

Abstract
Lidocaine produces voltage- and use-dependent inhibition of voltage-gated Na+ channels through preferential binding to channel conformations that are normally populated at depolarized potentials and by slowing the rate of Na+ channel repriming after depolarizations. It has been proposed that the fast-inactivation mechanism plays a crucial role in these processes. However, the precise role of fast inactivation in lidocaine action has been difficult to probe because gating of drug-bound channels does not involve changes in ionic current. For that reason, we employed a conformational marker for the fast-inactivation gate, the reactivity of a cysteine substituted at phenylalanine 1304 in the rat adult skeletal muscle sodium channel α subunit (rSkM1) with [2-(trimethylammonium)ethyl]methanethiosulfonate (MTS-ET), to determine the position of the fast-inactivation gate during lidocaine block. We found that lidocaine does not compete with fast-inactivation. Rather, it favors closure of the fast-inactivation gate in a voltage-dependent manner, causing a hyperpolarizing shift in the voltage dependence of site 1304 accessibility that parallels a shift in the steady state availability curve measured for ionic currents. More significantly, we found that the lidocaine-induced slowing of sodium channel repriming does not result from a slowing of recovery of the fast-inactivation gate, and thus that use-dependent block does not involve an accumulation of fast-inactivated channels. Based on these data, we propose a model in which transitions along the activation pathway, rather than transitions to inactivated states, play a crucial role in the mechanism of lidocaine action.
Keywords: local anesthetic, SkM1, antiarrhythmic, patch clamp, methanethiosulfonate
introduction
The gating of voltage-sensitive Na+ channels determines the time course of the rising phase of the action potential and the length of the refractory period in nerve, skeletal muscle, and heart. As a result, Na+ channels are the targets of several classes of drugs that modulate electrical excitability, including antiarrhythmics, local anesthetics, antimyotonics, and anticonvulsants. Among these, lidocaine and related local anesthetics have received a great deal of experimental attention because of their striking effects on Na+ channels: they induce a voltage-dependent inhibition of the peak current upon infrequent stimulation (tonic block), and they dramatically slow repriming of sodium channels after depolarizations (use-dependent block), thereby preventing the repetitive discharges that occur in cardiac arrhythmia, epilepsy, and myotonia (Butterworth and Strichartz, 1990).
Several experimental findings implicate a role for the Na+ channel fast inactivation mechanism in generating these effects: depolarization favors local anesthetic binding, many local anesthetics shift the steady state availability (h ∞) curve in the hyperpolarizing direction (Bean et al., 1983; Hille, 1977), and fast-inactivation– defective Na+ channels are more resistant to some of the effects of local anesthetics than are normal channels (Cahalan, 1978; Wang et al., 1987; Yeh and Tanguy, 1985).
However, a number of questions remain. Is there cooperativity, negative or positive, between lidocaine and the fast-inactivation gate? Does use-dependent block involve an accumulation of fast-inactivated sodium channels? Is there a direct, mutually stabilizing interaction between lidocaine binding and closure of the fast-inactivation gate? Answers to these questions have been difficult to obtain, primarily because gating transitions that occur in drug-bound channels do not involve changes in ionic current, and are thus electrophysiologically silent. Indeed, neither the ionic current nor the gating current provides direct information about the position of the fast-inactivation gate during local anesthetic block.
To circumvent this difficulty, we have employed a conformational marker for the position of the fast-inactivation gate, the reactivity of a cysteine substituted for phenylalanine 1304 in the rat adult skeletal muscle sodium channel α subunit with the thiol-modifying reagent [2-(trimethylammonium)ethyl]methanethiosulfonate (MTS-ET).1 Site 1304 lies in the sodium channel III–IV interdomain and plays a crucial role in fast inactivation (West et al., 1992). In a previous study, we have demonstrated that the reaction rate of the substituted cysteine with MTS-ET follows closely the voltage dependence of steady state fast inactivation (Vedantham and Cannon, 1998). This has enabled us to use this reaction rate as a measure of the fraction of channels whose fast-inactivation gates are shut under conditions of particular interest.
In this study, we have determined the position of the fast-inactivation gate in channels that are bound to the local anesthetic drug lidocaine under several experimental conditions. We found that lidocaine does not compete with closure of the fast-inactivation gate; on the contrary, the fraction of blocked channels that are fast inactivated increases with depolarization and with drug concentration. More surprisingly, our data show that recovery from fast inactivation precedes recovery of the ionic current in drug-bound channels and is just as fast as recovery in the absence of drug, demonstrating that use-dependent block does not involve an accumulation of fast-inactivated channels. Based on these findings, we propose a model in which lidocaine binding affinity is modulated by gating transitions along the activation pathway, without a direct interaction between lidocaine binding and fast inactivation.
materials and methods
Expression of Na+ Channels
The construction of cDNAs encoding F1304C and human Na+ channel β1 subunit in pGEMHE is described in Vedantham and Cannon (1998). RNA for F1304C and β1 subunit were all generated by in vitro translation of linearized plasmids (Message Machine kit; Ambion Inc.). Xenopus oocytes were harvested and coinjected with F1304C + human β1 RNA as described in Chen and Cannon (1995). Before electrophysiological recording, oocytes were incubated for 2–3 d at 18°C in ND-96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.6) supplemented with pyruvate (2.5 mM) and gentamicin (50 μg/ml).
Electrophysiology
Recording conditions and solution exchange were as described in Vedantham and Cannon (1998). The pipette solution contained (mM): 100 NaCl, 10 HEPES, 2 CaCl2, 1 MgCl2, pH 7.6. The bath contained 100 KCl, 10 HEPES, 5 EGTA, 1 MgCl2, pH 7.6. Lidocaine powder (hydrochloride salt; Sigma Chemical Co.) was added to the bath solution in appropriate amounts to obtain a final concentration of 0.5, 1, 2, 4, or 8 mM lidocaine. Stock solutions (2 mM) of MTS-ET (Toronto Research Chemicals Inc.) were prepared from the solid in 1 ml of distilled, deionized H2O and placed on ice at the beginning of each recording day. Appropriate amounts were diluted into 10 ml of bath solution (to a final concentration of 8 μM) after suitable patches were obtained and immediately before use. MTS-ET solutions were never used for >10 min after dilution from the stock. Our method for determining the fidelity of solution exchange is described in Vedantham and Cannon (1998), with one change: patches in which the seal formed without suction were included in the data set even if they did not last long enough for a switching test (such patches invariably exhibited rapid exchange kinetics).
Data Analysis
Curve fitting was performed off line using a custom AxoBasic analysis program (Axon Instruments, Inc.) or SigmaPlot (Jandel Scientific Co.). Steady state fast inactivation, h ∞, and the voltage dependence of the modification rate were fitted to Boltzmann curves with maximum values, I max, and nonzero pedestals, c, calculated as I/I peak = [I max − c]/{1 + exp[(V − V1/2)/k]} + c, where V1/2 is the voltage at half maximum, and k is the slope factor. Error bars indicate means ± SEM.
For modification experiments, the fraction modified after a given pulse of MTS-ET was calculated by averaging the value of the Na+ current between 3 and 3.5 ms after depolarization to −20 mV. For each experiment, the fraction modified (F) versus cumulative exposure time (t exp) were fit to a monoexponential: F = (I max − F o)[1 − exp(−t exp/τmod)] + F o, where τmod is the reciprocal of the reaction rate, F o is the mean value of the current between 3 and 3.5 ms before any exposure has occurred, and I max, the maximum value of the mean current between 3 and 3.5 ms, was a free parameter in the fit.
results
Modification of F1304C by MTS-ET in the Presence of Lidocaine
All experiments were conducted in excised inside-out patches pulled from Xenopus oocytes coinjected with rat adult skeletal muscle sodium channel α subunit F1304C and human β1 subunit RNA. Fig. 1 A shows macroscopic current traces elicited by depolarization from −120 to −20 mV before and after a 5-s application of 8 μM MTS-ET to the intracellular side. As reported previously for this channel (Vedantham and Cannon, 1998) and for the rat brain IIA homologue (Kellenberger et al., 1996), MTS-ET modification causes an increase in the peak current and a dramatic disruption of fast inactivation, consistent with the importance of F1304 (F1489 in the brain IIA channel) for fast inactivation (West et al., 1992).
Figure 1.
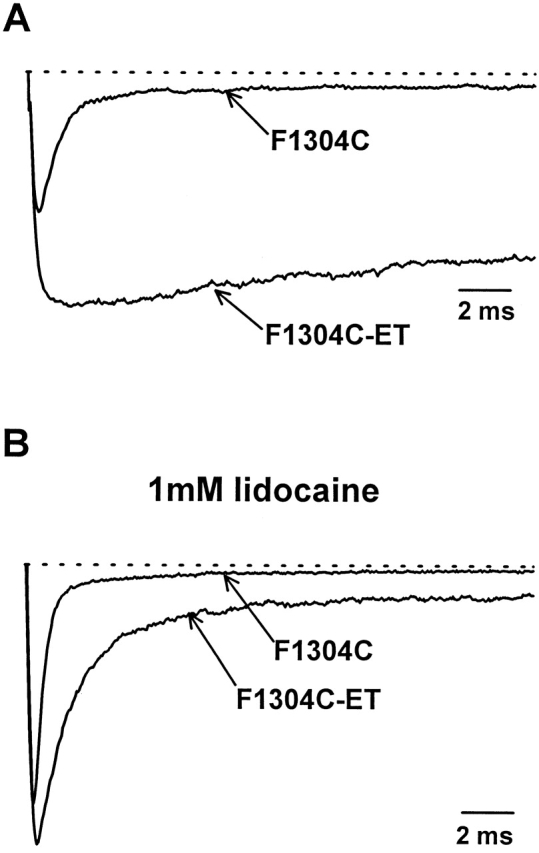
Modification of F1304C with MTS-ET in the presence of lidocaine. (A) Na+ currents recorded from inside-out patches excised from Xenopus oocytes injected with μ1 F1304C and human β1 subunit are shown before and after 5 s of exposure to 8 μM MTS-ET. Each trace is an average of 10 individual depolarizations from −120 to −20 mV. F1304C-ET has an increased peak current (62%) and a dramatic failure of inactivation as compared with unmodified F1304C. B shows experiments performed as in A with 1.0 mM lidocaine added to the bath. Under these conditions, MTS-ET causes a modest increase in peak current (22%). Lidocaine reduces the steady state current of unmodified F1304C and causes partial current decay of the noninactivating F1304C-ET current.
As in the absence of lidocaine, MTS-ET increases the peak current and disrupts fast inactivation in the presence of 1.0 mM lidocaine (Fig 1 B). However, block by lidocaine reduces the apparent fraction of channels that fail to inactivate and attenuates the increase in peak current associated with MTS-ET modification. These effects of lidocaine on F1304C-ET are similar to its effects on fast inactivation–defective Na+ channels studied in other contexts (Balser et al., 1996; Bennett et al., 1995; Wang et al., 1987). When lidocaine is subsequently washed out, the current traces were indistinguishable from the case when no lidocaine was present during the modification (data not shown), demonstrating that lidocaine does not prevent the modification reaction from reaching completion.
Measurement of the Modification Rate of in the Presence of Lidocaine
For accurate measurement of the rate of modification of F1304C by MTS-ET, we used a rapid perfusion system (Vedantham and Cannon, 1998) to apply controlled, brief exposures of MTS-ET to the intracellular face of inside-out patches. Fig. 2 A shows an example of such an experimental protocol: a series of 20-ms exposures to 8 μM MTS-ET at −120 mV, with a test pulse used to assay the macroscopic current between each exposure. The rate of modification was determined by averaging the value of the macroscopic current between 3.0 and 3.5 ms after depolarization (Fig. 2 B), plotting the averages from successive traces against cumulative exposure time, and fitting the resulting curve with a monoexponential (Fig. 2 C). For the experiment shown in Fig. 2, the time constant of this curve was 0.16 s, giving a rate of 0.781 μmol−1 s−1.
Figure 2.
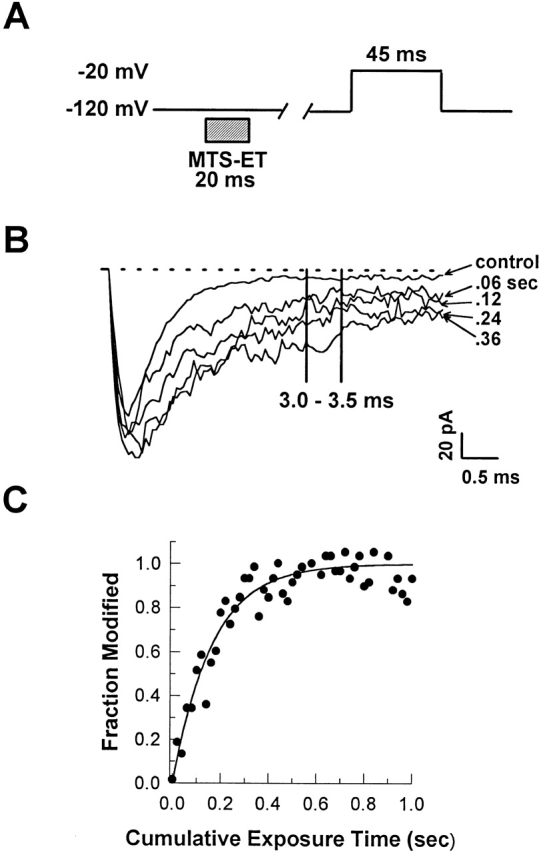
Measurement of the rate of MTS-ET modification of F1304C in the presence of lidocaine. The experimental protocol shown in A consists of a series of 20-ms exposures of excised inside-out patches to 8 μM MTS-ET with test pulses between each exposure. 1.0 mM lidocaine was present at all times. In B, selected traces from the modification experiment described in A are superimposed. To determine the degree of modification after each trace, the value of the macroscopic current between 3 and 3.5 ms after depolarization was averaged. Rates were determined by fitting the degree of modification of each trace as a function of cumulative exposure time with a monoexponential containing a maximum value after complete modification, a nonzero initial value before modification, and a time constant as free parameters. C shows these averages, normalized to the difference between the maximum value and the initial value, plotted against cumulative exposure time, with the normalized curve fit superimposed.
Concentration Dependence of Site 1304 Accessibility in Lidocaine-bound Channels
We estimated tonic block in F1304C by measuring the peak current elicited from excised inside-out patches by infrequent depolarization from −120 to −20 mV under control conditions, in the presence of a fixed concentration of lidocaine applied to the intracellular face (0.1, 0.5, 1.0, 2.0, 4.0, or 8.0 mM), and then back in control solution. The fraction of tonic block was obtained by dividing the peak current measured in the presence of lidocaine by the average of the peak current measured before exposure to lidocaine and after washout. These data (Fig. 3, •) were fit to a binding curve with a hill coefficient of 1.0, yielding a K d of 1.9 mM.
Figure 3.

Effect of tonic block on accessibility of site 1304. Tonic block of F1304C (•) was evaluated by dividing the peak current obtained from infrequent depolarizations to −20 from −120 mV in the presence of 0.1, 0.5, 1.0, 2.0, 4.0, or 8.0 mM lidocaine by the peak current in the absence of drug (n = 4 for each point). These data were fit to a binding curve with a hill coefficient of 1.0, giving a K d of 1.9 mM. Also shown in the graph is the modification rate of F1304C with 8 μM MTS-ET at −120 mV in the presence of 0.5 (n = 4), 1.0 (n = 10), 2.0 (n = 8), or 4.0 (n = 10) mM lidocaine divided by the rate measured without lidocaine present (n = 4) (○). The rates were measured using the protocol shown in Fig. 2 A. A fit to the modification rate data gives a K a of 6.6 mM.
We compared the relative accessibility of site 1304 to the degree of tonic block at specific lidocaine concentrations by dividing the modification rate in the presence of 0.5, 1.0, 2.0, or 4.0 mM lidocaine by the rate measured with no lidocaine present (Fig. 3, ○). We found that the accessibility of site 1304 was less sensitive to lidocaine than the peak current (K a = 6.6, as compared with 1.9), indicating that, at −120 mV and lidocaine concentrations below the K d, the majority of tonic block is caused by lidocaine binding to noninactivated Na+ channels. However, as drug concentration is increased, the fraction of inactivated channels appears to increase as well, although we cannot rule out the possibility that nonspecific effects cause the reduction of reaction rate at such high drug concentrations.
The Voltage Dependence of Site 1304 Accessibility Is Shifted in the Presence of Lidocaine
We confirmed that lidocaine causes a hyperpolarizing shift in the steady state availability curve for F1304C using 1.0 mM lidocaine. In five experiments, the steady state availability curve was measured with 200-ms prepulses, first in control solution, and then in 1.0 mM lidocaine. Each curve was fit independently to a Boltzmann (Fig. 4). We observed a hyperpolarizing shift in the half-maximal voltage (10.8 ± 2.6 mV), and a reduction in the maximum value of 22 ± 6%.
Figure 4.

Effect of lidocaine on the steady state availability curve of F1304C. h ∞ curves were recorded with 200-ms prepulses, followed by a test pulse to −20 mV. For each patch, the h ∞ curve was measured in the absence and presence of 1.0 mM lidocaine (n = 5) in rapid succession. In other experiments without lidocaine, we did not observe significant left shifts in gating after patch excision on the time scale of these experiments. Data from each curve was fit with a Boltzmann with maximal value (I max), half-maximal voltage (V1/2), slope (k), and nonzero plateau (c) as free parameters. The graph shows data normalized to the maximum value for curves measured in the absence of lidocaine, and then averaged across trials. Without lidocaine, V1/2 = −78.9 ± 1.9 mV, k = 6.2 ± 0.7 mV, and c = 0.11 ± 0.02. In 1.0 mM lidocaine, V1/2 = −89.7 ± 1.8 mV, k = 7.1 ± 0.4 mV, c = 0.08 ± 0.02, and I max in 1.0 mM lidocaine was 78 ± 6% of I max in the absence of lidocaine. This represents a hyperpolarizing shift in the apparent h ∞ curve by 10.8 ± 2.6 mV.
Although such a shift strongly suggests a stabilizing effect of lidocaine on fast-inactivated channels, it is in principle possible that the decrease in available channels due to lidocaine does not involve closure of the fast-inactivation gate, but rather to the intrinsic properties of drug-block. Indeed, a hyperpolarizing shift in the apparent h ∞ curve does not preclude the possibility of competition between lidocaine binding and closure of the fast-inactivation gate (for example, if lidocaine and the fast-inactivation gate compete for a single binding site). In the case of a stabilizing interaction, the voltage dependence of site 1304 accessibility should shift in the same direction as the steady state availability curve, while in the case of competition it would not.
We therefore measured the modification rate at a variety of conditioning voltages using the protocol shown in Fig. 5 A in the absence of lidocaine (Fig 5 B, •), and in the presence of 1.0 mM lidocaine (Fig. 5 B, ○). The data were fit with Boltzmanns containing maximum rates (R max), minimum rates (R min), slope (k), and half-maximal voltage (V1/2) as free parameters. With no lidocaine present, R max = 0.71 μmol−1 s−1, R min = 0.12 μmol−1 s−1, k = 3.3 mV, and V1/2 = −79.7 mV; while in 1.0 mM lidocaine, R max = 0.68 μmol−1 s−1, R min = 0.08 μmol−1 s−1, k = 6.6 mV, and V1/2 = −89.9 mV.
Figure 5.
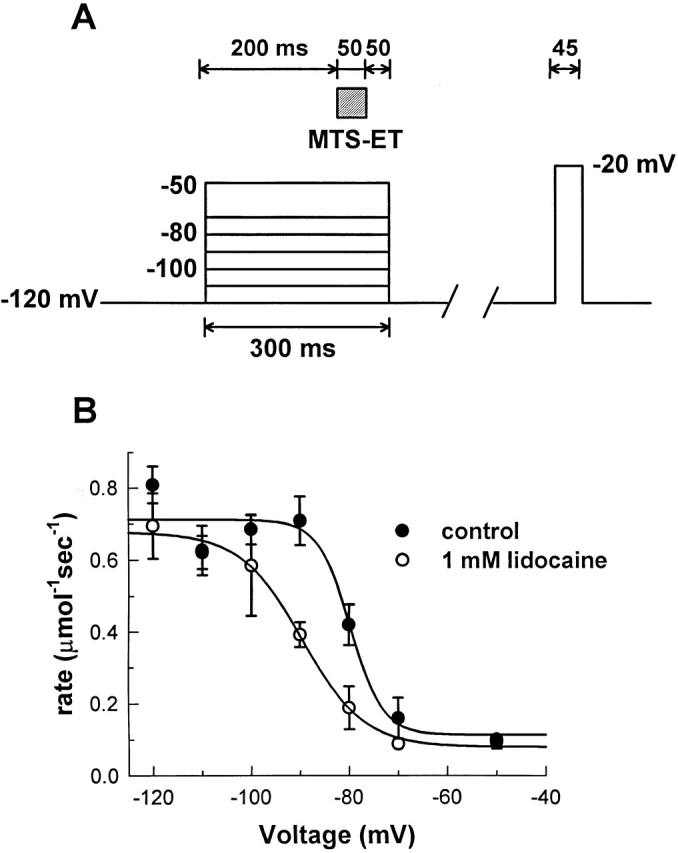
Effect of lidocaine on the voltage dependence of site 1304 accessibility. The rate of MTS-ET modification of F1304C was measured at a variety of voltages, as shown in A. Excised, inside-out patches underwent a series of 300-ms depolarizations: 200 ms to achieve steady state fast inactivation, followed by 50 ms for exposure to 8.0 μM MTS-ET, and a final 50-ms period after exposure to insure complete washout of MTS-ET before repolarization. After each 300-ms depolarization, the patch was maintained at −120 mV for enough time to insure complete recovery (2–8 s, depending on the conditioning voltage) before assaying macroscopic current with a test pulse to −20 mV. Modification rates are shown in B when the experiments were conducted in the absence (•) and presence (○) of 1.0 mM lidocaine. (On average, n = 6, n = at least 3 for each point.)
The voltage at half-maximal modification is shifted by 10.2 mV in the hyperpolarizing direction, similar to the 10.8-mV shift seen in the steady state availability curves (Fig. 2 A). Thus, depolarization increases the fraction of blocked channels that are inactivated: at −120 mV, very few of the blocked channels are inactivated, while at −90 mV, nearly all of the blocked channels (∼40–50% of the total) are inactivated. Both the direction of the voltage shift and the fact that R min is unchanged demonstrate that lidocaine does not compete with the fast-inactivation gate. Rather, it tends to favor closure of the fast gate in a voltage-dependent manner. Also, the R max was not significantly different for control and 1.0 mM lidocaine. To confirm this convergence of R max values, further experiments were performed at conditioning voltages of −160 and −200 mV. We found no significant difference between control and 1.0 mM lidocaine conditions (data not shown).
Lidocaine Does Not Impede Recovery of Site 1304 Accessibility
Use-dependent block results from the slowing of the recovery of Na+ channel availability, and explains the ability of lidocaine to prevent rapid, high-frequency discharges in excitable tissues (Bean et al., 1983). We measured this effect in F1304C inside-out macropatches using a two-pulse recovery protocol with a 20-ms conditioning pulse to 0 mV, a variable recovery period, and a test pulse to 0 mV. The peak current from the test pulse divided by the peak current from the conditioning pulse is a measure of the amount of repriming that takes place during the recovery period. Experiments were performed in control solution (Fig. 6, •), and in 1 mM lidocaine (Fig. 6, ○). Consistent with previous studies, lidocaine dramatically slowed recovery of Na+ channel availability—by ∼100-fold under our experimental conditions.
Figure 6.
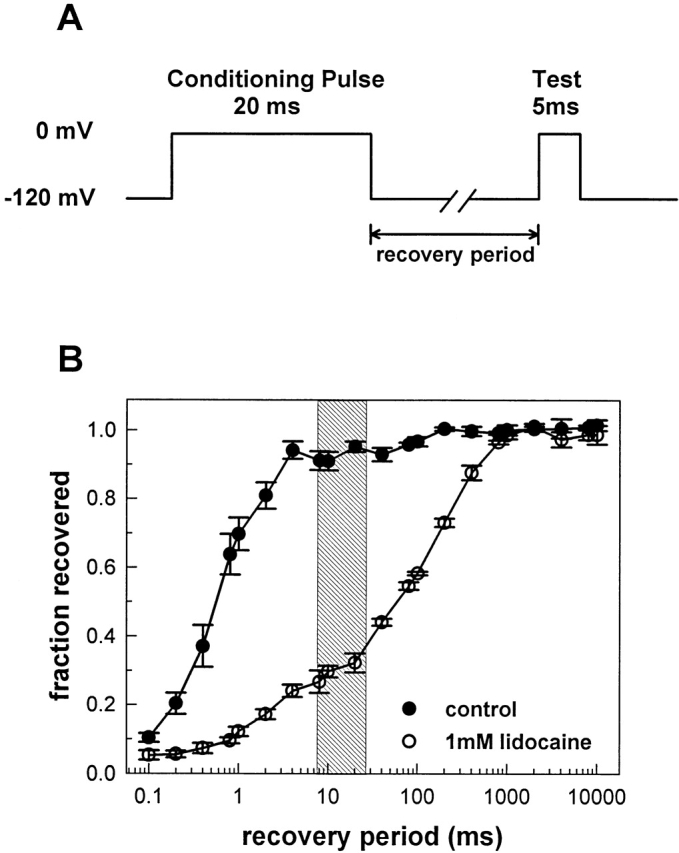
Lidocaine slows the repriming of Na+ channels after brief depolarizations. A two-pulse recovery protocol was used to assess the rate of repriming of F1304C channels after a 20-ms depolarization to 0 mV. The peak current measured during the test pulse was divided by the peak current measured during the conditioning pulse, and plotted as a function of the recovery period. In the absence of lidocaine, the current recovered almost completely within 10 ms, while in the presence of 1.0 mM lidocaine, the time constant of recovery is roughly 100-fold slower. The shaded area indicates the duration of MTS-ET exposure relative to Na+ channel repriming for the protocol shown in Fig. 7 A.
If use-dependent block involves accumulation of fast-inactivated channels, then the accessibility of site 1304 should be reduced for hundreds of milliseconds after a brief depolarization, in accordance with the reduction in Na+ channel availability. However, if recovery from fast inactivation is unaffected by lidocaine, then the accessibility of site 1304 after a short depolarization should not change in the presence of lidocaine, even though Na+ channel availability is reduced.
The protocol we used to resolve this issue (Fig. 7 A) consisted of a series of 20-ms conditioning pulses to 0 mV, each followed by a brief, experimentally measured 7.5-ms delay at −120 mV, and then a 20-ms exposure to 8 μM MTS-ET, also at −120 mV. Relative to the time course of Na+ channel repriming, the duration of MTS-ET exposure corresponds to the shaded area of Fig. 6 B. After each depolarization and MTS-ET exposure, sufficient time was allowed for complete recovery before assaying the macroscopic current. Experiments were performed in control solution and in 1.0 mM lidocaine. Averages of modification time courses, analogous to the individual time course in Fig. 2 C, are shown in Fig. 7 B (•, control; ○, 1.0 mM lidocaine). The reaction rates were not significantly different for the two conditions: 0.63 ± 0.07 μmol−1 s−1 for control, and 0.60 ± 0.07 μmol−1 s−1 for 1.0 mM lidocaine, and are within 10–14% of R max, the maximum rate of modification estimated from the data of Fig. 5. The dashed line shows the modification time course that would be expected if site 1304 accessibility mirrored the availability of Na+ current (∼0.13 μmol−1 s−1; see Fig. 7 legend for calculation). These data demonstrate that recovery from fast inactivation is not significantly affected by lidocaine, and thus that use-dependent block does not involve the accumulation of fast-inactivated Na+ channels.
Figure 7.
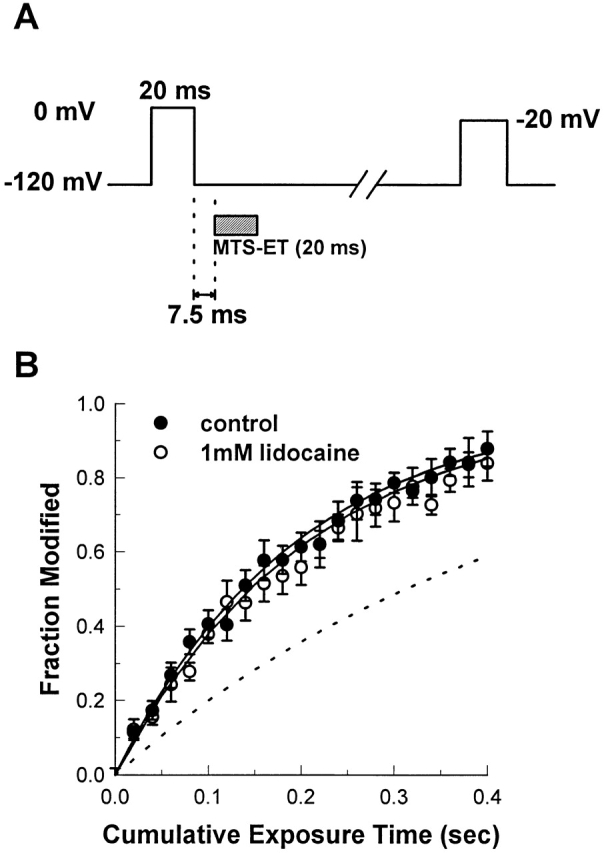
Lidocaine does not slow the return of site 1304 accessibility after brief depolarizations. In the protocol diagrammed in A, 20-ms exposures to 8 μM MTS-ET were timed to occur 7.5 ms after 20-ms depolarizations to 0 mV. The macroscopic current was assayed between each exposure, and modification rates were measured as shown in Fig. 2. The 7.5 ms reflects the experimentally measured delay between the voltage command to the piezoelectric stack (which occurred exactly at the end of the conditioning pulse) and the commencement of solution exchange (for details, see Vedantham and Cannon, 1998). B shows the average of several modification experiments conducted with or without 1.0 mM lidocaine. The solid lines are exponentials whose time constants are the mean values of the time constants obtained from fits to data from individual experiments. The dashed line reflects the curve that would be expected if the accessibility of site 1304 paralleled the degree of Na+ channel availability in 1.0 mM lidocaine during the exposure. The fraction of current recovered in the presence of 1.0 mM lidocaine after 8-, 10-, and 20-ms recovery times was averaged (corresponding to the shaded area in Fig. 6), giving 0.2962. An accessibility of 0.30 predicts a rate of 2.1 s−1 at 8 μM MTS-ET (predicted rate = 0.30 × (R max − R min) + R min), or 0.26 μmol−1 s−1.
discussion
Accessibility of Site 1304 During Lidocaine Block
The major findings of this study are that (a) lidocaine does not compete with the fast-inactivation gate, (b) lidocaine potentiates the degree to which depolarization favors closure of the fast-inactivation gate, and (c) lidocaine does not measurably affect the rate of recovery of the fast-inactivation gate. These observations were made possible by our ability to follow the position of the fast-inactivation gate with a conformational marker, the reactivity of site 1304 with MTS-ET, characterized in detail in a previous study (Vedantham and Cannon, 1998).
In the first set of experiments, we determined the position of the fast-inactivation gate as a function of lidocaine concentration during tonic block, the inhibition of peak sodium current that occurs with infrequent depolarization. Our results indicate that at −120 mV, for lidocaine concentrations below the K d for block, the majority of blocked channels are not fast- inactivated. Above the K d for block, the data suggest that lidocaine favors closure of the fast-inactivation gate, although the certainty of this conclusion is undermined by the possibility of nonspecific effects interfering with the reaction between MTS-ET and site 1304 at such high drug concentrations. (Our data on the voltage dependence of the reaction rate show that nonspecific reduction of the reaction rate is not occurring at 1.0 mM lidocaine: the reaction rates in 1.0 mM lidocaine and control conditions are equal at very hyperpolarized voltages.)
Assuming that the modification rate faithfully reports the position of the fast-inactivation gate even above the K d for tonic block, our observations on concentration dependence are consistent with state-dependent binding of lidocaine to channel conformations that are populated significantly only at depolarized potentials in the absence of drug. As the lidocaine concentration is increased, the population of channels that are in these “depolarized” conformations will increase by mass action, even at −120 mV. Because depolarized states favor closure of the fast-inactivation gate, increasing lidocaine concentration should also favor closure of the fast gate.
That the modification rate is reduced by 40–50% in the presence of 4.0 mM lidocaine at −120 mV predicts a dramatically altered h ∞ curve: at −120 mV, a significant fraction of channels must be unavailable. We found, consistent with our data, that in 4.0 mM lidocaine, availability at −120 mV is somewhere on the steep portion of the h ∞ curve, although we could not accurately estimate the relative availability at −120 mV because patches do not survive the strong hyperpolarizations (less than −140 mV) that would be required to determine the maximum availability (data not shown).
Our next set of experiments on the voltage dependence of site 1304 accessibility in the presence of lidocaine showed a 10.2-mV hyperpolarizing shift of the half-maximal modification rate, similar to the 10.8-mV hyperpolarizing shift of the V1/2 of the h ∞ curve. However, R max and R min were not significantly changed, even though the maximum value of the h ∞ curve was reduced by 22% in 1.0 mM lidocaine.
Most state-dependent models predict that block at very hyperpolarized voltages reflects binding of drug to noninactivated channels. R max, the limiting modification rate at such hyperpolarized voltages, reflects the position of the fully accessible fast-inactivation gate and should not, according to a state-dependent model, be reduced in the presence of 1.0 mM lidocaine, even if 22% of the channels are blocked. As the channels are depolarized, however, a state-dependent mechanism favors binding to channels further along in the activation pathway and predicts that the fraction of blocked channels that are fast-inactivated will increase. This explains the observed left shift of the voltage dependence of the modification rate. R min, which reflects the maximal degree of gate closure in F1304C, is not significantly changed in the presence of lidocaine, a finding that is also predicted by a state-dependent mechanism favoring inactivation.
The final set of experiments was directed at the effect of lidocaine on the recovery of site 1304 accessibility after brief depolarizing pulses. We first confirmed that 1.0 mM lidocaine dramatically slows the recovery of F1304C availability at −120 mV after a 20-ms depolarization to 0 mV. In the absence of lidocaine, the time constant of recovery is on the order of 1–2 ms, while in 1.0 mM lidocaine, it is ∼100–200 ms. This effect produces use-dependent block, a frequency-dependent, cumulative inhibition of sodium current with repetitive depolarizations. Between 7.5 and 37.5 ms, only ∼20– 30% of channels recover in the presence of lidocaine, whereas >90% recover with no lidocaine present. By contrast, the modification rate was not changed at all in the presence of 1.0 mM lidocaine, demonstrating that lidocaine does not significantly alter the kinetics of recovery from fast inactivation.
A Possible Mechanism of Lidocaine Action
At first glance, the results of these experiments seem to be in conflict: on the one hand, lidocaine shifts the h ∞ curve in a way that favors fast inactivation, suggesting a stabilizing interaction between lidocaine block and fast-inactivated channels. On the other hand, lidocaine has no measurable effect on the off rate of the fast-inactivation particle, suggesting that it does not preferentially stabilize fast inactivation.
One model that reconciles our results is shown in Fig. 8. Following Kuo and Bean (1994), we employ a model for sodium channel gating consisting of several closed, noninactivated states, each in equilibrium with a fast-inactivated state (Fig. 8 A). For convenience, only a few such equilibria are depicted. Horizontal equilibria represent the voltage-dependent transitions along the activation pathway, with depolarization favoring a rightward shift in the distribution of populated states. The vertical transitions, by contrast, are voltage independent, and the rightmost equilibria favor inactivated, rather than noninactivated, channels. According to this model, depolarization moves the distribution of channels to the right and down, while hyperpolarization tends to shift the distribution to the left and up.
Figure 8.
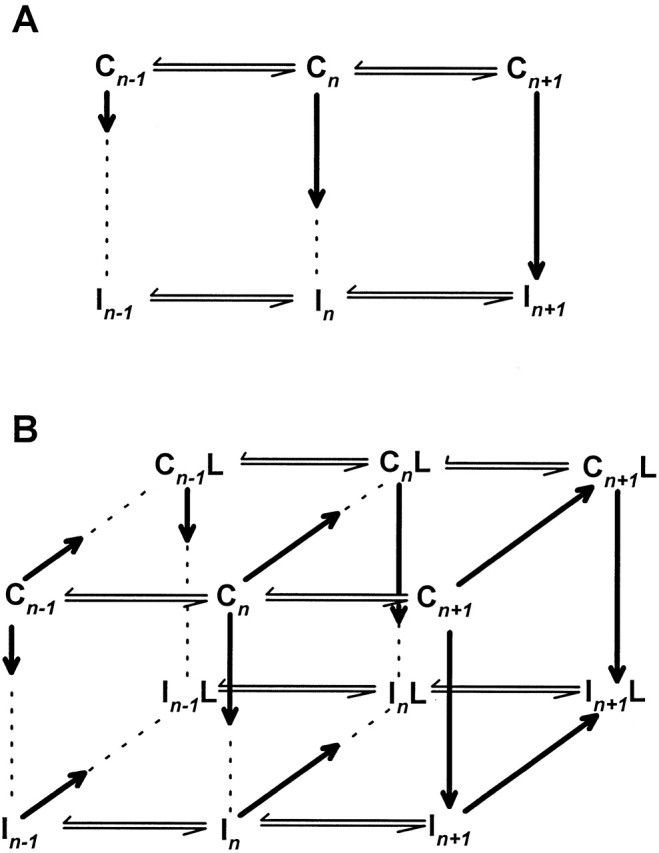
A model for lidocaine action. In A, a section of the activation pathway for sodium channels is shown, in which each noninactivated state (Cn) is connected to an inactivated state (In). The length of the vertical arrows between inactivated and noninactivated states indicate the degree to which the equilibrium favors inactivated channels: the longer the arrow, the greater the fraction of inactivated channels. Thus, depolarization causes rightward movement and increases the fraction of inactivated channels. In B, a set of states is added to the model that incorporate lidocaine binding. The arrows that move between unbound (Cn or In) and lidocaine-bound states (CnL or InL) indicate the degree to which the equilibrium favors lidocaine binding: the longer the arrow, the greater the fraction of lidocaine-bound channels. Thus, as for the case of inactivation, depolarization favors lidocaine block as well as inactivation. The model implies that addition of lidocaine causes a rightward shift in the distribution of channels owing to coupling between activation and lidocaine binding, while the vertical equilibria experience no such coupling (explaining why recovery from fast inactivation is not altered in lidocaine-bound channels). The rightward movement of the distribution will tend to increase the fraction of channels that are inactivated, thereby causing a shift in the h ∞ curve. The slowing of repriming is a result of the slow dissociation of lidocaine from the CnL states.
Fig. 8 B presents a qualitative model for how lidocaine affects the states depicted in Fig. 8 A. We assume that each state can bind lidocaine, since our data suggest that both inactivated and noninactivated channels may experience block. We incorporate state dependence by postulating that lidocaine binds more favorably to channels that are further along in the activation pathway (towards the right), regardless of whether they are noninactivated or inactivated. In other words, lidocaine is sensitive to position along the horizontal, voltage-dependent axis of the state diagram, but not the vertical, voltage-independent axis. In this model, lidocaine does not directly affect the equilibrium constants between inactivated and noninactivated channels (the equilibrium distributions for Cn ↔ In and CnL ↔ InL are equal). Consequently, lidocaine binding does not affect the rate of recovery from fast inactivation by very much, in agreement with our findings on the recovery of accessibility of site 1304. However, the voltage-dependent equilibria in the activation pathway are altered in lidocaine-bound channels, shifting the overall distribution of channels to the right in Fig. 8 B.
The model also explains why lidocaine causes a hyperpolarizing shift in the h ∞ curve. By mass action, addition of lidocaine at any given voltage will tend to shift the distribution of channels towards the right in the state diagram of Fig. 8 B. Since the vertical equilibria will favor fast-inactivated states as the distribution of channels moves sufficiently rightward along the activation pathway, the addition of lidocaine will indirectly promote fast inactivation. This phenomenon also explains our tonic block measurements: the greater the lidocaine concentration, the greater the rightward shift along the activation pathway, and hence the greater the fraction of inactivated channels.
The model also predicts a reciprocal effect of fast inactivation on lidocaine action: the presence of the fast-inactivation gate promotes block, because (like lidocaine) the fast-inactivation particle binds more tightly to the rightmost states on the activation pathway. This would partly explain why channels with disrupted fast inactivation show a reduction in sensitivity to lidocaine effects (Cahalan, 1978; Yeh, 1978; Bennett et al., 1995; Balser et al., 1996). We need not attribute this reduction in sensitivity to an essential role played by inactivation in the mechanism of lidocaine action.
Use-dependent block, in our model, is a consequence of a slow off rate of drug from the drug-bound, non–fast-inactivated states. Recall that at depolarized potentials, our data show that both lidocaine and the fast-inactivation particle are bound (i.e., the back, lower row in Fig 8 B is populated), and that on repolarization the fast-inactivation particle dissociates rapidly, populating the back, upper row of Fig. 8 B. The transitions from the back, upper row to the front, upper row, along with full leftward movement along the activation pathway, is rate limiting and slow (100-fold slower than recovery from fast inactivation), and generates use- dependent block when further depolarization occurs before full recovery.
A remaining question concerns the kinetics of leftward movement along the activation pathway upon repolarization. Because inactivation is not intrinsically voltage dependent, but derives its voltage dependence from activation, some leftward movement along the activation pathway must precede recovery from inactivation. In other words, some inward charge movement must occur if recovery from inactivation is to occur. Unfortunately, whether and to what extent lidocaine impedes inward charge movement upon repolarization has not been examined carefully. Our results predict that some component of the gating charge must remain relatively free to move even in lidocaine-bound channels, and that inward movement of this fraction must be sufficient for complete recovery of the fast- inactivation gate. Further experiments will be required to elucidate the details of the coupling between inactivation and gating charge movement in the presence of lidocaine, and thereby to determine how far the distribution of channels must move to the left on repolarization for full recovery from inactivation to occur.
Relation to Previous Work on Lidocaine
Our model is a version of the modulated receptor hypothesis (Hille, 1977; Hondeghem and Katzung, 1977), in which the affinity of a single receptor site for lidocaine is altered by the conformational state of the channel. Our model differs from Hille's original presentation and from that of Bean et al. (1983) by not treating the inactivated state as the high-affinity state. Instead, we propose that transitions along the activation pathway (outward movement of S4 segments and/ or opening of the activation gate) affect the affinity of lidocaine for its receptor, following the proposals of Wang et al. (1987), Strichartz and Wang (1986), and Yeh and Tanguy, 1985. Several lines of evidence support our hypothesis.
First, numerous studies have shown a reduction in the potency of local anesthetics in fast-inactivation defective sodium channels (Cahalan, 1978; Yeh, 1978; Bennett et al., 1995; Balser et al., 1996). However, despite the loss of potency, local anesthetics do retain their ability to generate tonic and use-dependent block in these channels (Shepley et al., 1983; Strichartz and Wang, 1986; Wang et al., 1987). As noted above, this is consistent with the predictions of our model: the inactivation gate potentiates the effects of local anesthetics, but is not necessary to generate those effects. There is also evidence that at least some local anesthetic molecules can be trapped by closure of the activation gate, suggesting a possible mechanism for use-dependent block that does not involve the fast-inactivation gate (Strichartz, 1973; Yeh and Tanguy, 1985).
Gating-current studies have revealed that lidocaine can produce a hyperpolarizing shift in the Q/V curve (Hanck et al., 1994; Josephson and Chi, 1994) along with a reduction in the total amount of on-gating current. A possible interpretation of this finding is that some of the voltage sensors of drug-bound channels move outward at less depolarized potentials than normal. This would entail, at any given voltage, a drug- induced rightward shift in the distribution of channels along the activation pathway diagrammed in Fig. 8, as our model predicts.
Finally, site-directed mutagenesis has placed the receptor for lidocaine roughly in the middle of the S6 transmembrane segment (Ragsdale et al., 1994). Extrapolation to Na+ channels of a recent substituted cysteine accessibility study in segment S6 of Shaker K+ channels (Liu et al., 1997) suggests that the position of the activation gate is likely to be very close to the local anesthetic binding site. Thus, it would not be surprising if the primary action of lidocaine is to interact with activation gating, perhaps by stabilizing the channel in the open conformation.
We wish to emphasize that our results are not sufficient to determine uniquely our particular model of lidocaine action. Although our results do suggest a very limited role for fast inactivation in generating use- dependent block, it is still possible that the affinity of lidocaine for its receptor is increased by closure of the fast-inactivation gate in the intact channel (i.e., with a phenylalanine at site 1304). Another possibility is that the Na+ channel slow inactivation mechanism plays a role in lidocaine action. Our finding that recovery from fast inactivation precedes recovery of the ionic current in the presence of lidocaine parallels an earlier finding that recovery from fast inactivation precedes recovery from slow inactivation (Vedantham and Cannon, 1998), and raises the possibility that the two slowly recovering states are related in some way. For example, lidocaine might accelerate the rate of entry into slow-inactivated states.
Also, the mechanism of lidocaine action might vary among sodium channel isoforms. Our experiments were conducted in skeletal muscle sodium channels, which have a lower apparent lidocaine affinity that cardiac channels (Hille, 1978; Nuss et al., 1995). However, most of this difference is attributable to relative shifts in voltage-dependent gating between the two isoforms, rather than to differences in the putative binding site (Wright et al., 1997), suggesting that our results with skeletal muscle channels will probably hold for cardiac channels as well. We should also emphasize that our results may not hold for all local anesthetics, which exhibit considerable variation at the chemical level as well as in their effects on sodium channels (Hille, 1977).
These uncertainties aside, our data do enable us to place important new constraints on the possible forms that models for lidocaine action can take. Any such model must involve cooperativity between lidocaine binding and fast inactivation, and must incorporate a state that is slowly recovering, but not fast inactivated, to explain use-dependent block.
Acknowledgments
We thank Adriana Pechanova for assistance with mRNA preparation. We are also grateful to Bruce Bean, Jim Morrill, and Masanori Takahashi for comments on the manuscript.
This work was supported by the National Institutes of Health (AR-42703), the Harvard-Mahoney Neuroscience Institute (V. Vedantham) and the Esther A. and Joseph Klingenstein Fund, Inc. (S.C. Cannon).
Abbreviation used in this paper
- MTS-ET
[2-(trimethylammonium)ethyl]methanethiosulfonate
references
- Balser JR, Nuss HB, Orias DW, Johns DC, Marban E, Tomaselli GF, Lawrence JH. Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation-deficient rat skeletal muscle sodium channels. J Clin Invest. 1996;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Valenzuela C, Chen LQ, Kallen RG. On the molecular nature of the lidocaine receptor of cardiac Na+channels. Modification of block by alterations in the alpha-subunit III–IV interdomain. Circ Res. 1995;77:584–592. doi: 10.1161/01.res.77.3.584. [DOI] [PubMed] [Google Scholar]
- Butterworth JF, Strichartz GR. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990;72:711–734. doi: 10.1097/00000542-199004000-00022. [DOI] [PubMed] [Google Scholar]
- Cahalan MD. Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophys J. 1978;23:285–311. doi: 10.1016/S0006-3495(78)85449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CF, Cannon SC. Modulation of Na+ channel inactivation by the β1subunit: a deletion analysis. Pflügers Arch. 1995;431:186–195. doi: 10.1007/BF00410190. [DOI] [PubMed] [Google Scholar]
- Hanck DA, Makielski C, Sheets MF. Kinetic effects of quaternary lidocaine block of cardiac sodium channels: a gating current study. J Gen Physiol. 1994;103:19–43. doi: 10.1085/jgp.103.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 1978. Local anesthetic action on inactivation of the Na channel in nerve and skeletal muscle. In Biophysical Aspects of Cardiac Muscle. M. Morad, editor. Academic Press, Inc., New York. 55–74.
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Time and voltage dependent interaction of antiarryhthmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:397–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- Josephson IR, Chi Y. Voltage- and concentration- dependent effects of lidocaine on cardiac Na channel and Ca channel gating charge movements. Pflügers Arch. 1994;428:485–491. doi: 10.1007/BF00374569. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, Scheuer T, Catterall WA. Movement of the Na+channel inactivation gate during inactivation. J Biol Chem. 1996;271:30971–30979. doi: 10.1074/jbc.271.48.30971. [DOI] [PubMed] [Google Scholar]
- Kuo C, Bean BP. Sodium channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Tomaselli GF, Marban E. Cardiac sodium channels (hH1) are intrinsically more sensitive to block by lidocaine than are skeletal muscle (μ1) channels. J Gen Physiol. 1995;106:1193–1209. doi: 10.1085/jgp.106.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Shepley MP, Strichartz GR, Wang GK. Local anaesthetics block non-inactivating sodium channels in a use-dependent manner in amphibian myelinated axons. J Physiol (Lond) 1983;341:62P. [Google Scholar]
- Strichartz, G., and G.K. Wang. 1986. The kinetic basis for phasic local anesthetic blockade of neuronal sodium channels. In Molecular and Cellular Mechanisms of Anesthetics. S.H. Roth and K.W. Miller, editors. Plenum Publishing Corp., New York. 217–225.
- Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V, Cannon SC. Slow inactivation does not affect movement of the fast-inactivation gate in voltage-gated Na+channels. J Gen Physiol. 1998;111:83–93. doi: 10.1085/jgp.111.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Brodwick MS, Eaton DC, Strichartz GR. Inhibition of sodium currents by local anesthetics in chloramine-T-treated squid axons. The role of channel activation. J Gen Physiol. 1987;89:645–667. doi: 10.1085/jgp.89.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SN, Wang S, Kallen RG, Wang GK. Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophys J. 1997;73:779–788. doi: 10.1016/S0006-3495(97)78110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JZ. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophys J. 1978;24:569–574. doi: 10.1016/S0006-3495(78)85403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JZ, Tanguy J. Na channel activation gate modulates slow recovery from use-dependent block by local anesthetics in squid giant axons. Biophys J. 1985;47:685–694. doi: 10.1016/S0006-3495(85)83965-5. [DOI] [PMC free article] [PubMed] [Google Scholar]