” with |ΔΔG o| < 1 kcal/mol. The remaining 9 “high-impact” positions (E283, T284, C286, I287, W289, F290, E293, L294, and A300) have properties substantially different from wild type. This binary cut between the two types of residues is of course arbitrary, but it naturally falls out of the results; our overall conclusions do not change if we double or halve this cutoff value. In most channels with altered equilibrium activation properties, the kinetic parameters were also changed greater than twofold (Fig. 3, B and C). We emphasize that the intention of this gating analysis is empirical, not mechanistic: to identify positions at which mutations produce obvious and gross changes in gating, not to understand the steps in the activation pathway that are altered by the mutations.
Figure 2.

Gating of tryptophan-substituted S2 mutants. (A) Two-electrode voltage clamp recordings of wild-type channels expressed in Xenopus oocytes. Currents in response to a standard pulse protocol were recorded in KD98 medium. (B and C) Similar recordings taken for mutations I287W and E293W. Because of the left shift in activation and slowed kinetics of E293W, test potentials ranged from –80 to +30 mV, and pulse duration was increased to 100 ms. (D) Voltage-activation curves for wild-type, I287W, and E293W mutant channels, calculated from tail-current analysis. Solid curves are Boltzmann fits to the equilibrium activation data. Scale bars represent 10 ms, 1 μA in all data panels.
Table I.
Electrophysiological Properties of Individual Trp or Ala Mutant Channels
| Construct | V0 | z | ΔG o | ΔΔG o | t o | τd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mv | kcal/mol | kcal/mol | ms | |||||||||
| WT | −21 ± 5 | 2.5 ± 0.6 | −1.3 ± 0.6 | — | 2.7 ± 0.7 | 1.2 ± 0.4 | ||||||
| P278W | −13 ± 5 | 3.9 ± 0.6 | −1.2 ± 0.6 | −0.1 | 4.9 ± 2.2 | 1.3 ± 0.4 | ||||||
| F279W | −23 ± 4 | 2.3 ± 0.5 | −1.3 ± 0.5 | 0 | 2.1 ± 0.4 | 1.3 ± 0.1 | ||||||
| F280W | −13 ± 2 | 1.7 ± 0.1 | −0.5 ± 0.1 | −0.8 | 1.2 ± 0.3 | 0.6 ± 0.1 | ||||||
| L281W | −12 ± 3 | 1.9 ± 0.2 | −0.5 ± 0.2 | −0.8 | 2.2 ± 0.7 | 1.0 ± 0.2 | ||||||
| I282W | −16 ± 4 | 3.2 ± 0.6 | −1.2 ± 0.6 | −0.1 | 6.2 ± 1.7 | 1.4 ± 0.5 | ||||||
| E283W | 7 ± 6 | 2.9 ± 0.3 | 0.5 ± 0.4 | −1.8 | 249 ± 38 | 5.1 ± 0.9 | ||||||
| T284W | 29 ± 1 | 2.5 ± 0.4 | 1.8 ± 0.3 | −3.0 | 11.1 ± 4.8 | ∼4.1 | ||||||
| L285W | −26 ± 6 | 2.5 ± 0.4 | −1.6 ± 0.7 | 0.3 | 2.8 ± 0.8 | 2.1 ± 0.9 | ||||||
| C286W | 23 ± 3 | 1.7 ± 0.3 | 0.9 ± 0.2 | −2.2 | 3.4 ± 0.4 | 0.6 ± 0.2 | ||||||
| I287W | 6 ± 5 | 1.9 ± 0.2 | 0.3 ± 0.2 | −1.6 | 1.8 ± 0.2 | 0.7 ± 0.1 | ||||||
| I288W | −24 ± 4 | 2.7 ± 0.5 | −1.5 ± 0.3 | 0.2 | 3.9 ± 0.7 | 2.0 ± 0.8 | ||||||
| W289A | −37 ± 3 | 5.5 ± 0.9 | −5.0 ± 1.0 | 3.7 | 12.3 ± 7.5 | 5.4 ± 2.3 | ||||||
| F290W | −65 ± 3 | 6.1 ± 1.6 | −9.5 ± 2.7 | 8.2 | 61 ± 46 | 14.5 ± 1.9 | ||||||
| T291W | −17 ± 9 | 2.2 ± 0.5 | −1.0 ± 0.6 | −0.3 | 2.4 ± 1.7 | 1.7 ± 0.7 | ||||||
| F292W | −19 ± 5 | 2.3 ± 0.3 | −1.1 ± 0.4 | −0.2 | 2.7 ± 0.7 | 1.4 ± 0.1 | ||||||
| E293W | −43 ± 5 | 6.2 ± 1.9 | −6.5 ± 2.7 | 5.2 | 103 ± 57 | >13 | ||||||
| L294W | ∼0 | — | ∼0 | >−1.3 | ∼1.4 | <1 | ||||||
| T295W | −27 ± 5 | 2.8 ± 0.6 | −1.9 ± 0.8 | 0.6 | 4.2 ± 1.9 | 2.5 ± 1.0 | ||||||
| V296W | −28 ± 5 | 2.4 ± 0.5 | −1.7 ± 0.7 | 0.4 | 3.9 ± 1.2 | 2.6 ± 0.1 | ||||||
| R297W | ∅ | ∅ | ∅ | ∅ | ∅ | ∅ | ||||||
| F298W | −29 ± 6 | 3.0 ± 0.8 | −2.2 ± 0.9 | 0.9 | 5.0 ± 2.0 | 3.9 ± 1.7 | ||||||
| L299W | −22 ± 6 | 2.2 ± 0.4 | −1.2 ± 0.4 | −0.1 | 2.0 ± 0.6 | 2.0 ± 0.9 | ||||||
| A300W | −37 ± 3 | 4.0 ± 0.8 | −3.6 ± 1.0 | 2.3 | 5.8 ± 1.0 | 3.6 ± 0.6 |
Data from individual activation curves, obtained from 3–12 oocytes, were fit to a Boltzmann function. V0 is the half-maximal activation voltage. Values for t o, the half-time for activation, were obtained by measuring activation kinetics at a test voltage near V0. Values for τd, the deactivation time constant, were obtained by fitting tail currents with an exponential function. Values are mean values ± SD. For L294W, the tail kinetics were too fast to permit accurate determination of activation curves. ∅, no expression of current.
Figure 3.

Gating parameters of S2 tryptophan scan. Changes of empirical gating parameters brought about by point Trp mutations are displayed vs. residue number. (A) Intrinsic free energy of opening with respect to wild type, ΔΔG o. The shaded region (|ΔΔG o| < 1 kcal/mol) represents the range of values defining tolerant residues. (B) Mutant-to-wild type ratio of activation times, t o, determined at the half point on the activation curve, Vo. (C) Mutant-to-wild type ratio of deactivation time constant at −70 mV, τd.
Most of the tolerant positions bear large, hydrophobic residues (Fig. 1), and so their mutation to Trp causes only moderate changes in side chain volume. We therefore challenged these positions with more radical changes of side chain chemistry. We constructed a set of single Asn substitutions that would alter both side chain volume and polarity of Trp-tolerant positions: L285N, I288N, F292N, V296N, F298N, and L299N. Remarkably, all six mutants expressed channels exhibiting electrophysiological properties similar to those of the wild-type channel (Fig. 4 and Table II).
Figure 4.
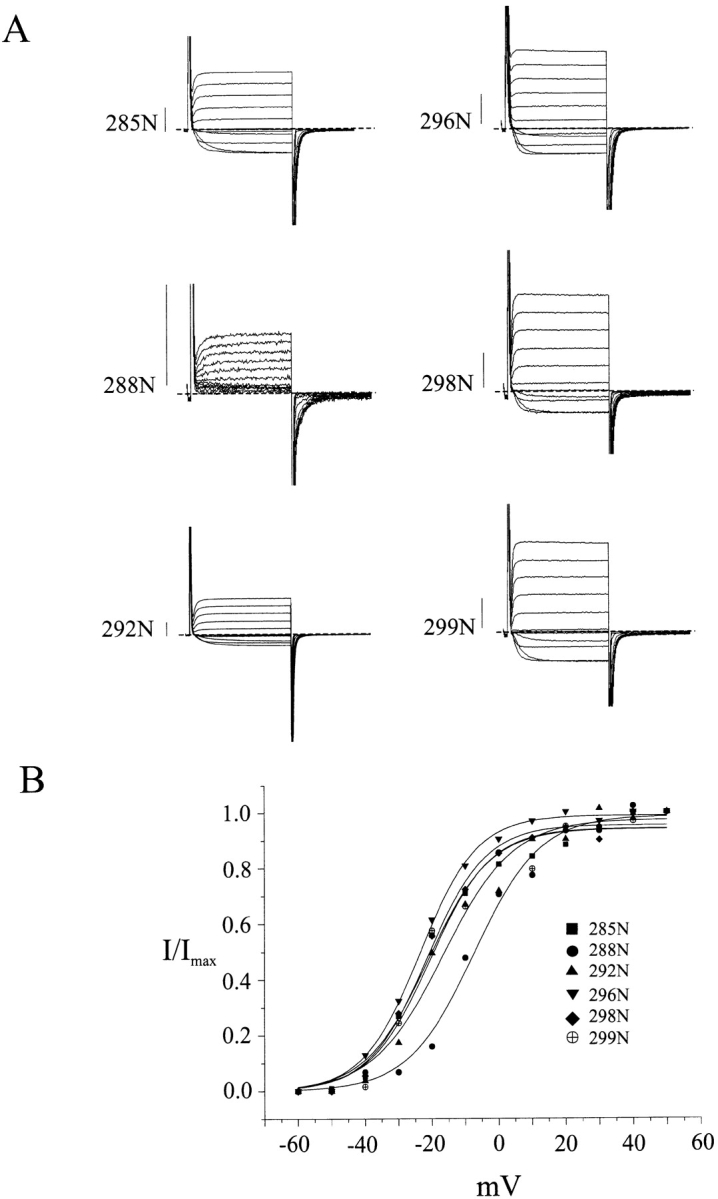
Small polar residues are tolerated on the low-impact face. Two-electrode voltage clamp recordings (A) and activation curves (B) of channels carrying the Asn substitutions shown. Conditions and scale bars are as in Fig. 2. Electrophysiological parameters are reported in Table II.
Table II.
Electrophysiological Properties of Single Asn Mutant Channels
| Construct | V0 | z | ΔG o | ΔΔG o | t o | τd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mV | kcal/mol | kcal/mol | ms | ms | ||||||||
| WT | −21 ± 5 | 2.5 ± 0.6 | −1.3 ± 0.6 | — | 2.7 ± 0.7 | 1.2 ± 0.4 | ||||||
| L285N | −21 ± 4 | 2.9 ± 0.4 | −1.2 ± 0.3 | −0.10 | 1.9 ± 0.8 | 1.0 ± 0.2 | ||||||
| I288N | −8 ± 6 | 3.1 ± 0.7 | −0.6 ± 0.5 | −0.7 | <1 | 2.7 ± 0.6 | ||||||
| F292N | −17 ± 3 | 2.7 ± 0.8 | −1.1 ± 0.5 | −0.2 | 2.7 ± 1.3 | 1.0 ± 0.4 | ||||||
| V296N | −24 ± 4 | 1.8 ± 0.7 | −1.1 ± 0.5 | −0.2 | 0.8 ± 0.2 | 1.6 ± 0.2 | ||||||
| F298N | −20 ± 2 | 2.7 ± 0.2 | −1.3 ± 0.3 | 0 | 3.2 ± 1.1 | 1.4 ± 0.4 | ||||||
| L299N | −22 ± 3 | 2.4 ± 0.4 | −1.3 ± 0.2 | 0 | 3.3 ± 1.2 | 1.1 ± 0.2 |
Parameters are as described in Table I.
discussion
The S2 transmembrane segment has been proposed to participate in two ways in the charge movements underlying voltage-dependent gating in Kv-type K+ channels: first by providing a helical foundation from which two negative residues, E283 and E293, form salt bridges with positive residues in the charge-carrying S4 sequence (Papazian et al., 1995; Tiwari-Woodruff et al., 1997), and second by moving at least one of these negative residues inward during gating; i.e., in the direction opposite to S4 movement (Seoh et al., 1996). These proposals are intriguing, but neither can be considered established. Indeed, there has been no evidence demonstrating that S2 (or any of the first four transmembrane sequences) is helical, and even the accepted topology identifying S2 and S3 as crossing the membrane is unsupported by any experimental results on K+, Na+, or Ca2+ channels.
A Pattern of Tryptophan Toleration
In this study, we used the tryptophan-perturbation strategy previously applied to three other membrane proteins: MotA, a component of the E. coli flagellar motor complex (Sharp et al., 1995), and two inward rectifier-type K+ channels, RomK1 (Choe et al., 1995) and Kir2.1 (Collins et al., 1997). This approach exploits the Janus-like nature of transmembrane α-helices in integral membrane proteins. Transmembrane helices often have two distinct faces: one exposed to bilayer lipid, which is expected to accommodate tryptophan, the other packed at more structurally stringent protein– protein interfaces. Not only does the Trp-scanning strategy make intuitive sense, but it now may be placed on a firm empirical foundation (Fig. 5): comparison to the structure of KcsA (Doyle et al., 1998). Here, we map the Trp-tolerant residues of the first transmembrane helix found previously in RomK1 (Choe et al., 1995) onto the equivalent positions of KcsA. The figure demonstrates an impressive correspondence between the willingness of positions to accommodate Trp and their disposition on the outer, lipid-facing surface of the channel protein. This agreement enhances our confidence in a Trp-perturbation scan as a probe of helical orientation in membrane proteins.
Figure 5.

Structural verification of a previous tryptophan scan. The KcsA structure is represented in RASMOL images, showing residues identified as tryptophan tolerant in ROMK1 (Choe et al., 1995) mapped onto equivalent positions in KcsA. (Left) Ribbon diagrams, with the second transmembrane helix, M2, in red and the selectivity filter, GYG, in cyan. (Upper image) Longitudinal view, with external side of the pore oriented towards the top of the figure. (Lower image) View along the pore axis from the cytoplasmic side. Yellow spheres represent β carbons of residues equivalent to those scored as tolerant of tryptophan substitution in ROMK1. (Right) Space-filling image in longitudinal view, showing Trp-tolerant M2 side chains in yellow; natural tryptophan and tyrosine residues are colored violet to indicate the lipid bilayer-water interface. Residues in ROMK1 corresponding to KcsA residues were lined up according to Doyle et al. (1998).
We carried out an initial scan of 23 point mutations along the S2 segment of a Shaker K+ channel. Had we assayed only the expression of functionally active protein, we would have failed to discern any pattern of Trp toleration whatsoever: all mutants save one expressed voltage-dependent K+ channels. The fact that all these mutants were properly folded and faithfully delivered to the plasma membrane is in itself a notable result for several reasons. First, it corroborates the well-known flexibility of proteins in adjusting local structure to accept mutations even in closely-packed regions (Matthews, 1995). Second, two of the functionally competent mutants are at the absolutely conserved glutamate residues (E283, E293) thought to be critical for proper folding and for salt-bridging to positive charges in S4 (Papazian et al., 1995; Tiwari-Woodruff et al., 1997). We were surprised that alterations as outrageous as Glu → Trp simultaneously in all four subunits would be tolerated, but our results are in harmony with other studies (Papazian et al., 1995) showing that these residues are not strictly essential for channel function.
A clear pattern of response to Trp substitution emerges upon examination of empirical gating characteristics of the mutant channels (Fig. 3), such as the intrinsic free energy of opening, ΔGo. Between E283 and A300, the tolerant and high-impact residues follow an α-helical pattern, with the two types of positions segregating on opposite sides of a helical wheel diagram (Fig. 6), the single exception being T284. The two absolutely conserved glutamates (E283, E293), which have been proposed to pack against the S4 segment, are located squarely within the high-impact face. Moreover, the tolerant residues coincide with the region of highest natural sequence variability (Fig. 1 C), a hallmark of lipid exposure. These considerations argue that S2 is in fact α-helical from position 283 to 300, that the tolerant face of the helix projects side chains into membrane lipid, and that the high-impact face packs against other membrane-spanning parts of the Shaker protein.
Figure 6.

Lateral asymmetry between high-impact and tolerant residues. A helical wheel projection of the S2 segment is shown with residues scored as either not expressed (shaded triangle), high impact (shaded circles), or tolerant (open circles), according to criteria described in the text.
Side Chain Chemistry and the Lipid-exposed Face of S2
It is both notable and satisfying that a clean helical pattern emerges from a technique such as Trp substitution scanning, which is at best a diagnostic bludgeon. However, we worried that this pattern might be an artifact arising from the nature of the S2 sequence itself. The tolerant face of the proposed helix tends towards larger and more hydrophobic residues than the high-impact face, and so it could be argued that Trp substitution would be inherently less perturbing on the former side of the helix than on the latter. This possible objection is weak, however, since the differences are slight in both side chain volume (98 vs. 84 Å3 mean volume per residue) and polarity (six hydrophobic/three uncharged-polar on the tolerant side, six hydrophobic/ three charged-polar on the high-impact side), and since the channel responds differently to the same mutations (I, L, F → W) made on the different sides of the putative helix. Nevertheless, the pattern of Trp toleration is so fundamental to our interpretation that we sought to test further whether the tolerant face remains tolerant if subjected to a wider range of substitutions.
For this reason, we attempted to abuse the channel by introducing residues that would simultaneously alter side chain polarity and volume. Six hydrophobic residues on the tolerant face, L285, L288, F292, V296, F298, and L299, were substituted singly by asparagine, a small, polar side chain that is not found in any of the S2 sequences we surveyed and is almost never observed in lipid-exposed domains of membrane proteins (Donnelly et al., 1993; Wallin et al., 1997). We might therefore expect that these mutations would be disruptive, but this was not the case; all six Asn mutants expressed K+ channels with properties close to wild type, a result that further illustrates the permissive character of the Trp-accepting positions. (We note, however, that double, triple, and quadruple alanine mutations on the tolerant face did lead to channels with significantly left-shifted voltage-activation curves.)
Conformational Stabilization by S2 Substitutions
The Shaker channel's toleration of Trp substitutions is in harmony with the proposal that the tolerant face of the S2 helix is lipid exposed. However, its similar compliance towards Asn residues appears to contradict this idea. It seems alarming that the channel structure readily admits these polar groups into the hydrocarbon-rich bilayer interior. This fact would be disquieting if our assay for the impact of a substituted residue had been the ability of the protein to fold properly in the membrane; i.e., if we had measured the standard-state free energy of Shaker folding from the denatured state. But no such measurement exists; instead, our assay quantifies a process entirely different from folding and assembly: the relative thermodynamic stability of the conducting vs. nonconducting conformations.
Nearly all our mutants assemble well enough to produce immediately recognizable K+ channels. It may be unexpected that Shaker polypeptides bearing lipid-exposed Asn residues behave in so forgiving a manner, but that is the experimental fact; our assay has nothing to say about the channel's thermodynamic comfort once it is in the membrane. Instead, the assay identifies mutations having strong differential effects on the stability of the open compared with the closed conformations; this circumstance is expected to occur where close-packed parts of the protein move upon gating. In contrast, residues that remain lipid-exposed in both open and closed conformations should be functionally unresponsive to substitution regardless of side chain chemistry, as long as the protein achieves a transmembrane insertion in the first place. This is the situation that we observe on the tolerant face of the S2 helix.
Closer inspection of the high-impact residues hints at a picture of S2 movement during gating. We notice (Fig. 3 A) that the high-impact residues in the first half of the Trp-scan (positions 278–287) tend to destabilize, while those in the latter half (289–300) tend to stabilize the open state. Assuming that most Trp substitutions destabilize a protein interface, this pattern suggests that upon opening, S2 moves so that the external part of the helix packs against the rest of the protein more tightly, while the cytoplasmic end of the helix loosens its interaction with other protein regions. These could be subtle motions, since the free energies involved are small.
Conclusions
The helical periodicity observed in this mutational perturbation scan suggests a structural picture of Shaker S2, a picture that most likely applies to all Kv-type K+ channels. We conclude that S2 is indeed an α helix, as envisioned in most models of voltage-gated ion channels. Our results are also consistent with an orientation of S2 that places the variable residues in the sequence projecting into the membrane lipid. Finally, the present results position the two conserved glutamates in the core of a protein–protein interface, a picture consistent with earlier proposals (Papazian et al., 1995) of a salt bridge between S2 and S4.
The impressive correspondence between sequence variability and Trp-toleration in S2 prompts more confidence in a sequence-based analysis of the other “outer” helices S1 and S3. A BLAST search on S1 and S3 identified 120 and 135 sequences, respectively, and the conserved and variable positions were identified. These residues display a clear segregation on helical wheel projections (Fig. 7). In analogy to the S2 results, we therefore consider it likely that S1 and S3 are indeed helical, with their variable sides facing membrane bilayer lipid. We note that the putative lipid-exposed face of S1, like that of S2, covers approximately half the helical surface, while that of S3 is much smaller. This leads to the speculation that S1 and S2 are positioned towards the periphery of the tetrameric channel, while S3 is more buried within the protein complex.
Figure 7.
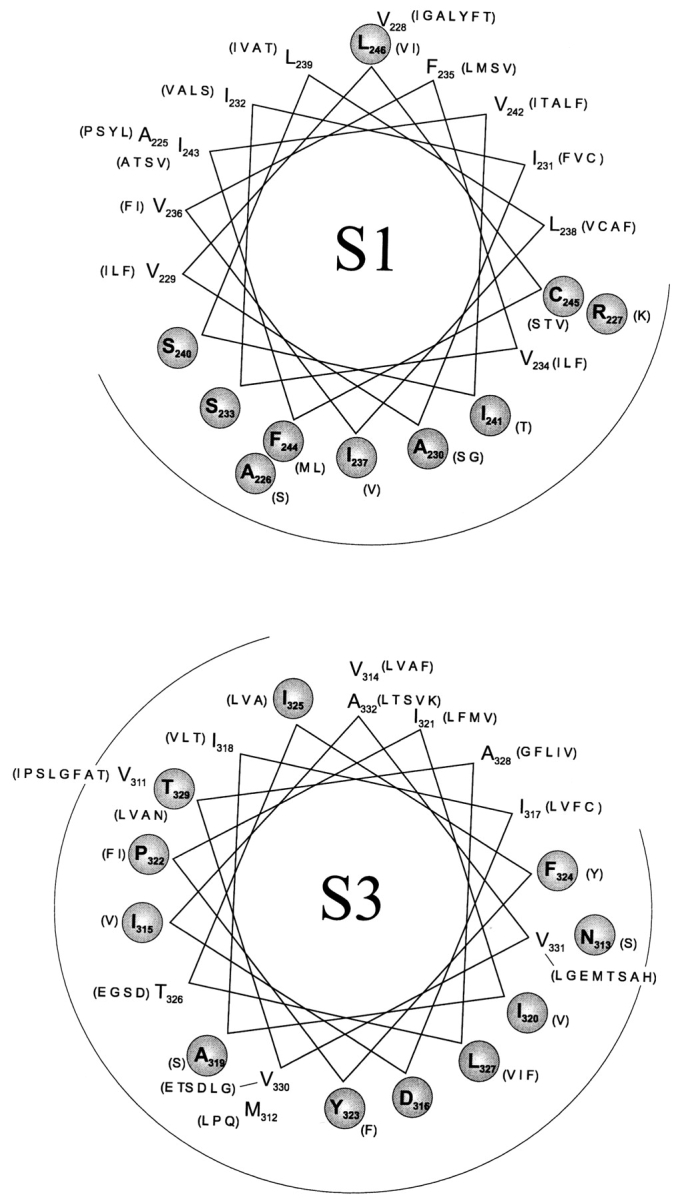
Sequence variability in S1 and S3. BLAST searches were carried out on S1 and S3, as for S2 (Fig. 1 C), using Shaker S1 and S3 as query sequences. Conserved (circled, bold) and variable residues are distinguished as in Fig. 1 C.
Acknowledgments
We thank L. Heginbotham, M. Maduke, R. Blaustein, I. Levitan, and M. Kono for comments on the manuscript, J. Mindell for unceasing computer assistance, and L. Kolmakova-Partensky for help with DNA constructs. We also are grateful to the unsung graduate students who suffered through their first-year rotations by constructing several of the mutant channels used here: K.H. Hong, M. LeMasurier, D. Zhang, and C. McClure.
Footnotes
D.J. Needleman was the recipient of a HHMI undergraduate studentship. This study was supported in part by National Institutes of Health grant GM-31768.
references
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A, Bezanilla F. Characterizing voltage-dependent conformational changes in the Shaker K+channel with fluorescence. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Choe S, Stevens CF, Sullivan JM. Three distinct structural environments of a transmembrane domain in the inwardly rectifying K+channel ROMK1 defined by perturbation. Proc Natl Acad Sci USA. 1995;92:12046–12049. doi: 10.1073/pnas.92.26.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A, Chuang H, Jan YN, Jan LY. Scanning mutagenesis of the putative transmembrane segments of Kir2.1, an inward rectifier K+channel. Proc Natl Acad Sci USA. 1997;94:5456–5460. doi: 10.1073/pnas.94.10.5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly D, Overington JP, Ruffle SV, Nugent JH, Blundell TL. Modeling alpha-helical transmembrane domains: the calculation and use of substitution tables for lipid-facing residues. Protein Sci. 1993;2:55–70. doi: 10.1002/pro.5560020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the K+ channel: molecular basis of K+conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Miller C. A point mutation in a Shaker K+channel changes its charybdotoxin binding site from low to high affinity. Biophys J. 1992;62:5–7. doi: 10.1016/S0006-3495(92)81760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle PC, Hinkle PV, Kaback HR. Information content of amino acid residues in putative helix VIII of the lac permease from Escherichia coli. . Biochemistry. 1990;29:10989–10994. doi: 10.1021/bi00501a017. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker K+channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the Shaker K+channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated K+channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+channels. Science. 1990;250:276–279. doi: 10.1126/science.2218530. [DOI] [PubMed] [Google Scholar]
- Matthews BW. Studies on protein stability with T4 lysozyme. Adv Protein Chem. 1995;46:249–278. doi: 10.1016/s0065-3233(08)60337-x. [DOI] [PubMed] [Google Scholar]
- Papazian DM, Shao XM, Seoh SA, Mock AF, Huang Y, Wainstock DH. Electrostatic interactions of S4 voltage sensor in Shaker K+channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Rees DC, DeAntonio L, Eisenberg D. Hydrophobic organization of membrane proteins. Science. 1989;245:510–513. doi: 10.1126/science.2667138. [DOI] [PubMed] [Google Scholar]
- Santacruz-Toloza L, Huang Y, John SA, Papazian DM. Glycosylation of Shaker K+ channel protein in insect cell culture and in Xenopusoocytes. Biochemistry. 1994;33:5607–5613. doi: 10.1021/bi00184a033. [DOI] [PubMed] [Google Scholar]
- Schwarz TL, Tempel BL, Papazian DM, Jan YN, Jan LY. Multiple K+-channel components are produced by alternative splicing at the Shakerlocus in Drosophila. Nature. 1988;331:137–142. doi: 10.1038/331137a0. [DOI] [PubMed] [Google Scholar]
- Seoh SA, Sigg D, Papazian DM, Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- Sharp LL, Zhou J, Blair DF. Features of MotA proton channel structure revealed by tryptophan-scanning mutagenesis. Proc Natl Acad Sci USA. 1995;92:7946–7950. doi: 10.1073/pnas.92.17.7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ. Voltage gating of ion channels. Q Rev Biophys. 1994;27:1–40. doi: 10.1017/s0033583500002894. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Jones DT, Green NM. A method for α-helical integral membrane protein fold prediction. Proteins Struct Funct Genet. 1994;18:281–294. doi: 10.1002/prot.340180309. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff SK, Schulteis CT, Mock AF, Papazian DM. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+channel subunits. Biophys J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E, Tsukihara T, Yoshikawa S, von Heijne G, Elofsson A. Architecture of helix bundle membrane proteins: an analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci. 1997;6:808–815. doi: 10.1002/pro.5560060407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G, Jurman M, Abramson T, Mackinnon R. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+channel. Science. 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker K+channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]