

Abstract
To study the role of the inositol 1,3,4,5-trisphosphate–binding protein GAP1IP4BP in store-operated Ca2+ entry, we established a human erythroleukemia (HEL) cell line in which the expression of GAP1IP4BP was substantially reduced by transfection with a vector containing antisense DNA under control of a Rous Sarcoma virus promoter and the Escherichia coli LacI repressor (AS-HEL cells). Control cells were transfected with vector lacking antisense DNA (V-HEL cells). GAP1IP4BP protein, which is a member of the GTPase-activating protein (GAP1) family, was reduced by 85% in AS-HEL cells and was further reduced by 96% by treatment with isopropylthio-β-d- galactoside to relieve LacI repression. The loss of GAP1IP4BP was associated with both a membrane hyperpolarization and a substantially increased Ca2+ entry induced by thrombin or thapsigargin. The activation of intermediate conductance Ca2+-activated K+ channels in AS-HEL cells (not seen in V-HEL cells) was responsible for the membrane hyperpolarization and the enhanced Ca2+ entry, and both were blocked by charybdotoxin. Stimulated V-HEL cells did not hyperpolarize and basal Ca2+ influx was unaffected by charybdotoxin. In V-HEL cells hyperpolarized by removal of extracellular K+, the thapsigargin-stimulated Ca2+ influx was increased. Expression of mRNA for the human Ca2+-activated intermediate conductance channel KCa4 was equivalent in both AS-HEL and V-HEL cells, suggesting that the specific appearance of calcium-activated potassium current (IK(Ca)) in AS-HEL cells was possibly due to modulation of preexisting channels. Our results demonstrate that GAP1IP4BP, likely working through a signaling pathway dependent on a small GTP-binding protein, can regulate the function of K(Ca) channels that produce a hyperpolarizing current that substantially enhances the magnitude and time course of Ca2+ entry subsequent to the release of internal Ca2+ stores.
Keywords: GAP1IP4BP; inositol 1,3,4,5-tetrakisphosphate; store-operated calcium entry; calcium-activated potassium channel
introduction
Many electrically nonexcitable cells respond to agonist stimulation with an increase of intracellular free calcium ([Ca2+]i), which results from an inositol 1,4,5-trisphosphate (InsP3)1–induced release of calcium from internal calcium stores followed by calcium entry from the extracellular space. It is now widely accepted that, in many cases, activation of calcium entry is controlled by emptying of InsP3-sensitive stores, although the cellular mechanisms that relay the signal from the internal calcium stores to the channels in the plasma membrane are poorly understood. Nevertheless, a number of molecular species have been proposed to play a role in store-operated calcium entry (reviewed by Irvine, 1990, 1992; Putney and Bird, 1993; Berridge, 1995; Clapham, 1995; Parekh and Penner, 1997). Among these are small GTPase protein(s) and inositol 1,3,4,5-tetrakisphosphate (InsP4), both of which have been proposed to participate in the activation and regulation of store-operated calcium entry (Irvine, 1992; Bird and Putney, 1993; Fasolato et al., 1993).
Bird and Putney (1993) reported that microinjection of the nonhydrolyzable analogues of GTP, GTPγS, or GDPβS into mouse lacrimal acinar cells blocked calcium entry induced by thapsigargin. The effect of these guanine nucleotide analogues could be reversed by including GTP in the injection pipette. Fasolato et al. (1993) reported that introduction of GTPγS via a patch pipette blocked the activation of the calcium release– activated calcium current (ICRAC) by ionomycin or photoreleased caged InsP3 in rat basophilic leukemia cells. Interestingly, once ICRAC had been activated, GTPγS no longer had an inhibitory effect, suggesting that GTP was required during a step before the activation of ICRAC. The two groups independently reached a similar conclusion, that activation of calcium entry or ICRAC involved hydrolysis of GTP, most likely by a small GTP-binding protein. Additionally, small GTPase proteins (e.g., Ras) may influence calcium entry indirectly by controlling the expression of K(Ca) channels (Huang and Rane, 1994; Draheim et al., 1995). When [Ca2+]i rises, activation of K(Ca) channels will hyperpolarize the plasma membrane and thereby facilitate calcium entry (Parekh and Penner, 1997). If small GTPase proteins participate in the activation or regulation of ICRAC, it is possible that a GTPase-activating protein may play a role in the process because of the slow intrinsic GTP hydrolysis rate of small GTP-binding proteins (Boguski and McCormick, 1993).
InsP4 is produced from InsP3 by Ins(1,4,5)P3-3-kinase (Batty et al., 1985; Irvine et al., 1986). Whether InsP4 is involved in activation of calcium entry remains controversial. There is a body of evidence suggesting that InsP4 may activate or facilitate calcium entry (Irvine and Moor, 1986; Morris et al., 1987; Changya et al., 1989a,b; Guse et al., 1992; Luckhoff and Clapham, 1992; O'Rourke et al., 1996). Others find no evidence for the involvement of InsP4 in activation of calcium entry (Bird et al., 1991; Fasolato et al., 1993). In addition, Bird and Putney (1996) reported that microinjection of a high concentration InsP4 in mouse lacrimal cells blocked calcium entry induced by inositol-2,4,5 trisphosphate. However, InsP4 activated a K(Ca) channel in a smooth muscle cell line (Molleman et al., 1991) that may indirectly influence calcium entry by hyperpolarizing the plasma membrane.
Multiple InsP4 binding sites have been found in different tissues, and several InsP4-binding proteins have been identified (Bradford and Irvine, 1987; Enyedi and Williams, 1988; Donie et al., 1990; Donie and Reiser, 1991; Theibert et al., 1991; Koppler et al., 1994, 1996; Cullen et al., 1995a,b; Fukuda and Mikoshiba, 1996; O'Rourke et al., 1996). Among them is GAP1IP4BP, a member of the GAP1 protein family (Cullen et al., 1997). GAP1IP4BP has the potential to integrate the inositol phosphate pathway and the small GTP-binding protein pathways, both of which have been suggested to participate in the activation or regulation of calcium entry (see above). To examine whether GAP1IP4BP is involved in calcium entry, a human erythroleukemia (HEL) cell line was established in which the expression of GAP1IP4BP protein was substantially reduced using antisense techniques. In this cell line, calcium influx was increased in response to thrombin and thapsigargin. Our findings indicate that the increased calcium entry resulted from membrane hyperpolarization caused by intermediate conductance K(Ca) channels. Based on previous measurements of the voltage dependence of ICRAC in HEL cells, we suggest that the increased calcium influx results from an increase in ICRAC due to the hyperpolarization. We also suggest a possible mechanism by which reduced expression of GAP1IP4BP might cause an increase in the function of the K(Ca) channels.
materials and methods
Establishment of GAP1IP4BP Antisense-transfected Cell Lines
The antisense strategy employed the “Lac Switch” mammalian expression system in which expression of the antisense cDNA occurs from a Rous Sarcoma virus (RSV) long terminal repeat promoter that contains the Escherichia coli lacI repressor. Release from repression can be induced by isopropylthio-β-d-galactoside (IPTG). All molecular biological procedures were performed following standard protocols (Sambrook et al., 1989; Ausubel et al., 1994) unless specified otherwise. The cDNA fragment corresponding to nucleotides 80–454 (375 nucleotides) of GAP1IP4BP was amplified in a reverse transcription PCR using a primer pair to which a Not1 restriction enzyme cutting site was attached (sense primer: 5′GAAAAAAGCGGCCGCTGAAGATCAAGATCGGTGAAGCC3′; antisense primer: 5′GAAAAAAGCGGCCGCTGTCTGTGATGACCTCGCTCAG3′; Not1 cutting site underlined). The PCR protocol was: 94°C, 5 min; 94°C, 45 s; 58°C, 1 min; 72°C, 1 min; 30 cycles. The PCR product was gel purified and digested with Not1 restriction enzyme at 37°C for 12 h. The plasmid pOPRSVICAT (Stratagene Inc.) was digested with the Not1 and the 5.6-kb vector backbone (pOPRSVICAT) was recovered by gel purification. The purified GAP1IP4BP fragment and pOPRSVICAT were ligated yielding a plasmid (pOPRSVIGAP1IP4BPAS) with the 375-nucleotide GAP1IP4BP cDNA in antisense orientation. Religated plasmid backbone pOPRSVICAT was used as a transfection control.
HEL cells were first transfected with plasmid p3′SS, which carries E. coli lacI and a hygromycin resistance gene (Stratagene Inc.), using the calcium phosphate precipitation method. Transfected cells were selected using 400 μg/ml hygromycin and further cloned by limiting dilution. The cloned p3′SS transfected cells (LacI-HEL cells) were cotransfected with pOPRSVIGAP1IP4BPAS or pOPRSVICAT, which carry a geneticin resistance gene, to establish the GAP1IP4BP antisense (AS-HEL) and vector transfected (V-HEL) control cell lines. The cotransfected cells were selected using 300 μg/ml geneticin and maintained in Hybrimax (Sigma Chemical Co.) supplemented with 10% fetal bovine serum with the above antibiotics. A primer pair, in which one primer binds to the GAP1IP4BP insert and the other binds to the vector backbone, was used to amplify genomic DNA to verify the presence of the pOPRSVIGAP1IP4BPAS plasmid in the genome of the transfected cell line. The primer pair was: 5′GAAAAAAGCGGCCGCTGAAGATCAAGATCGGTGAAGCC3′, 5′GTTCAAAGAACTGCTCCTCAGGG3′.
Western Blot of GAP1IP4BP
We produced polyclonal antisera from rabbits raised against the COOH-terminal 20 amino acid residues of GAP1IP4BP and also used an antiserum obtained from Dr. Peter Cullen (Cullen et al., 1995b) to determine the expression of the protein in the cell lines by quantitative Western blot analysis as described by O'Rourke et al. (1996).
Solutions
The following external solutions were used for the calcium imaging and electrophysiological experiments. (1) HEPES-buffered physiological salt solution (HPSS) contained (mM): 120 NaCl, 5.3 KCl, 1.0 MgSO4, 1.8 CaCl2, 5.5 glucose, 20 HEPES-Na, pH 7.4 (with NaOH). (2) Ca2+-free HPSS was made by omitting CaCl2 and adding 0.5 mM EGTA. (3) External solution for patch clamp experiments (mM): 150 NaCl, 4.5 KCl, 1.0 MgCl2, 10 d-glucose, 1.8 CaCl2, and 10 HEPES, pH 7.4 with NaOH. (4) Calcium-free solution derived from external solution 3 by replacing CaCl2 with the same amount of MgCl2 and with 0.5 mM EGTA added. (5) For measurement of the reversal potential of the calcium-activated potassium current, we used external solution 3 with K+ concentrations of 4.5, 45, and 140 mM K+, and NaCl was adjusted accordingly to maintain osmolarity.
The following pipette solutions were used in the electrophysiological experiments. (1) For measurement of calcium-activated potassium current (IK(Ca)), the internal solution contained (mM): 140KCl, 1.0 MgCl2, 0.1 BAPTA, and 10 HEPES, pH 7.2 with KOH. (2) For current clamp experiments, the internal solution contained (mM): 140 K aspartate, 8.0 NaCl, 1.0 MgCl2, 1 EGTA, 0.37 CaCl2, 10 HEPES, pH 7.2 with KOH. The calculated free calcium concentration of the solution is 100 nM. (c) To dialyze cells with intracellular solutions with different buffered free calcium concentrations, the internal solution contained (mM): 140 KCl, 10 EGTA, 10 HEPES, pH 7.4 with KOH. Total Mg2+ and total Ca2+ concentrations were calculated using previously published equations (Fabiato and Fabiato, 1979) to achieve a constant free Mg2+ concentration of 1.0 mM and a given free Ca2+ concentration.
Measurement of [Ca2+]i Using Fura-2
Measurement of [Ca2+]i using single cell ratio imaging of fura-2 fluorescence was performed as described previously (Tertyshnikova and Fein, 1997). In brief, cells were plated on glass coverslips and incubated with 2.5 μM fura-2/AM in HPSS for 30 min. Then extracellular fura-2/AM was washed away and cells were incubated for an additional 1 h. Fluorescence images at 340 and 380 nm were collected at a rate of 1 Hz using an intensified CCD camera (Ionoptix) and analyzed using the program Ionwizard 4.1 (Ionoptix). To determine the rate of Mn2+ influx, fura-2 fluorescence was excited at 360 nm and fluorescence emission was measured at 510 nm. In the experiments measuring [Ca2+]i or Mn2+ influx, cells were initially incubated in 200 μl of the indicated solution and stimulation of the cells was achieved by adding 800 μl of new solution that brought the final concentration of reagents and/or [Ca2+]o to the indicated concentration.
Electrophysiology
Standard whole cell patch clamp techniques were employed (Hamill et al., 1981). Recording pipettes were pulled from borosilicate filament glass (Sutter Instrument Co.) using a multistep puller (Sutter Instrument Co.). Pipettes were coated with Sylgard™ near the tip and were fire-polished. Pipette resistance was 2–5 MΩ. An Axopatch 1D amplifier (Axon Instruments Inc.) was used for recording from the pipette under voltage or current clamp. The output signal was lowpass filtered at 2 kHz and digitized at a rate of 10 kHz using a TL-1 interface. Experimental data were acquired and analyzed using pClamp 6.0 software (Axon Instruments Inc.) on an IBM-compatible 80486 computer. Extracellular solution changes were made using a DAD-6 computer-controlled local superfusion system (ALA Scientific Instruments). All patch-clamp experiments were carried out at room temperature (22–25°C).
Measurement of the Cell Membrane Potential Using the Potentiometric Dye Tetramethylrhodamine ethyl ester
The potentiometric dye tetramethylrhodamine ethyl ester (TMRE) was used to determine the plasma membrane potential following the protocol described by Loew (1993). Cells were plated on glass coverslips and incubated in HPSS buffer containing 0.1 μM TMRE (a gift from Dr. Loew's laboratory, University of Connecticut Health Center) for 10 min. A series of confocal fluorescent images of the cells were collected using a confocal microscope (CLSM 410; Carl Zeiss, Inc.) at the rate of 0.1 Hz and were saved as TIF files. Mean intracellular fluorescence intensity was measured within the perinuclear membrane area of each cell using the software program NIH Image (http://rsb.info.nih.gov/nih-image/download.html). After subtraction of background fluorescence, the mean intracellular and extracellular fluorescence intensity was used to calculate the plasma membrane potential using following equation: 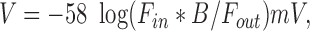 where F
in and F
out are the intracellular and extracellular fluorescence intensity, respectively. B represents the ratio of F
out/F
in obtained when the cell is depolarized with a solution containing 140 mM K+ to correct for nonpotentiometric binding of TMRE.
where F
in and F
out are the intracellular and extracellular fluorescence intensity, respectively. B represents the ratio of F
out/F
in obtained when the cell is depolarized with a solution containing 140 mM K+ to correct for nonpotentiometric binding of TMRE.
Various K+ channel antagonists, tetraethylammonium (TEA), quinine, charybdotoxin (CTX), d-tubocurarine, clotrimazole, and apamin were purchased from Sigma Chemical Co.
Reverse Transcription–PCR of Intermediate Conductance K(Ca) Channel hKCa4 in HEL Cells
Total RNA was isolated from V-HEL and AS-HEL cells by lysing 5 × 106 cells in 1 ml TRIAZOL reagent following the method described in the kit (Life Technologies, Inc.). RNA purity was estimated by spectrophotometry using the A260/A280 ratio and the concentration was estimated at A260, as described by Ausubel et al. (1994). Total RNA extracts were also separated on a denaturing formaldehyde agarose gel (wide range/standard 3:1; Sigma Chemical Co.) to further assess their purity. RNA (5 μg) from V-HEL and AS-HEL cells was reverse transcribed by the method described (kit manual; Clontech). cDNA (2–5 μg) was amplified by PCR using primers designed to amplify a 201-bp fragment of the intermediate conductance calcium-activated potassium channel hKCa4 (Logsdon et al., 1997). The primer sequences were: forward primer, 5′-TTGCTGGAGCAGGAGAAGTCT; backward primer, 5′-GACCTCTTTGGCATGAAAGGC. The PCR protocol was: (a) 94°C, 3 min; (b) 58°C, 1 min; (c) 72°C, 1 min; for 40 cycles, followed by 72°C for 10 min and cooling to 4°C using a minicycler (MJ Research, Inc.). β-Actin primers (amplimer set; Clontech) were used to quantify the amount of cDNA used for each PCR amplification. The amplified products were separated on a 1.3% agarose gel and detected by ethidium bromide. The φχ174rf/dna/haeiii fragments (Life Technologies, Inc.) were used as molecular weight markers. The PCR products were purified using a column (QIAGEN Inc.) followed by precipitation to concentrate the cDNA, which was resuspended in water at a concentration of 50 ng/μl and sequenced at the UCHC Molecular Core Facility using the forward primer described above.
results
Reduced Expression of the GAP1IP4BP in Antisense-transfected Cells
HEL cells were first transfected with the p3′SS plasmid. One clone (LacI-HEL) was used for the cotransfection with pOPRSVICAT or pOPRSVIGAP1IP4BPAS plasmids (see materials and methods). 10 colonies of pOPRSVIGAP1IP4BPAS plasmid-transfected cells as well as three colonies of pOPRSVICAT plasmid-transfected cells were selected at a geneticin concentration of 300–400 μg/ml and were cloned. The presence of the pOPRSVIGAP1IP4BPAS plasmid in genomic DNA was verified by PCR reaction using a pOPRSVIGAP1IP4BPAS plasmid-specific primer pair (data not shown). The expression of the GAP1IP4BP protein in the 10 clones of pOPRSVIGAP1IP4BPAS-transfected cells were compared with that of pOPRSVICAT-transfected cells using Western blot analysis. Among the pOPRSVIGAP1IP4BPAS-transfected cells, one clone, designated AS-HEL, showed 87 ± 1.5% (mean ± SEM, n = 3) reduction of the GAP1IP4BP protein without induction by IPTG (Fig. 1) while the other nine clones did not exhibit a significant difference from the control cells (data not shown). IPTG caused further reduction of GAP1IP4BP (96%). The AS-HEL clone, maintained in culture without induction by IPTG, and a pOPRSVICAT-transfected clone (V-HEL) were used to study the effect of reduced expression of GAP1IP4BP on calcium entry.
Figure 1.

Western blot demonstrating reduced expression of the GAP1IP4BP in AS-HEL cells. V-HEL and AS-HEL lanes were loaded with 40 μg of the cell lysate from V-HEL and AS-HEL cells and separated by SDS PAGE. A polyclonal anti– GAP1IP4BP antiserum was used as a probe. A platelet plasma membrane fraction served as a positive control for the antibody staining (Platelet).
Effect of Reduced GAP1IP4BP Expression on Calcium Entry
HEL cells respond to thrombin stimulation with a rise in [Ca2+]i resulting from an initial release of Ca2+ from internal stores followed by store-operated calcium entry (Somasundaram et al., 1997). First, we established that thrombin-stimulated calcium mobilization in LacI-HEL cells, the parental cell line of AS-HEL and V-HEL cells, was similar to that of nontransfected HEL cells (Somasundaram et al., 1997) (data not shown). When V-HEL and AS-HEL cells were stimulated with 0.5 U/ml thrombin in the presence of 1.8 mM [Ca2+]o, a rapid increase of [Ca2+]i was observed in both cell lines. The time course of [Ca2+]i in thrombin-stimulated V-HEL cells (Fig. 2 a) is similar to that in the LacI cells (data not shown). However, in AS-HEL cells the rise of [Ca2+]i was prolonged for ∼100 s compared with V-HEL cells (Fig 2). [Ca2+]i in AS-HEL cells began to decline with an initial slope similar to that of V-HEL cells, but in contrast to the latter did not return to baseline for at least 10 min. These findings demonstrate that calcium mobilization in AS-HEL cells is greatly prolonged relative to that of V-HEL cells, which could be due to an effect on release of internal stores and/or calcium entry.
Figure 2.

Thrombin-stimulated calcium mobilization in AS-HEL and V-HEL cells. (a) Typical V-HEL and AS-HEL single cell [Ca2+]i rise induced by thrombin in HPSS (1.8 mm [Ca2+]o). (b) Average [Ca2+]i response of 45 V-HEL and 65 AS-HEL cells stimulated with thrombin in HPSS are shown as mean ± SEM.
Thapsigargin, an inhibitor of the endoplasmic reticulum (ER) Ca2+-ATPase pump, has been used to deplete calcium stores, which activates store-operated calcium entry without the production of InsP3 (Takemura et al., 1989). When AS-HEL and V-HEL cells were stimulated with 5 μM thapsigargin in calcium free HPSS, a similar slow and prolonged rise in [Ca2+]i was observed in both cell types (Fig. 3). The rise of [Ca2+]i is caused by leakage of calcium from the internal stores when the ER Ca2+-ATPase pump is inhibited by thapsigargin. In the absence of extracellular calcium, the amplitude and time course of the rise in [Ca2+]i were similar, indicating that the rate of calcium leaking from intracellular stores and the rate of calcium extrusion from cytoplasm are similar in AS-HEL and V-HEL cells (Fig. 3). At 400 s, when [Ca2+]i had returned to baseline, extracellular calcium was restored to 1.8 mM and a second rise of [Ca2+]i, which resulted from calcium entry, was observed in both cell types. The amplitude of the second [Ca2+]i peak was substantially greater in AS-HEL than in V-HEL cells, indicating that calcium entry was specifically enhanced in AS-HEL cells.
Figure 3.

Calcium entry is enhanced in AS-HEL cells. AS-HEL (n = 78) and V-HEL (n = 93) were stimulated with 5 μM thapsigargin in the calcium-free HPSS solution to empty the Ca2+ stores. Calcium influx was initiated 400 s later by restoring [Ca2+]o to 1.8 mM.
Since the amplitude of the second peak of [Ca2+]i upon restoring calcium to the extracellular medium is influenced by both the rate of Ca2+ entry and the rate of Ca2+ extrusion, we used Mn2+ to assess the rate of calcium entry in AS-HEL cells. Manganese is widely used as a surrogate cation to monitor calcium entry (reviewed by Fasolato et al., 1994) because it is believed to enter cells through the same pathways used by Ca2+. Therefore, the rate of quenching of fura-2 fluorescence by Mn2+ can be used to estimate the rate of calcium entry. As shown in Fig. 4, fura-2–loaded AS-HEL and V-HEL cells were stimulated with 5 μM thapsigargin in calcium-free HPSS solution for 400 s, and then 1 mM MnCl2 was added to the extracellular medium. The rate of Mn2+ quenching of fura-2 fluorescence is shown as the percentage of remaining fluorescence compared with that at the time when Mn2+ was added. Note that the initial rate of quenching was only slightly greater in AS-HEL cells, although the findings in Fig. 3 indicate that the reduction in GAP1IP4BP protein is associated with much enhanced Ca2+ entry.
Figure 4.

Manganese influx in AS-HEL and V-HEL cells. Fura-2– loaded cells were stimulated with 5 μM thapsigargin in calcium-free HPSS for 400 s. Then the cells were challenged with 1 mM Mn2+ in calcium-free HPSS (time 0). The quenching of fura-2 fluorescence is shown as the percentage of remaining fluorescence of that at the time when Mn2+ was added.
Lanthanum has been used to block a variety of calcium channels including voltage-gated calcium channels (McDonald et al., 1994) and the channels responsible for capacitative calcium entry (Zhu et al., 1996). Therefore, AS-HEL cells were treated with 5 μM thapsigargin for 400 s in Ca2+-free HPSS solution, followed by addition of 1.8 mM extracellular calcium in the presence of different concentrations of La3+. As shown in Fig. 5, La3+ blocked calcium entry in AS-HEL cells in a dose-dependent manner, supporting the notion that calcium entry in AS-HEL cells is via the capacitative calcium entry pathway.
Figure 5.

Blockade of calcium entry by lanthanum in AS-HEL cells. The paradigm for the experiment was identical to that of Fig. 3 except that varying concentrations of lanthanum were added along with Ca2+. The data are shown as mean ± SEM (25–65 cells in each group).
Electrophysiological Basis for Enhanced Calcium Entry in AS-HEL Cells
The current designated ICRAC is believed to be mainly responsible for calcium entry in the HEL cells (Somasundaram et al., 1997), so that an increase in either the number or the activity of the channels could increase calcium entry. The plasma membrane potential may also influence store-operated calcium entry (reviewed in Parekh and Penner, 1997). Hyperpolarization of the plasma membrane will facilitate calcium entry carried by ICRAC because of the inward rectification of ICRAC. In addition, hyperpolarization would also increase the electrical driving force for calcium entry. The discrepancy between calcium entry determined by [Ca2+]i measurement (Fig. 3) and Mn2+ influx (Fig. 4) either indicates that Ca2+ and Mn2+ enter the cell via different pathways or, alternatively, that enhanced calcium entry in AS-HEL cells requires the presence of extracellular calcium. Many cells have K(Ca) or chloride channels that will hyperpolarize cells upon calcium mobilization. To determine which of the two mechanisms described above is responsible for the enhanced calcium entry observed in AS-HEL cells, whole cell patch clamp recording experiments were performed.
AS-HEL Cells Have a Calcium-activated Current
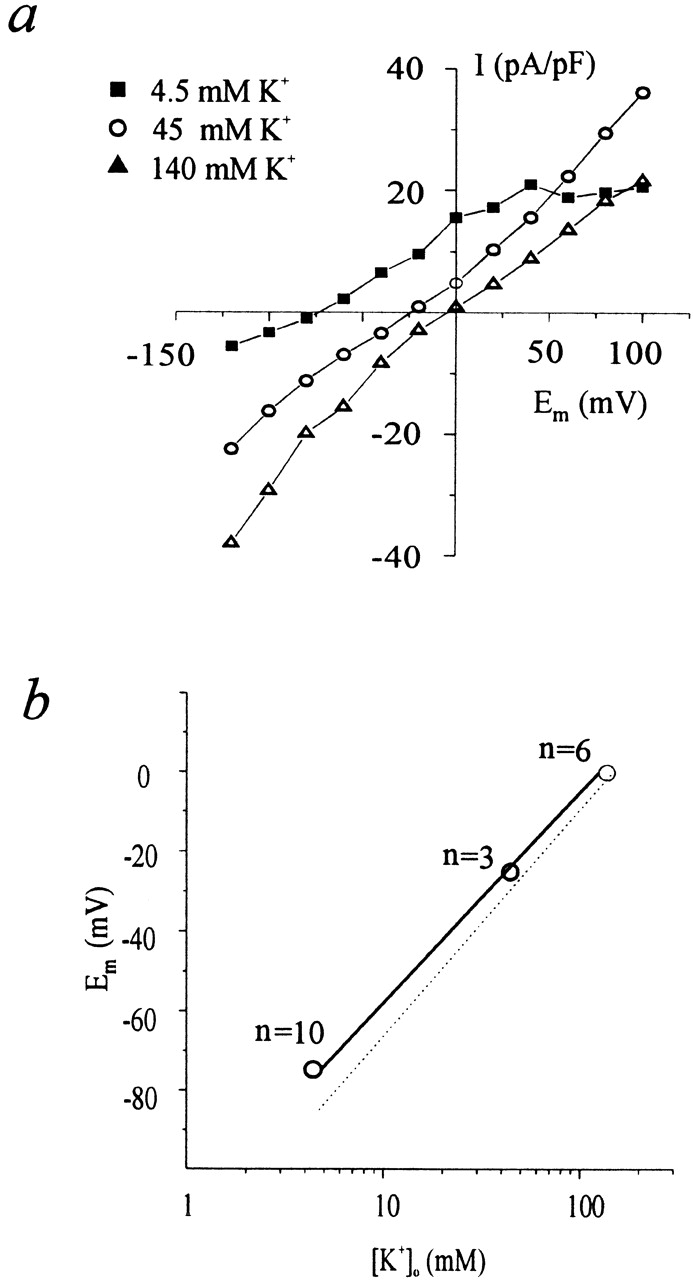
To determine whether any calcium-activated current was present in AS-HEL or V-HEL cells, whole cell recording was performed with internal solution 1, which allows for distinguishing between potassium currents and chloride currents based on their different reversal potentials. After establishment of whole cell recording, a voltage ramp ranging from −120 to +100 mV with a duration of 100 ms, which was applied every 2 s (Fig. 6 a), elicited a small and almost linear current in AS-HEL cells (Fig. 6 b, 1). Then the cell was stimulated with 2 μM thapsigargin and a current gradually developed over 100 s with a reversal potential of around −75 mV (Fig. 6 b, 2). To determine the current–voltage relationship more accurately, a set of voltage steps ranging from −120 to +100 mV in 20-mV increments and with a duration of 100 ms were applied at the indicated times (Fig. 6 d). The current appears to be activated by calcium influx because it diminished slowly when the extracellular solution was switched to a calcium-free external solution (solution 4) and it recovered again when extracellular calcium was restored (Fig. 6, b, 3 and c). After being activated, the current gradually declined to baseline over the next 600 s. Similar results were observed in five other AS-HEL cells. In contrast, V-HEL cells failed to develop a similar current (n = 10 cells, data not shown). These findings show that stimulated AS-HEL cells exhibit a calcium-activated potassium current (IK(Ca), see below) that was either not present or not activated in V-HEL cells.
Figure 6.

Calcium-activated current in AS-HEL cells. (a) Voltage ramp protocol applied every 2 s to monitor the current–voltage relationship. (b) Whole-cell current induced by the voltage ramp (1) before, (2) 100 s after addition of thapsigargin, and (3) in the presence of calcium free external solution (c, ▴). (c) Whole-cell current at 90 ms into the ramp (a, ○) is plotted as a function of time. Addition of thapsigargin is indicated by the line at the bottom. Switching of external solutions from one containing 1.8 mM Ca2+ (solid bar) to one containing 0 mM Ca2+ with 0.5 mM EGTA (open bar) is indicated at the top. (d) Voltage steps from −120 to +100 mV in 20-mV increments that elicited the currents shown in e and f. (e and f) Whole-cell current induced immediately after 1 and 2 in c, respectively.
V-HEL Cells Lack IK(Ca)
There are two possible explanations for the lack of IK(Ca) in V-HEL cells stimulated with thapsigargin: either the cells do not have functional K(Ca) channels, or the cells have such channels but the calcium influx in the V-HEL cells was not sufficient for their activation. In the latter case, similar IK(Ca) should be induced in both AS-HEL and V-HEL cells when the cells are dialyzed with the same concentration of Ca2+ in the pipette solution. Accordingly, AS-HEL and V-HEL cells were dialyzed with an internal solution in which free Ca2+ was buffered to a given concentration using an EGTA-Ca/EGTA buffering system and the current–voltage relationship in the presence of each Ca2+ concentration was determined using voltage steps (Fig. 7). In V-HEL cells, no appreciable Ca2+-dependent current was observed (Fig. 7 a), whereas in the AS-HEL cells IK(Ca) is clearly present, and the activation of the current is dependent on the free Ca2+ concentration in the pipette solution (Fig. 7 b). When LacI-HEL cells were dialyzed with a pipette solution containing 1 μM Ca2+, no appreciable IK(Ca) was detected (n = 5 cells, data not shown). These results indicate that LacI-HEL and V-HEL cells lack the IK(Ca) that is present in AS-HEL cells.
Figure 7.
Membrane current of AS-HEL and V-HEL cells dialyzed with internal solutions containing different concentrations of free calcium. (a and b) I–V relationship of V-HEL and AS-HEL cells dialyzed with internal solutions containing the indicated free calcium concentration. Current elicited by the voltage steps (see Fig. 6) is measured at 95 ms into the voltage step and is plotted as the mean ± SEM (five to nine cells in each group). (c) The data from b for the membrane current elicited by the voltage step to +20 mV are replotted as a function of the free calcium concentration in the pipette solution. In addition, the membrane current from cells dialyzed with 0.01 μM Ca2+ (n = 5) are also plotted. Data are given as the mean ± SEM and were fit with the Hill equation: I = I max/[1 + (C 1/2/C)n] with n = 4 and C 1/2 = 0.4 μM.
The activation of IK(Ca) in AS-HEL cells by Ca2+ was further analyzed by plotting the current induced at the voltage step to +20 mV (from Fig. 7 b) with the addition of data from cells dialyzed with 10 nM Ca2+, as a function of the free calcium concentration. As shown in Fig. 7 c, the data were fit with the Hill equation: I = (I max)/[1 + (C 1/2/C)n] in which the IK(Ca) is half maximally activated at a calcium concentration of C 1/2 = 0.4 μM with a Hill coefficient of n = 4.
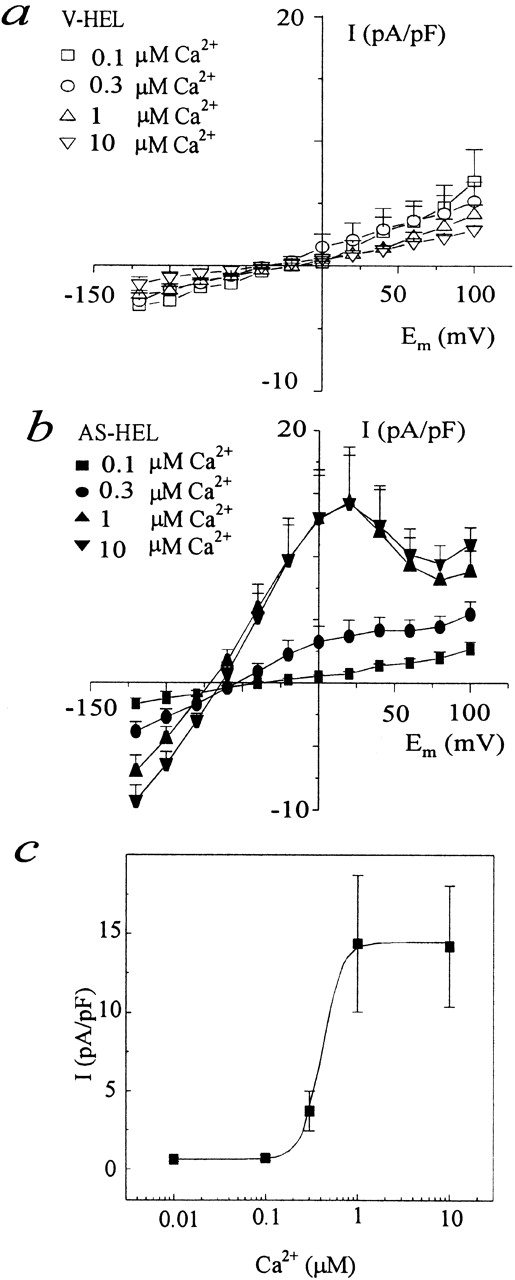
Potassium Is the Carrier for the Calcium-dependent Current of AS-HEL Cells
We suggested that the calcium-activated current in AS-HEL cells was IK(Ca) because it had a reversal potential of −75 mV, which is close to the equilibrium potential of potassium (E k = −86 mV) and far from the equilibrium potential of Cl− (E Cl = 0 mV). To confirm that potassium is the carrier of the current, AS-HEL cells were dialyzed with a pipette solution containing 140 mM K+ and 1 μM Ca2+ to activate the calcium-dependent current. The current–voltage (I–V) relationship was determined in external solutions containing either 4.5, 45, or 140 mM K+ using the voltage step protocol shown in Fig. 6. The reversal potential of the calcium-activated current was determined by plotting the I–V relationship for each K+ concentration (Fig. 8 a). The data in Fig. 8 b show that the reversal potential of the current is dependent on [K+]o and that a 10-fold change of [K+]o results in a 50.1-mV shift in reversal potential, a value close to the expected shift (58 mV/10-fold change of [K+]o) for a perfectly selective potassium conductance. The results confirm that the current is a calcium-activated potassium current IK(Ca).
Figure 8.
Potassium dependence of the reversal potential of the calcium-activated current of AS-HEL cells. (a) An AS-HEL cell was dialyzed with a pipette solution containing 140 mM K+ and 1 μM Ca2+ to activate the calcium-dependent current. The I–V relationship in the presence of different [K+]o was determined using the voltage protocol in Fig 6. (b) Potassium dependence of the reversal potential of the calcium-dependent current of AS-HEL (○ and solid line) and theoretical reversal potential for a perfectly selective K+ channel (dashed line). Mean reversal potentials of the indicated number of cells are shown. SEM is not shown because it is smaller than the symbol used to plot the mean.
Time Course of the IK(Ca)
IK(Ca) develops ∼30–60 s after the cell is stimulated with thapsigargin and reaches its peak at ∼100 s, and then gradually declines almost to baseline in ∼500–600 s (Fig. 6 c). A similar time course was observed in five other cells. The time course of the disappearance of IK(Ca) in AS-HEL cells may follow the course of disappearance of calcium entry, or alternatively it may reflect a rundown process as suggested by others (Mahaut-Smith and Schlichter, 1989; Huang and Rane, 1993; Draheim et al., 1995). To test this, AS-HEL cells were dialyzed with 1 μM Ca2+ and the time course of appearance and disappearance of the current was monitored using voltage ramps. As shown in Fig. 9, IK(Ca) activated by dialysis with internal Ca2+ has a similar course of appearance and disappearance as that seen when IK(Ca) is activated by exposure to thapsigargin, indicating that the decay of IK(Ca) reflects a rundown process. Dialysis of four other cells with 1 μM Ca2+ resulted in a similar time course of the disappearance of the IK(Ca) but with variable onset time (data not shown), presumably due to differing rates of dialysis from the patch pipette.
Figure 9.
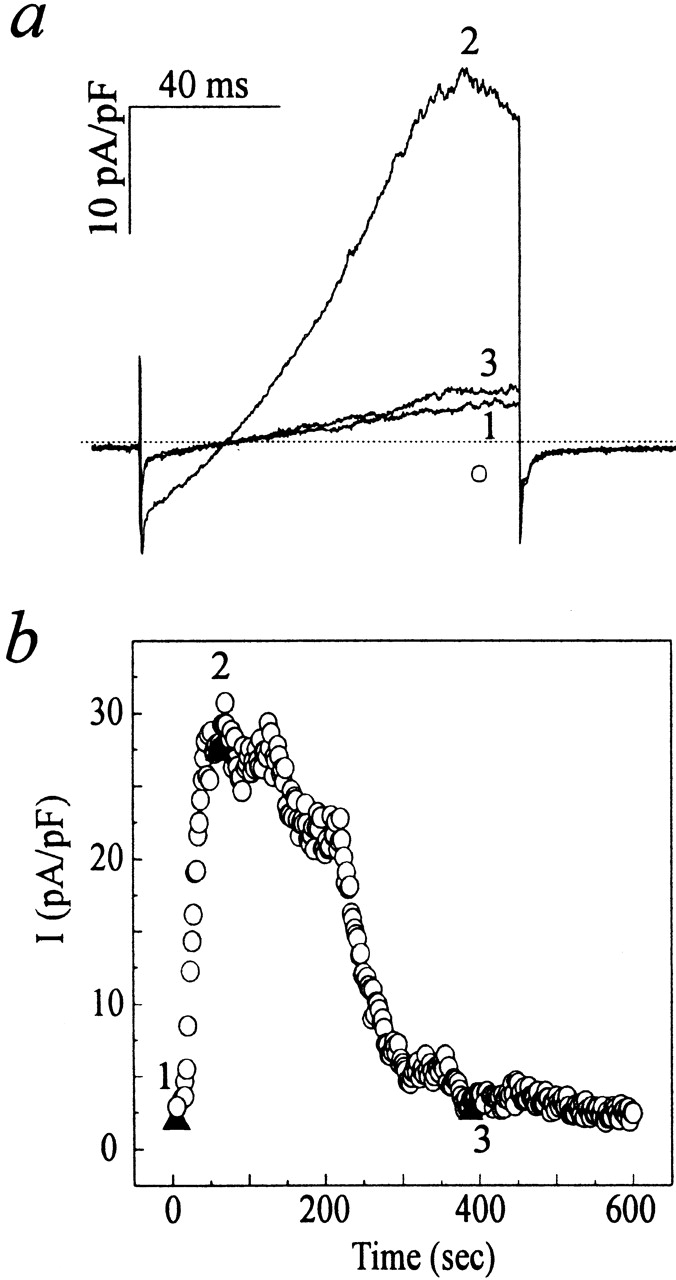
Rundown of the IK(Ca) of AS-HEL cells. An AS-HEL cell was dialyzed with an internal solution containing 1 μM Ca2+ to activate IK(Ca). (a) Whole-cell current induced by a voltage ramp (see Fig. 6) at 10, 80, and 400 s (b, ▴) after establishment of whole cell recording. (b) Whole-cell current measured at 90 ms into voltage ramp (corresponding to 76 mV; a, ○) was plotted as a function of time. Throughout the time course, the series resistance of the whole cell recording was ∼15 MΩ.
IK(Ca) Is Not Regulated by InsP4
It was reported that InsP4 together with Ca2+ activated a K(Ca) channel in a smooth muscle cell line (Molleman et al., 1991). Since reduced expression of GAP1IP4BP protein in HEL cells is associated with the appearance of a calcium-activated potassium current, it is possible that GAP1IP4BP may have an inhibitory effect on the K(Ca) channels and that activation of these channels requires both free Ca2+ and InsP4, as suggested by Molleman et al. (1991). In AS-HEL cells, this postulated inhibitory effect of the GAP1IP4BP would be eliminated by the reduction of the GAP1IP4BP and therefore the IK(Ca) could be activated by Ca2+ alone, whereas in V-HEL cells the presence of the GAP1IP4BP might inhibit the channel and dialysis with Ca2+ alone would be unable to activate the channel. To test this possibility, AS-HEL and V-HEL cells were dialyzed with a pipette solution containing 50 μM InsP4 and 1 μM Ca2+, which is sufficient to activate IK(Ca) in AS-HEL cells. As can be seen in Fig. 10, the presence of 50 μM InsP4 in the pipette solution had no effect on the I–V relationship of either AS-HEL or V-HEL cells. Thus, we did not find evidence to support the idea that InsP4 directly regulates IK(Ca) in AS-HEL or V-HEL cells.
Figure 10.

Activation of IK(Ca) by Ca2+ is not affected by InsP4. AS-HEL (a) and V-HEL (b) cells were dialyzed with an internal solution containing 1 μM Ca2+ with or without 50 μM InsP4. The data are shown as mean ± SEM (6–15 cells for each group).
Calcium Mobilization Causes Hyperpolarization in AS-HEL Cells
When calcium is mobilized by thrombin or thapsigargin, the activation of IK(Ca) in AS-HEL cells should hyperpolarize the plasma membrane, increasing the amplitude of the inwardly rectifying ICRAC as well as providing a greater electrical driving force for Ca2+ entry. To examine the extent of membrane hyperpolarization during calcium mobilization, whole cell current clamp experiments were performed. Cells were incubated in external solution 3 and the whole cell recording configuration was obtained with internal solution 2. Afterwards, cells were superfused for 2 min with calcium-free external solution 4 containing 2 μM thapsigargin, and then challenged with 1.8 mM Ca2+ in external solution 3. In both V-HEL and AS-HEL cells, stimulation with 2 μM thapsigargin in the absence of extracellular calcium did not cause significant changes in membrane potential (Fig. 11 a), presumably because the EGTA-Ca/ EGTA in internal solution 2 buffered the slow rise of [Ca2+]i caused by thapsigargin. However, upon the restoration of extracellular calcium, the membrane potential of AS-HEL cells hyperpolarized from −40 to −70 mV while that of V-HEL cells remained relatively unchanged (Fig. 11 a). These results indicate that the presence of IK(Ca) in the AS-HEL cells leads to hyperpolarization of the plasma membrane during calcium entry.
Figure 11.
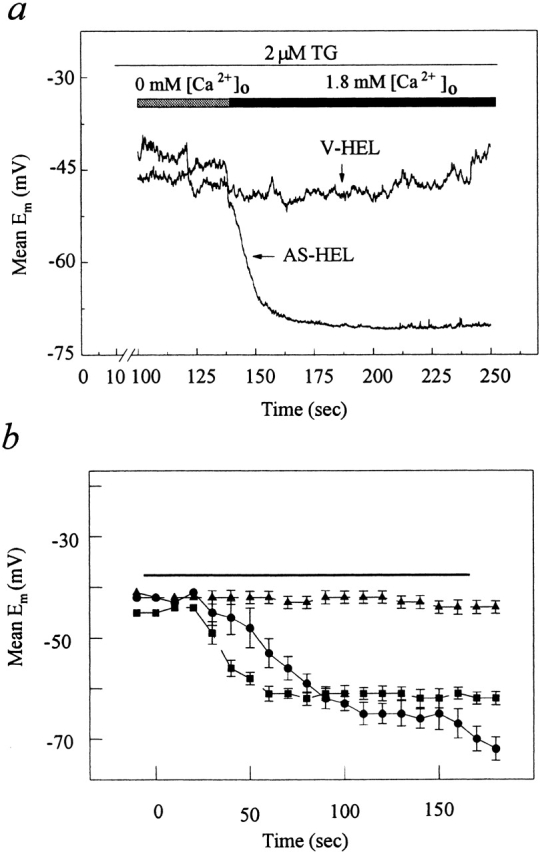
Change of membrane potential upon calcium mobilization by thapsigargin. (a) Mean membrane potential of AS-HEL (n = 10) and V-HEL (n = 7) cells were measured by whole-cell current clamp. Cells were stimulated with 2 μM thapsigargin in calcium-free external solution to empty the calcium stores and thereby activate calcium entry. Then cells were challenged with 1.8 mM calcium in the external solution. (b) Membrane potential was measured using the potentiometric dye TMRE (see materials and methods). The solid bar above the data indicates application of either 2 μM thapsigargin (▴, V-HEL; •, AS-HEL) or 1 U/ml thrombin (▪, AS-HEL). Membrane potentials are plotted as the mean ± SEM for 19 cells each.
We also used the potentiometric dye TMRE (see materials and methods) to independently confirm that the membrane potential of AS-HEL cells hyperpolarized during calcium mobilization. The use of TMRE to measure membrane potential has the advantage that cells remain intact during the measurement. As shown in Fig. 11 b, 2 μM thapsigargin hyperpolarized the membrane potential of AS-HEL cells, while that of V-HEL cells remained relatively unchanged. The AS-HEL cells also hyperpolarized when treated with thrombin but at a faster initial rate than with thapsigargin (0.9 vs. 0.33 mV/s).
Pharmacology of the IK(Ca) in AS-HEL Cells
The role of membrane hyperpolarization in calcium entry was studied using specific blockers of IK(Ca). To identify specific blockers of IK(Ca) in AS-HEL cells, we dialyzed cells with a pipette solution containing 1 μM Ca2+ to activate IK(Ca), and then the cells were superfused with known blockers for different K(Ca) channels (Kohler et al., 1996; Ishii et al., 1997). IK(Ca) was blocked by 1 μM clotrimazole and partially blocked by 10 mM TEA, but was not blocked by 10 μM quinine, 100 nM apamin, or 100 μM d-tubocurarine (Fig. 12 a). Furthermore, IK(Ca) was blocked by CTX with a half-maximal concentration (IC50) of 21 nM (Fig. 12, b and c). Fig. 12 b shows that 100 nM CTX effectively blocks IK(Ca) in AS-HEL cells and is sufficient to shift the reversal potential of the I–V curve from −70 mV to about −40 mV. The hyperpolarization (monitored with TMRE) caused by thapsigargin (n = 13 cells) and thrombin (n = 13 cells) was also inhibited by CTX, indicating the requirement of K(Ca) channels in the response to those stimuli (data not shown).
Figure 12.
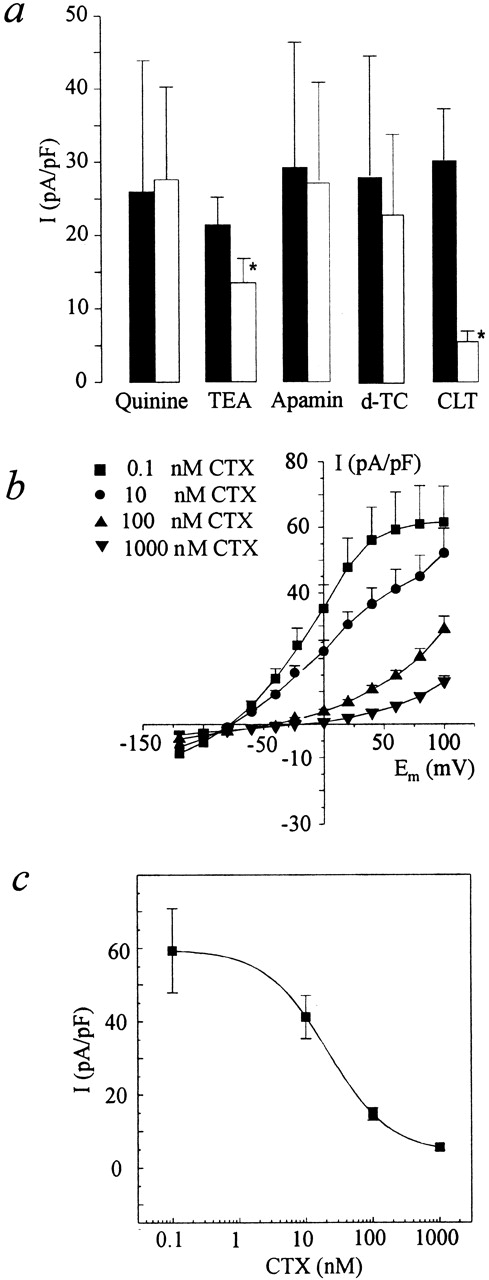
Effect of several potassium channel blockers on the IK(Ca) of the AS-HEL cells. IK(Ca) was activated by dialyzing AS-HEL cells with an internal solution containing 1 μM Ca2+. (a) Once IK(Ca) was activated, the cells were superfused with an external solution containing either 10 μM quinine, 10 mM TEA, 100 nM apamin, 100 μM d-tubocurarine (d-TC) or 1 μM clotrimazole (CLT). Data are shown as mean ± SEM (four cells in each group) of the outward current elicited by a voltage step to +60 mV from a holding potential of −80 mV, before (solid column) or during (open column) exposure to the indicated potassium channel blocker. IK(Ca) is significantly inhibited by 1 μM clotrimazole and 10 mM TEA (*P < 0.05, paired t test). (b) AS-HEL cells were superfused with different concentrations of CTX after activation of the IK(Ca). Current–voltage relationship shows the shift of reversal potentials upon CTX treatment (four to six cells in each group). (c) The data from b for the voltage step to +60 mV are replotted as a function of the concentration of CTX. The data were fit using the Hill equation: I = I max/[1 + (C 1/2/C)n] with n = 0.9 and C 1/2 = 21 nM.
Blockade of IK(Ca) by CTX Inhibits Enhanced Calcium Entry in AS-HEL Cells
Our results demonstrate that AS-HEL cells have a CTX-sensitive IK(Ca) that hyperpolarizes the cells upon calcium influx. The ability to block these channels with CTX enabled us to test whether hyperpolarization produced by IK(Ca) is necessary for the enhanced calcium entry observed in stimulated AS-HEL cells. Fura-2– loaded cells were stimulated with 5 μM thapsigargin in calcium-free HPSS, and 400 s later extracellular calcium was restored to 1.8 mM with or without CTX. Addition of 100 nM CTX reduced the enhanced calcium entry in AS-HEL cells back to a level similar to that observed in the V-HEL cells (Fig. 13), while CTX had no significant effect on calcium entry in V-HEL cells. These results indicate that the membrane hyperpolarization produced by IK(Ca) was necessary for the enhanced calcium entry seen in AS-HEL cells.
Figure 13.

Effect of 100 nM CTX on calcium entry in V-HEL and AS-HEL cells. AS-HEL and V-HEL cells were stimulated with 5 μM thapsigargin in calcium-free HPSS solution for 400 s, as in Fig. 3. Then the cells were challenged with 1.8 mM [Ca2+]o in HPSS solution with or without 100 nM CTX. Data are shown as mean ± SEM (20–35 cells in each group).
If membrane hyperpolarization is responsible for the enhanced Ca2+ entry in AS-HEL cells, it should be possible to demonstrate comparably enhanced Ca2+ entry in V-HEL cells simply by hyperpolarizing the membrane by lowering [K+]o. First we verified, using TMRE to monitor membrane potential, that V-HEL cells bathed in K+-free HPSS, in which NaCl was substituted for KCl, were hyperpolarized from a resting potential of −42 mV to a potential of −62 mV (SEM 1.9; n = 27 cells). Fura-2–loaded cells were stimulated with 5 μM thapsigargin to empty Ca2+ stores, and 400 s later extracellular Ca2+ was restored to 1.8 mM with or without K+ in the HPSS. Hyperpolarization of the membrane by lowering [K+]o to zero clearly enhanced Ca2+ entry in V-HEL cells (Fig. 14).
Figure 14.

Effect of removal of extracellular K+ on calcium entry in V-HEL cells. V-HEL cells were stimulated with 2 μM thapsigargin for 400 s in either calcium-free HPSS solution (▪) or potassium- and calcium-free HPSS (•) Then the cells were challenged with 1.8 mM [Ca2+]o in HPSS or potassium-free HPSS solution. Data are shown as mean ± SEM (n = 18 cells each).
Identity of the K(Ca) Channels in the AS-HEL Cells
K(Ca) channels can be grouped into three classes based on their unitary conductance, voltage dependence, and pharmacological characteristics (Latorre et al., 1989; Hille, 1992; Ishii et al., 1997). The large conductance K(Ca) channels (BK or maxiK channels) have a unitary conductance of 80–250 pS, the current is activated by the concerted action of Ca2+ and depolarization, and the channels are blocked by CTX. Small conductance (SK) Ca2+-activated potassium channels have a unitary conductance <20 pS and the current is activated by calcium alone, and may be blocked by apamin and/or d-tubocurarine. Intermediate conductance Ca2+-activated channels have a conductance of 20–80 pS, the current is activated by Ca2+ without a requirement for depolarization, and they are blocked by CTX. The K(Ca) channels of AS-HEL cells differ from BK channels in that their activation is only dependent on Ca2+ and does not require depolarization. The sensitivity of the K(Ca) channels in AS-HEL cells to CTX and clotrimazole, but not apamin or d-tubocurarine, distinguishes them from SK channels (Kohler et al., 1996). Recently, a human cDNA encoding an IK channel was cloned independently by three groups and designated as hIK1 (Ishii et al., 1997), hSK4 (Joiner et al., 1997), and hKCa4 (Logsdon et al., 1997). Like the cloned hIK1 channel and hKCa4 (Ishii et al., 1997; Logsdon et al., 1997), the K(Ca) channels of AS-HEL cells are blocked by both CTX and clotrimazole, and therefore their properties resemble the properties of those channels. One possible explanation for the presence of IK(Ca) in AS-HEL cells is that the level of transcription of mRNA for the channel is upregulated in AS-HEL cells.
mRNA for an Intermediate Conductance Ca2+-activated K+ Channel in HEL Cells
We therefore examined expression of mRNA for hKCa4, a human IK channel with similar physiological and pharmacological properties to the K(Ca) channels in AS-HEL cells. Primers were designed to amplify a 201-nucleotide fragment of the human intermediate conductance K(Ca) channel hKCa4 that is present in lymphocytes (Logsdon et al., 1997). We first amplified a fragment of the expected size by PCR of cDNA derived by reverse transcription from total RNA of several cultured cells of hematopoietic origin (i.e., HEL, Jurkat, and RPMI-288, a B cell). PCR of the cDNA from V-HEL and AS-HEL also amplified a fragment of the same size, which was sequenced and found to be identical to hKCa4 (Logsdon, 1997). The quantity of the amplified cDNA fragment for hKCa4 was similar in the V-HEL and AS-HEL cells (Fig. 15). The β-actin primers amplified the actin fragment in each cell transfectant to the same level, indicating that the starting amount of cDNA was equivalent in the V-HEL and AS-HEL cells. Thus, the amount of mRNA for hKCa4 was the same in both HEL cell transfectants. However, this does not rule out the possibility that a different gene might encode the K(Ca) channel present in AS-HEL cells.
Figure 15.

Agarose gel electrophoresis of cDNA products amplified by reverse transcription–PCR from mRNA of hKCa4. (1) φχ174rf/dna/haeiii standard. (2) no cDNA. (3) Jurkat hKCa4, 201-nucleotide fragment. (4) V-HEL β actin, 386-nucleotide fragment. (5) AS-HEL β actin. (6) V-HEL, hKCa4 (201 nucleotides). (7) AS-HEL, hKCa4.
discussion
The original goal of this study was to determine whether GAP1IP4BP was involved in the activation or regulation of calcium entry that occurs subsequent to unloading of internal calcium stores. A HEL cell line was successfully established in which the expression of GAP1IP4BP was markedly reduced using transfection with a viral vector containing antisense GAP1IP4BP DNA under control of an RSV promoter. We found that thrombin and thapsigargin caused much larger calcium entry in AS-HEL cells than in control V-HEL cells, and that AS-HEL cells selectively exhibited a hyperpolarization due to IK(Ca). When IK(Ca) was eliminated with CTX, calcium entry in AS-HEL cells was reduced back to that occurring in V-HEL cells. These results indicate that the basal activation of store-operated calcium entry in HEL cells is unaffected by the reduced expression of GAP1IP4BP in AS-HEL cells, but that deficiency of GAP1IP4BP results in an enhanced influx of Ca2 that results from membrane hyperpolarization caused by activation of K(Ca) channels.
We cannot conclude what role, if any, GAP1IP4BP plays in regulation of the basal calcium entry. Nor do our experiments address a possible role for InsP4 in calcium entry. Whether InsP4 and GAP1IP4BP are involved in regulation of calcium entry will require further study. However, it is clear that the presence of a K(Ca) channel-mediated current, which hyperpolarizes the plasma membrane upon calcium mobilization, occurred as a result of the loss of the GAP1IP4BP and was required for the enhanced calcium entry in the AS-HEL cells. Furthermore, our results demonstrate a close relationship between K(Ca) channels and calcium entry (see below).
Activation of IK(Ca) by Calcium Entry
With a weak calcium buffer in the pipette solutions, activation of IK(Ca) and hyperpolarization were apparently dependent on Ca2+ entry because they required the presence of extracellular Ca2+ (Figs. 6 and 11 a). However, since the Ca2+ EC50 for activation of IK(Ca) in AS-HEL cells was 400 nM, the K(Ca) channels might be activated by Ca2+ released from internal stores in the absence of Ca2+ buffers. Nevertheless, our results suggest that Ca2+ influx through the plasma membrane may play a more important role in the activation of the K(Ca) channels. Activation of IK(Ca) by calcium entry was also observed in mouse and rat lymphocytes. In mouse T lymphocytes, the membrane potential, measured using oxonol fluorescence, hyperpolarized when cells were stimulated with concanavalin A in a saline solution containing 1 mM Ca2+ but not in a Ca2+-free solution (Tsien et al., 1982). Using a similar method for measuring the membrane potential, Wilson et al. (1994) found that release of the intracellular calcium stores of rat T lymphocytes by thapsigargin caused hyperpolarization of the cells that was dependent on calcium entry because it was blocked by 5 mM Ni2+. Furthermore, K(Ca) channels may be preferentially activated by Ca2+ entry because the free Ca2+ concentration beneath the plasma membrane, where the store-operated calcium channels are located, will likely be higher than the whole cell average [Ca2+]i. In addition, a Ca2+-activated chloride current is also preferentially activated by Ca2+ entry through the store-operated pathway in Xenopus oocytes (Hartzell, 1996).
The Time Course of IK(Ca)
When AS-HEL cells were stimulated with thapsigargin in the presence of extracellular Ca2+, IK(Ca) was activated within 1 min, and then gradually declined close to its basal level in ∼10 min. The time course of IK(Ca) may reflect the kinetics of Ca2+entry, or alternatively it may reflect intrinsic regulation of the K(Ca) channel by some unknown mechanism. Our results support the latter hypothesis, because IK(Ca) activated by constant internal dialysis with Ca2+ disappeared with a similar time course as when activated by thapsigargin (Fig. 9). Similar results were observed in rat T lymphocytes and ras- and src-transformed fibroblasts (Mahaut-Smith and Schlichter, 1989; Huang and Rane, 1993; Draheim et al., 1995). Mahaut-Smith and Schlichter (1989) reported that single K(Ca) channel activity, recorded with cell- attached patch, could be activated by treating rat T lymphocytes with ionomycin plus extracellular calcium, but the channel activity decreased even in the continued presence of the ionophore. They also found that excised patches had a fewer number of activated channels and the percentage of time spent by channels in the open state was decreased, suggesting a possible desensitization of IK(Ca) with continued exposure to calcium. In src-transformed NIH3T3 cells, dialysis with a pipette solution containing 1 μM free Ca2+ activated an IK(Ca) that diminished within 5–10 min. The rundown of IK(Ca) was not due to the washout of intracellular factors because a similar time course for rundown of the current was observed when the IK(Ca) was measured using nystatin-perforated patch clamp recording with continuous exposure to A23187 in the presence of 2 mM [Ca2+]o (Draheim et al., 1995). In ras-transformed balb3T3 cells, single channel recordings of K(Ca) channels were obtained in the cell attached configuration while cells were stimulated with A23187 in presence of 1 mM [Ca2+]o (Huang and Rane, 1993). On exposure to A23187, channel activity appeared and eventually subsided and could not be reactivated by reapplication of A23187. However, K(Ca) channel activity was restored immediately when the patches were excised and exposed to a solution containing calcium, a result contrary to that in rat T lymphocytes (see above). In contrast, Grissmer et al. (1993) reported that IK(Ca) in human T lymphocytes was stable when activated by dialyzing with internal Ca2+ for a period of 10 min. Therefore, the time course of the IK(Ca) rundown may reflect regulation by unknown cell-specific mechanisms deserving further investigation. Such regulation may have a significant effect on the time course of calcium entry as discussed below.
Membrane Hyperpolarization Facilitates Calcium Entry
Our results demonstrate that activation of the IK(Ca) has a profound effect on calcium entry, presumably by hyperpolarizing the plasma membrane. The effect of plasma membrane hyperpolarization on calcium entry is due to both the electrophysiological characteristics of ICRAC and a greater electrical driving force for calcium entry. Somasundaram et al. (1997) reported that ICRAC is mainly responsible for calcium entry in HEL cells. ICRAC is a non– voltage-gated, low conductance, highly calcium selective current that can be activated by releasing internal calcium stores (reviewed by Parekh and Penner, 1997). Although activation of ICRAC is not controlled by plasma membrane potential, the channel activity is voltage-dependent because the current rectifies inwardly, especially in the voltage range below 0 mV (Hoth and Penner, 1992; Somasundaram et al., 1997). Therefore, once ICRAC is activated by release of internal stores, hyperpolarization of the plasma membrane will further increase the amplitude of the current, thereby causing more calcium entry.
Many nonexcitable cells have K(Ca) channels with similar electrophysiological characteristics as that of AS-HEL cells, which can be classified as intermediate conductance K(Ca) channels (Ishii et al., 1997; Joiner et al., 1997; Logsdon et al., 1997). K(Ca) channels in nonexcitable cells are believed to participate in regulation of cell volume and ion secretion (see references in Ishii et al., 1997; Joiner et al., 1997; Logsdon et al., 1997). It has also been proposed that K(Ca) channels may facilitate store-operated calcium entry (reviewed by Lewis and Cahalan, 1995; Parekh and Penner, 1997). Here we provide direct experimental evidence showing that activation of K(Ca) channels in AS-HEL cells has a profound effect on Ca2+ entry. Once Ca2+ entry is activated, K(Ca) channels may play a significant role in amplifying the magnitude and time course of Ca2+ entry. This mechanism provides a powerful tool for cells to regulate calcium homeostasis.
Does GAP1IP4BP Regulate the Expression or Activity of K(Ca) Channels?
One explanation for the presence of the IK(Ca) in AS-HEL but not V-HEL cells is that the K(Ca) channels are only expressed in AS-HEL cells. A CTX- and TEA-sensitive IK(Ca) current was present in ras-transformed fibroblasts, but not in control cells (Rane, 1991; Huang and Rane, 1993), and the activation of raf protein kinase, downstream of ras in the pathway, was necessary and sufficient for the induction of the current (Huang and Rane, 1994). Furthermore, IK(Ca) induced by growth factors in serum-starved fibroblasts was dependent on protein phosphorylation and protein synthesis, leading to the conclusion that the induction of IK(Ca) by ras transformation was due to activation of the ras/raf signaling cascade leading to the expression of the K(Ca) channels (Huang and Rane, 1994). Transformation of NIH3T3 cells with the src oncogene, which acts upstream from ras, also resulted in the expression of a similar IK(Ca) (Draheim et al., 1995). Also, the number of CTX-sensitive K(Ca) channels in human T-lymphocytes increased 20-fold after the mitogenic activation of the cells (Grissmer et al., 1993).
We found no difference between AS-HEL and V-HEL cells in hKCa4A mRNA expression. Although this does not rule out possible differences in the synthesis or membrane insertion of functional protein, or that a different K(Ca) channel may be present, we must also consider that in AS-HEL cells some posttranslational modification of the protein (e.g., phosphorylation state) accounts for the ability of K(Ca) channels in AS-HEL cells to be activated by Ca2+.
Since GAP1IP4BP is a GTPase-activating protein, its deficiency may cause a ras-like protein to spend more time in the active GTP-bound state leading to a downstream signal (e.g., ras/raf pathway) that modifies channel function.
Summary
We have established a human erythroleukemia cell line in which the expression of GAP1IP4BP protein is substantially reduced by expression of antisense DNA. This does not change the cell's intrinsic mechanism for store-operated Ca2+ entry, but leads to the appearance of an intermediate conductance K(Ca) channel that can hyperpolarize the cell and significantly influence the amplitude and time course of calcium entry subsequent to discharge of intracellular stores of Ca2+. We propose that in some cells GAP1IP4BP can function as a regulator of agonist-induced calcium influx by affecting the activity of a small GTP-binding protein involved in the expression and/or activity of calcium-activated potassium channels.
Acknowledgments
The authors thank Drs. Robin Irvine and Peter Cullen for providing antiserum against GAP1IP4BP and comments on the manuscript. We thank Drs. Hermes Yeh, Leslie Loew, and Achilles Pappano for their advice and technical support and Susan Kreuger at the Center for Biological Imaging Technology for her technical assistance.
Abbreviations used in this paper
- AS-HEL
antisense-transfected HEL
- CTX
charybdotoxin
- HEL
human erythroleukemia
- HPSS
HEPES-buffered physiological salt solution
- ICRAC
calcium release activated calcium current
- InsP3
inositol 1,4,5 trisphosphate
- InsP4
inositol 1,3,4,5 tetrakisphosphate
- IPTG
isopropylthio-β-d-galactoside
- I–V
current–voltage
- RSV
Rous Sarcoma virus
- TEA
tetraethylammonium
- TMRE
tetramethylrhodamine ethyl ester
- V-HEL
vector-transfected HEL
Footnotes
The experimental results described here have been submitted in the form of a dissertation as partial fulfillment for the degree of Doctor of Philosophy in Cellular and Molecular Pharmacology at the University of Connecticut Health Center.
Xinghua Lu's present address is Department of Pharmacology, University of Pittsburgh, Pittsburgh, PA 15260.
references
- Ausubel, F.M., R. Brent, R. Kingston, D.D. Moore, J.G., Seidman, J.A. Smith, and K. Struhl. 1994. Current Protocol in Molecular Biology. 2nd edition. Vol. 1–3. John Wiley & Sons Inc., New York.
- Batty IR, Nahorski SR, Irvine RF. Rapid formation of inositol 1,3,4,5-tetrakisphosphate following muscarinic receptor stimulation of rat cerebral cortical slices. Biochem J. 1985;232:211–215. doi: 10.1042/bj2320211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. Capacitative calcium entry. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GS, Putney JW., Jr Inhibition of thapsigargin- induced calcium entry by microinjected guanine nucleotide analogues. Evidence for the involvement of a small G-protein in capacitative calcium entry. J Biol Chem. 1993;268:21486–21488. [PubMed] [Google Scholar]
- Bird GS, Putney JW., Jr Effect of inositol 1,3,4,5-tetrakisphosphate on inositol trisphosphate-activated Ca2+signaling in mouse lacrimal acinar cells. J Biol Chem. 1996;271:6766–6770. doi: 10.1074/jbc.271.12.6766. [DOI] [PubMed] [Google Scholar]
- Bird GS, Rossier MF, Hughes AR, Shears SB, Armstrong DL, Putney JW., Jr Activation of Ca2+entry into acinar cells by a non-phosphorylatable inositol trisphosphate. Nature. 1991;352:162–165. doi: 10.1038/352162a0. [DOI] [PubMed] [Google Scholar]
- Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–653. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Bradford PG, Irvine RF. Specific binding sites for [3H]inositol(1,3,4,5)tetrakisphosphate on membranes of HL-60 cells. Biochem Biophys Res Commun. 1987;149:680–685. doi: 10.1016/0006-291x(87)90421-9. [DOI] [PubMed] [Google Scholar]
- Changya L, Gallacher DV, Irvine RF, Potter BV, Petersen OH. Inositol 1,3,4,5-tetrakisphosphate is essential for sustained activation of the Ca2+-dependent K+current in single internally perfused mouse lacrimal acinar cells. J Membr Biol. 1989a;109:85–93. doi: 10.1007/BF01870793. [DOI] [PubMed] [Google Scholar]
- Changya L, Gallacher DV, Irvine RF, Petersen OH. Inositol 1,3,4,5-tetrakisphosphate and inositol 1,4,5-triphosphate act by different mechanisms when controlling Ca++in mouse lacrimal acinar cells. FEBS Lett. 1989b;251:43–48. doi: 10.1016/0014-5793(89)81425-5. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Dawson AP, Irvine RF. Purification and characterization of an Ins(1,3,4,5)P4 binding protein from pig platelets: possible identification of a novel non-neuronal Ins(1,3,4,5)P4receptor. Biochem J. 1995a;305:139–143. doi: 10.1042/bj3050139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen PJ, Hsuan JJ, Truong O, Letcher AJ, Jackson TR, Dawson AP, Irvine RF. Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family. Nature. 1995b;376:527–530. doi: 10.1038/376527a0. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Loomis-Husselbeet J, Dawson AP, Irvine RF. Inositol 1,3,4,5 tetrakisphosphate and Ca2+ homeostasis: the role of GAP1IP4BP . Biochem Soc Trans. 1997;25:991–996. doi: 10.1042/bst0250991. [DOI] [PubMed] [Google Scholar]
- Donie F, Reiser G. Purification of a high-affinity inositol 1,3,4,5-tetrakisphosphate receptor from brain. Biochem J. 1991;275:453–457. doi: 10.1042/bj2750453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donie F, Julser E, Reiser G. High-affinity inositol 1,3,4,5-tetrakisphosphate receptor from cerebellum: solubilization, partial purification and characterization. FEBS Lett. 1990;268:194–198. doi: 10.1016/0014-5793(90)81006-a. [DOI] [PubMed] [Google Scholar]
- Draheim HJ, Repp H, Dreyer F. Src-transformation of mouse fibroblasts induces a Ca2+-activated K+ current without changing the T-type Ca2+current. Biochim Biophys Acta. 1995;1269:57–63. doi: 10.1016/0167-4889(95)00106-3. [DOI] [PubMed] [Google Scholar]
- Enyedi P, Williams GH. Heterogenous inositol tetrakisphosphate binding sites in the adrenal cortex. J Biol Chem. 1988;263:7940–7942. [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol (Paris) 1979;75:463–504. [PubMed] [Google Scholar]
- Fasolato C, Hoth M, Penner R. A GTP-dependent step in the activation mechanism of capacitative calcium influx. J Biol Chem. 1993;268:20737–20740. [PubMed] [Google Scholar]
- Fasolato C, Innocenti B, Pozzan T. Receptor-activated Ca2+ influx: how many mechanisms for how many channels? . Trends Pharmacol Sci. 1994;15:77–83. doi: 10.1016/0165-6147(94)90282-8. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Mikoshiba K. Structure-function relationships of the mouse Gap1m. Determination of the inositol 1,3,4,5-tetrakisphosphate-binding domain. J Biol Chem. 1996;271:18838–18842. doi: 10.1074/jbc.271.31.18838. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Cahalan MD. Calcium-activated potassium channels in resting and activated human T lymphocytes. Expression levels, calcium dependence, ion selectivity, and pharmacology. J Gen Physiol. 1993;102:601–603. doi: 10.1085/jgp.102.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse AH, Roth E, Emmrich F. D-myo-inositol 1,3,4,5-tetrakisphosphate releases Ca2+from crude microsomes and enriched vesicular plasma membranes, but not from intracellular stores of permeabilized T-lymphocytes and monocytes. Biochem J. 1992;288:489–495. doi: 10.1042/bj2880489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hartzell HC. Activation of different Cl currents in Xenopusoocytes by Ca liberated from stores and by capacitative Ca influx. J Gen Physiol. 1996;108:157–175. doi: 10.1085/jgp.108.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B.1992. Ionic channels of excitable membranes. 2nd edition. Sinauer Associates Inc., Sunderland, MA. 607 pp.
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Huang Y, Rane SG. Single channel study of a Ca2+-activated K+current associated with ras-induced cell transformation. J Physiol (Lond) 1993;461:601–618. doi: 10.1113/jphysiol.1993.sp019531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Rane SG. Potassium channel induction by the Ras/Raf signal transduction cascade. J Biol Chem. 1994;269:31183–31189. [PubMed] [Google Scholar]
- Irvine RF. ‘Quantal' Ca2+ release and the control of Ca2+entry by inositol phosphates—a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- Irvine RF. Inositol phosphates and Ca2+ entry: toward a proliferation or a simplification? . FASEB J. 1992;6:3085–3091. doi: 10.1096/fasebj.6.12.1325932. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Moor RM. Micro-injection of inositol 1,3,4,5-tetrakisphosphate activates sea urchin eggs by a mechanism dependent on external Ca2+ . Biochem J. 1986;240:917–920. doi: 10.1042/bj2400917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF, Letcher AJ, Heslop JP, Berridge MJ. The inositol tris/tetrakisphosphate pathway—demonstration of Ins(1,4,5)P33-kinase activity in animal tissues. Nature. 1986;320:631–634. doi: 10.1038/320631a0. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci USA. 1997;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner WJ, Wang LY, Tang MD, Kaczmarek LK. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc Natl Acad Sci USA. 1997;94:11013–11018. doi: 10.1073/pnas.94.20.11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Koppler P, Mersel M, Malviya AN. Subcellular localization of specific inositol 1,3,4,5-tetrakis([3H]phosphate) binding sites in rat liver membrane fractions: a comparative evaluation of pH sensitivity and binding characteristics. Biochemistry. 1994;33:14707–14713. doi: 10.1021/bi00253a008. [DOI] [PubMed] [Google Scholar]
- Koppler P, Mersel M, Humbert JP, Vignon J, Vincendon G, Malviya A. High affinity inositol 1,3,4,5-tetrakisphosphate receptor from rat liver nuclei: purification, characterization and amino-terminal sequence. Biochemistry. 1996;35:5481–5487. doi: 10.1021/bi9522918. [DOI] [PubMed] [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Annu Rev Physiol. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- Lewis RS, Cahalan MD. Potassium and calcium channels in lymphocytes. Annu Rev Immunol. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- Loew, L.M. 1993. Confocal microscopy of potentiometric fluorescent dye. In Methods in Cell Biology. Vol. 38. B. Matsumoto, editor. Academic Press, Orlando, FL. 194–209. [DOI] [PubMed]
- Logsdon NJ, Kang J, Togo JA, Christian EP, Aiyar J. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J Biol Chem. 1997;272:32723–32726. doi: 10.1074/jbc.272.52.32723. [DOI] [PubMed] [Google Scholar]
- Luckhoff A, Clapham DE. Inositol 1,3,4,5-tetrakisphosphate activates an endothelial Ca2+-permeable channel. Nature. 1992;355:356–358. doi: 10.1038/355356a0. [DOI] [PubMed] [Google Scholar]
- Mahaut-Smith MP, Schlichter LC. Ca2+-activated K+channels in human B lymphocytes and rat thymocytes. J Physiol (Lond) 1989;415:69–83. doi: 10.1113/jphysiol.1989.sp017712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer D. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Molleman A, Hoiting B, Duin M, Akker JVD, Nelemans A, Hertog AD. Potassium channels regulated by inositol 1,3,4,5-tetrakisphosphate and internal calcium in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1991;266:5658–5663. [PubMed] [Google Scholar]
- Morris AP, Gallacher DV, Irvine RF, Petersen OH. Synergism of inositol trisphosphate and tetrakisphosphate in activating Ca2+-dependent K+channels. Nature. 1987;330:653–655. doi: 10.1038/330653a0. [DOI] [PubMed] [Google Scholar]
- O'Rourke F, Matthews E, Feinstein MB. Isolation of InsP4 and InsP6 binding proteins from human platelets: InsP4 promotes Ca2+ efflux from inside-out plasma membrane vesicles containing 104 kDa GAP1IP4BPprotein. Biochem J. 1996;315:1027–1034. doi: 10.1042/bj3151027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Putney JW, Jr, Bird GS. The inositol phosphate-calcium signaling in nonexcitable cells. Endocr Rev. 1993;14:610–631. doi: 10.1210/edrv-14-5-610. [DOI] [PubMed] [Google Scholar]
- Rane SG. A Ca2+-activated K+current in ras-transformed fibroblasts is absent from nontransformed cells. Am J Physiol. 1991;260:C104–C112. doi: 10.1152/ajpcell.1991.260.1.C104. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd edition. Vol. 1–3. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Somasundaram B, Mason MJ, Mahaut-Smith MP. Thrombin-dependent calcium signalling in single human erythroleukemia cells. J Physiol (Lond) 1997;501:485–495. doi: 10.1111/j.1469-7793.1997.485bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura H, Hughes AR, Thastrup O, Putney JW., Jr Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J Biol Chem. 1989;264:12266–12271. [PubMed] [Google Scholar]
- Tertyshnikova S, Fein A. [Ca2+]i oscillations and [Ca2+]iwaves in rat megakaryocytes. Cell Calc. 1997;21:331–344. doi: 10.1016/s0143-4160(97)90026-9. [DOI] [PubMed] [Google Scholar]
- Theibert AB, Estevez VA, Ferris CD, Danoff SK, Barrow RK, Prestwich GD, Snyder SH. Inositol 1,3,4,5-tetrakisphosphate and inositol hexakisphosphate receptor proteins: isolation and characterization from rat brain. Proc Natl Acad Sci USA. 1991;88:3165–3169. doi: 10.1073/pnas.88.8.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY, Pozzan T, Rink TJ. T-cell mitogens cause early changes in cytoplasmic free Ca2+and membrane potential in lymphocytes. Nature. 1982;295:68–71. doi: 10.1038/295068a0. [DOI] [PubMed] [Google Scholar]
- Wilson OI, Marriott I, Mahaut-Smith MP, Hymel LJ, Mason MJ. Isolation and characterization of membrane potential changes associated with release of calcium from intracellular stores in rat thymic lymphocytes. J Membr Biol. 1994;137:159–168. doi: 10.1007/BF00233485. [DOI] [PubMed] [Google Scholar]
- Zhu X, Jaing M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. Trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+entry. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]