

Abstract
Multilocus sequence typing analysis of Streptococcus uberis has identified a cluster of isolates associated with clinical and subclinical mastitis and a cluster associated with cows with low somatic cell counts in their milk. Specific groups of genotypes (global clonal complex [GCC] sequence type 5s [ST5s] and GCC ST143s) were highly associated (P = 0.006) with clinical and subclinical mastitis and may represent a lineage of virulent isolates, whereas isolates belonging to GCC ST86 were associated with low-cell-count cows. This study has, for the first time, demonstrated the occurrence of identical sequence types (ST60 and ST184) between different continents (Australasia and Europe) and different countries (Australia and New Zealand). The standardized index of association and the empirical estimation of the rate of recombination showed substantial recombination within the S. uberis population in Australia, consistent with previous multilocus sequence type analyses.
Mastitis is the single most important cause of financial loss to the dairy industry worldwide (2, 39), with production losses amounting to more than US$2 billion annually (31, 59). The dairy industry in Australia has an annual production of approximately 10 billion liters, valued at A$3.3 billion (8) and is mainly concentrated in the southern states with 65% of milk production originating from the state of Victoria. Production losses in the Australian dairy industry as a result of mastitis are in excess of A$150 million per year (37), mainly due to decreased milk output and reduced milk quality payments. Dairy farmers also suffer losses due to increased veterinary intervention, labor costs, loss of income due to the need to withhold milk during treatment of clinical cases, and the premature culling of affected animals (2, 39, 51).
Although the introduction of mastitis control schemes has been successful in reducing the incidence of contagious mastitis caused by Streptococcus agalactiae and Staphylococcus aureus in dairy herds (32), these measures have had little impact on environmental pathogens, notably Streptococcus uberis and Escherichia coli (3, 4, 26, 29). In an earlier study, the incidence of clinical mastitis in Australian herds caused by S. uberis was estimated at 22.7% of cases (58). The ability to control these infections depends on a detailed knowledge of the epidemiology of the organism and the management of the herd and its environment.
Several molecular typing methods have been used to investigate the epidemiology of S. uberis. These methods include restriction fragment length polymorphism (23, 27), pulsed-field gel electrophoresis (PFGE) (9, 40, 57), randomly amplified polymorphic DNA fingerprinting (61), and multilocus sequence typing (MLST) (6, 42, 62). These studies suggested that transmission of S. uberis occurs from environmental sources (40, 61, 63) and that feces could be a reservoir (61, 63). An earlier MLST study identified specific sequence types (STs) belonging to major clonal complexes (CCs) from milk samples and the cow's environment and suggested that these STs have the ability to survive in the environment and to establish intramammary infections (42).
Several investigators have used PFGE to demonstrate that S. uberis is a highly diverse species (9, 36, 40, 57) with many different PFGE types present on a single farm, suggesting that the species is behaving as an opportunistic pathogen. However, chronically infected cows often harbor the same PFGE type (36, 40, 41, 57), and there has been a single report demonstrating (61) and others suggesting cow-to-cow transmission of a single clone (1, 40). These observations suggest the possibility that some strains are either hypervirulent, hypertransmissible between cows, or able to survive in a host, for example, by evading the host immune response. It has not been conclusively proven, however, whether certain clones with enhanced virulence are responsible for mastitis. In addition, despite numerous epidemiological studies using PFGE, no clear evidence with regard to the relatedness of S. uberis isolates from different herds has been reported in the literature. This lack of evidence can be attributed partly to the inherent difficulties associated with the interlaboratory comparison of PFGE-based epidemiological studies.
MLST differs fundamentally from PFGE and most other molecular typing methods, being based on nucleotide sequence data from approximately 500 bp of housekeeping genes that have been shown to accumulate sequence variation slowly. Accordingly, MLST, in contrast to PFGE, which uses whole genomic DNA, is much less affected by recent rearrangement of the genome by recombination. Consequently, MLST has greater utility for determining the recent ancestral lineage and the relatedness of individual strains. In addition, MLST utilizes multiple genes of different sizes for analysis, which provides substantial discriminatory power for subtyping. In some species, inclusion of one or more virulence-associated genes (multi-virulence-locus sequence typing) can be used to further increase the discriminatory power (33, 34, 54, 64). Both MLST and multi-virulence-locus sequence typing profiles are unambiguous and can be represented by a number of digits corresponding to the allelic number of each of the loci used. This greatly facilitates interlaboratory comparisons and the study of global epidemiology (12).
Two MLST methods have been developed for the typing of S. uberis. The initial method (62) used the pauA gene, which encodes a virulence factor (plasminogen activator A), and the gapC gene, which encodes a vaccine target (glyceraldehyde-3-phosphate dehydrogenase) (18). The pauA gene has been shown to exhibit a high ratio of nonsynonymous substitutions to synonymous substitutions (dN/dS) (>1.0) (62), indicating positive selection, and therefore may not provide accurate phylogenetic information (6). In addition, the initial MLST scheme uses only six loci (62), and as such, it may not provide sufficient resolution for a large data set compared with the seven loci used in the more recent MLST scheme (6). For these reasons, we chose to use the MLST scheme developed by Coffey et al. in 2006 (6).
In the present study, we performed MLST analyses on a collection of Australian S. uberis isolates from cases of clinical and subclinical mastitis and from cows with low somatic cell counts in their milk. The main aim of this study was to investigate the molecular epidemiology of S. uberis mastitis in Australia to ascertain whether there was any association of a particular MLST with mastitis. In addition, we investigated the evolutionary lineage of Australian isolates in relation to the S. uberis global population as a whole by comparing the MLST profiles of Australian isolates with those currently available in the MLST database from different countries.
MATERIALS AND METHODS
Bacterial isolates.
In total, 46 S. uberis isolates were obtained from quarter milk samples (n = 45) of dairy cattle (n = 36) in Victoria, Australia. Twenty isolates were from cows with clinical mastitis, 16 from subclinical cases, and 10 from cows with low somatic cell counts in their milk. Clinical mastitis was defined as the presence of clots in milk or visible signs of inflammation in the mammary glands. Cows were considered to have subclinical mastitis if the individual cow somatic cell count (performed by a herd testing service on pooled milk samples within 1 week of culture) was >250,000 cells/ml; otherwise, they were placed in the low-cell-count category (5). In all cases, our allocation was in accordance with previously published criteria (63), where the three individual cow somatic cell count results closest to the date of culture were used.
The geographic distribution and date of collection of the samples were as follows: 29 isolates were collected from 24 cows (28 quarters) from a dairy farm in Newry (eastern Victoria, Australia). Of these, 16 isolates were collected from a cow with clinical mastitis (n = 1), cows with subclinical mastitis (n = 5), and cows with low somatic cell counts in their milk (n = 10) in October 2004, and 13 isolates from cows with clinical mastitis were collected between November and December 2005. Twelve isolates from 12 quarters of seven cows in Timboon (western Victoria, Australia) were collected September 2004. Five isolates were obtained from a centralized veterinary diagnostic laboratory (Gippsland Vetnostics, Traralgon, Victoria, Australia) between April and May 2006. Consequently, the latter samples were originally isolated from quarter milk samples (n = 5) from dairy cattle (n = 5) from various parts of Victoria, Australia.
Culture of milk sample and identification of S. uberis.
Quarter milk samples were collected according to Australian guidelines (5). Samples were kept at 4 °C during transportation and cultured immediately on arrival at the laboratory. Milk was cultured by spreading 50 μl over the surface of one horse blood agar plate and one Edward's agar plate (Oxoid Ltd., Basingstoke, United Kingdom). Plates were incubated at 37°C for 48 h.
All isolates were initially identified as S. uberis on the basis of the following: Gram stain, catalase reaction, hydrolysis of esculin and hippurate, or API 20 Strep (bioMérieux Vitek, Hazelwood, MO). In addition, S. uberis species-specific PCRs (20) for both the 16S rRNA and 23S rRNA genes were performed for primary species identification. The PCR conditions and the primers used are listed in Table 1. To further confirm species identification, 16S rRNA gene sequencing was also performed as described previously (42). The 16S rRNA gene sequences were compared with the reference sequence of S. uberis, strain HN1 (AB023576.1) using BLASTN at the NCBI (http://www.ncbi.nlm.nih.gov/BLAST/), and the individual isolates were designated as S. uberis if the 16S rRNA sequence was 100% identical to the reference sequence.
TABLE 1.
Primers and PCR conditions
| Primer name | Target gene | Primer function(s) | PCR product (bp) | Primer sequence | PCR cycling conditiona | Reference |
|---|---|---|---|---|---|---|
| ub-I | 16S rRNA | PCR (species identification) | 445 | 5′-CGCATGACAATAGGGTACA-3′ | 1 | 20 |
| ub-II | 5′-GCCTTTAACTTCAGACTTATCA-3′ | |||||
| ub-23S-I | 23S rRNA | PCR (species identification) | 451 | 5′-CGTATTTAAAATTGACTTTAGCC-3′ | 1 | 20 |
| ub-23S-II | 5′-AATTTCTCCGCTACCCAC-3′ | |||||
| 16S forward | 16S rRNA | PCR and sequencing | 534 | 5′-GAGAGTTTGATCCTGGCTCAGGA-3′ | 2 | 42 |
| 16S reverse | (16S rRNA sequencing) | 5′-TTACCGCGGCTGCTGGCACGT-3′ | ||||
| arcC F | arcC | PCR and sequencing | 518 | 5′-GTTTGTGACGCAAAATCTTTATCGATAACA-3′ | 3a | 6 |
| arcC R | 5′-ACTCATGGTAACGGACCACAAGTTGGTAAC-3′ | |||||
| ddl F | ddl | PCR and sequencing | 503 | 5′-GTCTATATTGAAGGTAATGACTTGGAAGACTGT-3′ | 3b | 6 |
| ddl R | 5′-TACATGGACCACTGAGTGAATCCAGGCATAGTATTC-3′ | |||||
| gki F | gki | PCR and sequencing | 564 | 5′-GACCGGACCCAAAACACAGTCACAGGTGCTTTT-3′ | 3a | 6 |
| gki R | 5′-AAGAGAATCTGGATTTAGGATATTTGAAATATT-3′ | |||||
| recP F | recP | PCR and sequencing | 531 | 5′-AATTCAGGTCACCCTGGCTTACCAATGGGTGCAGCC-3′ | 3a | 6 |
| recP R | 5′-TGTGAAAGCCATTGATGTTGGACCATCAAGTGAAAT-3′ | |||||
| tdk F | tdk | PCR and sequencing | 793 | 5′-TATTTTCATTTCATAATAAGTTAGTGGATTTAGTAA-3′ | 3a | 6 |
| tdk R | 5′-TTGATCATATATATTCATGTTATGAATCGTTCTCCT-3′ | |||||
| tpi F | tpi | PCR and sequencing | 471 | 5′-GTTATTGGTCATTCAGAACGTCGTGATTAC TTC-3′ | 3a | 6 |
| tpi R | 5′-GTCAAGTAATGCTAAGAAGCTATCTGCTTCAAGTGA-3′ | |||||
| yqiL F | yqiL | PCR and sequencing | 574 | 5′-TTTCTTCTTTGAAACGATTATTTTTAAGTGCTTCAG-3′ | 3a | 6 |
| yqiL R | 5′-CAAGCTCTAAGAACACCAATTGGTGCATTCGGAGGA-3′ | |||||
| yqiL in 2F | yqiL | PCR (additional primers for yqiL) | 500 | 5′-GCCTTTTGTTGACTATTGAATGC-3′ | 3a | This study |
| yqiL in 2R | 5′-GCTTTTAAAGATGTTAATGCCGTTAC-3′ | |||||
| yqiL out 1F | yqiL | PCR (additional primers for yqiL) | 772 | 5′-CATTTCCAGCTGTAACAGTCC-3′ | 3a | This study |
| yqiL out 1R | 5′-TTTCTATTTATGCAAAGGAGTCG-3′ | |||||
| hasA-for | hasA | PCR | 319 | 5′-GAAAGGTCTGATGCTGATG-3′ | 1 | 17 |
| hasA-rev | 5′-TCATCCCCTATGCTTACAG-3′ | |||||
| hasC-for | hasC | PCR | 225 | 5′-TGCTTGGTGACGATTTGATG-3′ | 1 | 17 |
| hasC-rev | 5′-GTCCAATGATAGCAAGGTCAC-3′ |
PCR conditions are as follows: condition 1, 5 min at 95°C, 28 cycles (1 cycle consisting of 30 s at 95°C, 30 s at 58°C, and 2 min at 72°C), and 5 min at 72°C; condition 2, 5 min at 94°C, 35 cycles (1 cycle consisting of 30 s at 94°C, 60 s at 56°C, and 90 s at 72°C), and 6 min at 72°C; condition 3a, 5 min at 95°C, 40 cycles (1 cycle consisting of 30 s at 95°C, 30 s at 55°C, and 45 s at 72°C), and 7 min at 72°C; and condition 3b, 5 min at 95°C, 40 cycles (1 cycle consisting of 30 s at 95°C, 30 s at 63°C, and 45 s at 72°C), and 7 min at 72°C.
PFGE analysis.
Pulsed-field gel electrophoresis was based on the methods described previously (40, 47) with the following modifications. S. uberis isolates were incubated in 10 ml of Todd-Hewitt broth (Oxoid) for 16 h at 37°C with gentle shaking. Cells were harvested by centrifugation for 15 min at 3,382 × g and resuspended in 1 ml Pett IV buffer (10 mM Tris-HCl [pH 7.6], 1 M NaCl). An aliquot of the S. uberis suspension was then mixed with the same volume of 2.4% (wt/vol) low-melting-point agarose (Progen Industries, Darra, Australia) in Pett IV buffer and dispensed into plug molds. Plugs were incubated at 37°C for 16 h in lysis solution (0.2% sodium deoxycholate, 0.5% N-lauryl sarcosine, 1 mg/ml lysozyme, 6 mM Tris-HCl [pH 7.6], 1 M NaCl, 0.1 M EDTA [pH 7.6]). The plugs were subsequently incubated in ESP buffer (1% N-lauryl sarcosine, 1 mg/ml proteinase K, 0.5 M EDTA [pH 8.6]) for 16 h at 50°C and then incubated in TE buffer (10 mM Tris-HCl, 10 mM EDTA) with 1 mM phenylmethylsulfonyl fluoride (PMSF) for 2 h at 37°C, followed by four 15-min washes with TE buffer. For restriction endonuclease digestion, plugs containing S. uberis DNA were digested with 40 U of SmaI (MBI Fermentas, Hanover, MD) at 30°C overnight. The plugs were then loaded into a 1% (wt/vol) SeaKem Gold agarose gel (Cambrex Bio Science Rockland, Inc., Rockland, ME), and the SmaI-digested DNA fragments were separated by PFGE using a contour-clamped homogeneous electric field DR-II chamber (Bio-Rad, Hercules, CA) at 14°C with pulse times of 5 s to 15 s for 7.7 h and 15 s to 45 s for 9.5 h at 6 V/cm. Gels were stained with ethidium bromide (0.5 μg/ml), and the DNA bands were visualized using the Gel Doc 2000 imaging system (Bio-Rad, Hercules, CA).
The GelCompar II software (Applied Maths, Kortrijk, Belgium) was used to perform cluster analysis and construct a dendrogram using the unweighted pair group method using arithmetic averages (UPGMA) based on Dice coefficients using 0.75% optimization and 1% tolerance parameters. For analysis, PFGE patterns showing <80% similarity were assigned to different types, whereas patterns with similarity values of >80% and <100% were assigned to subtypes.
MLST analysis.
MLST was performed using the primers described by Coffey et al. in 2006 (6) and the modified cycling conditions as detailed in Table 1. The S. uberis reference strain, ATCC 700407, was used as a positive control. The allelic profiles and sequence types (STs) of the individual isolates were determined using the query functions implemented in the S. uberis MLST database (http://pubmlst.org/suberis). The clonal complex for each ST in the MLST database was assigned by the curator and in this study referred to as global clonal complex (GCC).
Data analysis.
Simpson's index of diversity (D) with 95% confidence intervals (95% CI) of the MLST and the PFGE techniques used in this study were determined as described previously (19, 24).
The dN/dS ratios, the G+C ratios, and the number and proportion of variable nucleotide sites for each of the seven loci were analyzed using Sequence Type Analysis and Recombinational Tests, version 2 (START2) (28).
Lineage analyses of Australian isolates were performed by constructing a UPGMA-based dendrogram using START2 (28). The eBURST version 3 program (http://eburst.mlst.net/) was used to investigate the population structure of the Australian S. uberis isolates and the relatedness of the Australian isolates in the global population of S. uberis. For eBURST analyses, an individual CC was defined as a group of isolates that shared alleles at six of the seven loci (12, 14, 49).
The standardized index of association (ISA) (21) was calculated using START2. ISA measures the degree of linkage equilibrium between allelic profiles and estimates the rate of recombination (30, 48, 50). The rates of recombination to mutation and the per site ratio of recombination to mutation parameter were determined as described previously (16). Statistical analyses were performed using chi-square tests using SPSS 13.0 software (SPSS Inc., Chicago, IL).
Investigation of yqiL-negative Australian isolates.
In an attempt to amplify the yqiL gene of the four Australian isolates which were PCR negative using the original primers (6), two additional primer pairs for the yqiL gene were designed using Primer3 (44). The primer specifications and amplification conditions used are detailed in Table 1.
To confirm the absence of the yqiL gene, Southern blot hybridization was performed. For Southern blot hybridization, 1 μg of genomic DNA was digested with HindIII (Promega, Madison, WI) and separated by agarose gel electrophoresis at 80 V for 90 min in a 1.5% (wt/vol) agarose gel. The DNA fragments were subsequently transferred by Southern blotting to a positively charged nylon membrane (Immobilon-Ny+; Amersham, Little Chalfont, United Kingdom) and hybridized with a yqiL gene probe using standard procedures. The yqiL gene probe was prepared using the digoxigenin DNA labeling kit (Roche Diagnostics, Basel, Switzerland) in accordance with the manufacturer's instructions with a yqiL PCR product generated from a reference yqiL-positive S. uberis isolate used as a positive control.
The flanking region of the yqiL gene was further investigated to determine whether a possible virulence factor gene or surface protein gene was present. Any such association would place the locus under high selective pressure making it unsuitable for MLST. The yqiL flanking region of the S. uberis genome sequence obtained from the S. uberis genome project (http://www.sanger.ac.uk./Projects/S_uberis/) was used for this analysis.
The S. uberis yqiL gene was queried against the S. uberis genome sequence using BLASTN and BLASTX to ascertain whether possible yqiL homologues that could possibly complement for yqiL-negative isolates were present.
Detection of hasA and hasC genes by PCR and Southern blot hybridization.
Amplification of the capsular genes, hasA and hasC, was performed using a modification of a previously described method (17). The PCR conditions and the primers used are listed in Table 1. The isolates that were negative for hasA by PCR and the strains that were positive for hasA by PCR (control strains) were further examined by Southern blot hybridization using a digoxigenin-labeled hasA gene probe generated from a reference S. uberis strain using the same procedure as that used for the yqiL probe described earlier.
Nucleotide sequence accession numbers.
The nucleotide sequences of the MLST alleles from the Australian S. uberis isolates have been deposited in GenBank as a data set with the accession numbers EF672733 to EF672747.
RESULTS
Identification and characterization of isolates.
Forty-six Australian isolates were identified and confirmed as S. uberis using biochemical tests and/or API Strep20 (bioMérieux Vitek, Hazelwood, MO). In addition, all isolates were positive using both 16S rRNA and 23S rRNA gene S. uberis species-specific PCRs (20). Nucleotide sequence determination of the 16S rRNA gene sequences of all isolates, except for two of the isolates (686-5 and 686-10), demonstrated that they were 100% identical to the reference sequence of S. uberis strain HN1 (accession number AB023576) in GenBank. Nevertheless, the 16S rRNA gene sequences of isolates 686-5 and 686-10 differed from the reference sequence by only 1 and 2 nucleotides (nt), respectively.
Statistical analysis based on the geographic origin of the samples against types of global clonal complex demonstrated that while more than 50% of our samples were isolated from farms in Newry, Australia, there was no significant difference (P = 0.57) in the distribution of strains by geographic location of sampling against types of GCC.
PFGE analysis.
Analysis of the Australian S. uberis isolates by PFGE clearly demonstrated that they exhibited considerable diversity. In total, 42 PFGE types (including subtypes) were identified from the 46 isolates (Table 2). There were four examples of identical PFGE types being isolated from different cows on the same farm (three cases) or from different quarters of the same cow (one case). In no instance were identical PFGE types observed for cows from different farms.
TABLE 2.
MLST profiles of the 46 Australian isolates
| Location of farm in Australia | STa | Isolate IDb | Allelec
|
GCCd | Disease statuse | PFGE typef | PCR resultg for:
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| arcC | ddl | gki | recP | tdk | tpi | yqiL | hasC | hasA | ||||||
| Newry | 60 | 2907-1 | 1 | 1 | 2 | 2 | 7 | 1 | 3 | 5 | C | 3 | + | − (+) |
| Newry | 60 | 2481-1 | 1 | 1 | 2 | 2 | 7 | 1 | 3 | 5 | C | 5-b | + | + (+) |
| Newry | 60 | 2956-4 | 1 | 1 | 2 | 2 | 7 | 1 | 3 | 5 | C | 5-c | + | + |
| Newry | 60 | 3209-4 | 1 | 1 | 2 | 2 | 7 | 1 | 3 | 5 | C | 6 | + | + |
| Timboon | 153 | 796-2 | 9 | 1 | 2 | 2 | 25 (N) | 1 | 3 | 143 | SC | 2 | + | + |
| Timboon | 155 | 684-1 | 1 | 19 (N) | 2 | 2 | 17 | 4 | 3 | 143 | SC | 8-a | + | + |
| Timboon | 155 | 684-6 | 1 | 19 (N) | 2 | 2 | 17 | 4 | 3 | 143 | SC | 8-b | + | + |
| Timboon | 155 | 684-9 | 1 | 19 (N) | 2 | 2 | 17 | 4 | 3 | 143 | SC | 8-a | + | + |
| Timboon | 155 | 816-5 | 1 | 19 (N) | 2 | 2 | 17 | 4 | 3 | 143 | SC | 8-a | + | + |
| Newry | 156 | 2893-1 | 18 | 1 | 2 | 2 | 7 | 3 | 3 | 143 | C | 28 | + | + (+) |
| Newry | 184 | 2874-4 | 1 | 1 | 2 | 2 | 17 | 1 | 3 | 143 | C | 31-a | + | − (+) |
| Newry | 194 | 2042-5 | 32 (N) | 1 | 20 (N) | 1 | 17 | 1 | 3 | − | SC | 4 | + | + |
| Korumburra | 194 | 5838-3 | 32 (N) | 1 | 20 (N) | 1 | 17 | 1 | 3 | − | C | 24 | + | + |
| Newry | 216 | 2407-7 | 1 | 1 | 20 (N) | 2 | 17 | 1 | 3 | 143 | SC | 7 | + | + |
| Newry | 217 | 2655-5 | 3 | 2 | 21 (N) | 3 | 3 | 2 | 3 | 86 | SC | 22 | + | − (−) |
| Timboon | 251 | 796-3 | 1 | 1 | 5 | 2 | 3 | 1 | 3 | − | SC | 13 | + | + |
| Timboon | 252 | 853-4 | 9 | 12 | 4 | 2 | 17 | 1 | 3 | − | C | 30 | + | + |
| Timboon | 253 | 816-2 | 4 | 1 | 5 | 1 | 17 | 1 | 3 | − | SC | 23 | + | + |
| Timboon | 253 | 883-4 | 4 | 1 | 5 | 1 | 17 | 1 | 3 | − | C | 23 | + | + |
| Timboon | 254 | 868-4 | 37 (N) | 2 | 4 | 1 | 13 | 12 (N) | 26 (N) | − | SC | 1 | + | + |
| Newry | 255 | 2690-2 | 3 | 19 (N) | 3 | 2 | 36 (N) | 3 | 3 | − | SC | 27 | + | + |
| Newry | 256 | 3147-4 | 1 | 1 | 13 | 4 | 2 | 3 | 3 | − | L | 11-a | + | + |
| Newry | 257 | 3217-1 | 19 | 1 | 5 | 2 | 8 | 8 | 19 | − | L | 15 | + | − (−) |
| Newry | 258 | 2581-2 | 19 | 1 | 5 | 2 | 37 (N) | 1 | 3 | − | SC | 20 | + | + |
| Newry | 258 | 3333-3 | 19 | 1 | 5 | 2 | 37 (N) | 1 | 3 | − | L | 20 | + | + |
| Newry | 259 | 3327-3 | 3 | 1 | 3 | 2 | 6 | 1 | 3 | 86 | L | 32 | + | − (−) |
| Newry | 260 | 3147-1 | 4 | 1 | 4 | 3 | 13 | 4 | 3 | − | L | 21 | + | + |
| Newry | 261 | 3147-5 | 1 | 1 | 13 | 4 | 17 | 3 | 13 | − | L | 16 | + | + |
| Newry | 262 | 3217-2 | 3 | 2 | 3 | 3 | 13 | 4 | 3 | 86 | L | 18 | + | − (−) |
| Newry | 263 | 3217-5 | 3 | 2 | 3 | 3 | 5 | 1 | 3 | 86 | L | 31-b | + | + (+) |
| Newry | 264 | 3133-3 | 3 | 2 | 3 | 2 | 17 | 4 | 3 | 143 | L | 17 | + | − (−) |
| Moe | 272 | 5851 | 18 | 1 | 2 | 1 | 13 | 1 | 3 | 5 | C | 33 | + | + |
| Camperdown | 273 | 5867 | 20 | 1 | 2 | 2 | 7 | 1 | 3 | 143 | SC | 9 | + | + |
| Maffra | 275 | 6104 | 1 | 1 | 5 | 2 | 4 | 2 | 3 | − | C | 35 | + | + |
| Newry | 276 | 2565-2 | 4 | 4 | 4 | 2 | 17 | 1 | 10 | − | C | 29 | + | − (+) |
| Newry | 277 | 2169-2 | 1 | 1 | 13 | 2 | 7 | 3 | 3 | − | C | 11-b | + | + |
| Newry | 278 | 2285-1 | 1 | 1 | 2 | 2 | 4 | 1 | 3 | 5 | C | 5-a | + | + |
| Newry | 279 | 2520-1 | 18 | 1 | 2 | 2 | 7 | 1 | 3 | 143 | C | 26 | + | + |
| Newry | 280 | 2530-2 | 1 | 1 | 3 | 4 | 17 | 1 | 13 | − | C | 10-b | + | + |
| Newry | 281 | 2730-5 | 1 | 1 | 3 | 2 | 7 | 3 | 3 | − | C | 25 | + | + |
| Newry | 282 | 2988-1 | 1 | 1 | 13 | 2 | 17 | 3 | 3 | 143 | C | 5-d | + | + |
| Maffra | 283 | 6093 | 9 | 1 | 2 | 1 | 40 (N) | 3 | 7 | − | C | 34 | + | + |
| Newry | 594h | 3064-1 | 1 | 1 | 3 | 2 | 1 | 1 | −i | − | C | 12 | + | + |
| Newry | 595h | 3217-3 | 3 | 2 | 28 (N) | 2 | 6 | 4 | −i | − | L | 19 | + | + |
| Timboon | 596h | 686-5 | 6 | 14 | 4 | 15 (N) | 22 | 13 (N) | −i | − | SC | 14 | + | − (−) |
| Timboon | 597h | 686-10 | 21 | 21 | 18 | 11 | 41 (N) | 6 | −i | − | SC | 10-a | + | − (−) |
ST, sequence type.
Isolate ID, isolate identification.
(N), new allele that had not been previously recorded in the MLST database and was identified in Australia for the first time.
Global clonal complex (GCC) for each ST has been assigned by the S. uberis MLST database. −, does not belong to any GCC.
Disease status is indicated as follows: C, clinical mastitis; S, subclinical mastitis; L, low somatic cell count.
PFGE type and subtype are represented with Arabic numbers and letters.
PCR results for hasC and hasA genes are shown as follows: +, positive by PCR; −, negative by PCR; (+), positive by Southern blotting; (−), negative by Southern blotting.
The yqiL-negative isolates have yet to be assigned a ST by the MLST database and are indicated by their strain IDs in the ST column.
−, yqiL negative.
MLST analysis. (i) MLST analysis of Australian isolates.
Multilocus sequence typing revealed 33 STs and four yqiL-negative isolates in the 46 Australian isolates (Table 2). While five of the STs consisted of multiple isolates, the remaining 28 STs and the four yqiL-negative isolates consisted of single isolates. Simpson's index of diversity of MLST data (D = 98.6%; 95% CI, 97.1% to 100%) was comparable to that obtained using PFGE typing (D = 99.5%; 95% CI, 97.8% to 100%).
The most common GCC was GCC ST143 (26%), followed by GCC ST5 (13%) and GCC ST86 (9%); 24 of the isolates (52%) were not assigned to a specific GCC (Table 2). Thirty-one out of 33 STs, 4 yqiL-negative isolates, and 15 of the alleles had not been previously recorded in the MLST database and were identified in Australia for the first time.
The STs with the largest number of isolates were ST60 and ST155, with both consisting of four isolates (8.7%) (Table 2). All four ST60 isolates had different PFGE types (including subtypes), whereas three ST155 isolates had an identical PFGE type (PFGE type 8a), and the fourth was a different subtype (type 8b) (Table 2). The ST of four Australian ST60 isolates was identical to the ST of isolates originally identified in the United Kingdom (41). One ST184 isolate had a MLST profile identical to that of an isolate from New Zealand (42). In addition, two ST194 isolates recovered from different farms located 100 km apart in Victoria, Australia, were shown by PFGE analysis not to be clones.
(ii) Analysis of the loci of Australian isolates.
Analysis of the STs (n = 33) identified in the Australian data set (excluding the four yqiL-negative isolates, as START2 cannot perform locus analysis using partial allelic profiles) showed the dN/dS ratios for each locus were substantially <1.0, ranging from 0.000 (gki) to 0.139 (tpi). The G+C ratios for each locus ranged from 35.7% (tdk) to 45.6% (recP). The average number of alleles at each locus was 7.1, and the proportion of variable nucleotide sites in each locus ranged from 1.1% (recP) to 4.4% (tdk). The dN/dS ratios, the G+C ratios, and the proportions of variable nucleotide sites were comparable to those in a previous study (6).
(iii) Lineage analysis using UPGMA and eBURST.
The majority of the Australian isolates (85%) were resolved (<0.3 linkage distance) into three clusters (Fig. 1). The level of 0.3 linkage distance was arbitrarily set. This is not an absolute limit, but it distinguished two main clusters and an association between GCC and disease status. Cluster I (n = 31) consisted of two-thirds of the Australian isolates, and 90% of the isolates in this cluster were from cows with either clinical or subclinical mastitis. There were two possible clonal complexes in cluster I identified by eBURST analysis, and these were termed local clonal complex ST60 (LCC ST60) and ST277 (LCC ST277) (Fig. 2). ST60 was the founder of LCC ST60, which consisted of seven isolates, four single-locus variants (SLVs) and two double-locus variants, while LCC ST277 consisted of three isolates and two SLVs (Fig. 2). The majority of STs (80%) belonging to the two LCCs belonged to GCC ST5 and GCC ST143 (Fig. 1).
FIG. 1.

Dendrogram showing genetic relatedness of the 46 Australian S. uberis as determined by the unweighted pair grouping method using arithmetic means. Clusters are denoted by the bars and Roman numerals. Strains belonging to local clonal complexes are indicated with a bar and by the ST which corresponded to the founder of the LCC. The disease status is indicated as C (clinical mastitis), S (subclinical), and L (low somatic cell count). The global clonal complexes assigned to the isolates are given. The PFGE types and subtypes are represented with Arabic numbers and letters, respectively, with the number of isolates if more than one is given in parentheses. The countries of origin of previously reported STs are listed as United Kingdom (UK) and New Zealand (NZ). The yqiL-negative [yqiL (−)] isolates (which have yet to be assigned a ST by the MLST database) are indicated in the Note column and by their strain identifications in the sequence type column.
FIG. 2.
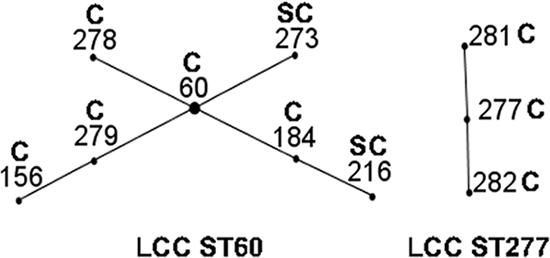
eBURST analysis of 46 Australian S. uberis isolates. The founder of a clonal complex is situated at the center of the chart. Disease status was indicated as C (clinical mastitis) and SC (subclinical mastitis) with numbers which correspond to ST. Single-locus variants are joined by lines.
In contrast, cluster III consisted of six isolates, which were mainly obtained from cows with low somatic cell counts (n = 4), and a few (n = 2) were from cows with subclinical mastitis (Fig. 1). Notwithstanding the fact that the number of isolates in cluster III was small, all of the GCC ST86 isolates identified in this study belonged to cluster III.
All six isolates in the GCC ST5 and approximately 90% of the isolates of GCC ST143 were associated with clinical and subclinical mastitis (Table 3). In contrast, none of the isolates of GCC ST86 were associated with clinical mastitis, and 75% of these isolates were associated with cows with low somatic cell counts. Chi-square analysis of the association of disease status and GCC found that it was statistically significant (P = 0.006) (Table 3).
TABLE 3.
Association between disease status and global clonal complexesa
| Disease statusb | No. (%) of isolates in GCC
|
No. (%) of isolates
|
|||
|---|---|---|---|---|---|
| ST5 | ST143 | ST86 | Otherc | Total | |
| C | 6 (100) | 4 (33) | 0 | 10 (42) | 20 (43) |
| SC | 0 | 7 (58) | 1 (25) | 8 (33) | 16 (35) |
| L | 0 | 1 (8) | 3 (75) | 6 (25) | 10 (22) |
| Total no. of isolates | 6 | 12 | 4 | 24 | 46 |
Analysis with the chi-square test showed statistical significance (P = 0.006) in the association between disease status and GCC.
C, clinical mastitis; SC, subclinical mastitis; L, low somatic cell count.
Isolates not belonging to any GCC.
eBURST analysis of Australian isolates and all isolates in the S. uberis MLST database (n = 593).
The three GCCs (ST5, ST143, and ST86) and the Australian cluster are shown by light shaded ovals in Fig. 3. Australian isolates belonging to LCCs were distributed between GCC ST5 and GCC ST143, and eBURST analysis clearly demonstrated that GCC ST86 was distantly related to GCC ST5 and GCC ST143 (Fig. 3).
FIG. 3.

Snapshot of global population structure of S. uberis generated by eBURST. The three GCCs (ST143, ST5, and ST86) and the Australian cluster are represented by light shaded ovals. The Australian isolates are indicated by numbers corresponding to their ST. The sizes of the dots are in direct proportion to the size of the population. Single-locus variants are joined by lines.
Recombination analysis. (i) Standardized index of association.
An ISA analysis performed on 42 Australian isolates (excluding the four yqiL-negative isolates) demonstrated linkage disequilibrium (ISA = 0.0981; P < 0.001). The ISA of all isolates in the S. uberis MLST database as of February 2007 was comparable (n = 593; ISA = 0.0995; P < 0.001) to that of the Australian data set.
(ii) Ratio of recombinational to mutational events and the per site ratio of recombination to mutation parameter.
All of the MLST STs available as of February 2007 in the S. uberis MLST database (n = 593) were used for this analysis. A total of 9 possible CCs and 60 SLVs were identified following BURST analysis (Fig. 4). While 59 of the SLVs arose by recombination, surprisingly, only one variant yqiL allele arose by point mutation (rate of recombinational to mutational events of 59:1) (Fig. 4; Table 4) (16). While 21 variant alleles differed at a single nucleotide position from corresponding alleles in founders of the CCs, 20 of these were assigned as arising by recombinational events, since the variant alleles were present in unrelated STs, which differed at three or more loci, in the S. uberis MLST database (Fig. 4; Table 4) (16).
FIG. 4.

Assignment of single-locus variants and clonal complexes by BURST for empirical estimation of rate of recombination to mutation. The founder of a CC is shown in the center circle, and SLVs are in the outer circle. Small circles with a black background indicate that the SLV arose by recombination, whereas a circle with a white background indicates that the SLV arose by point mutation. The numbers in the small circles show the number of nucleotide differences between a variant allele of the SLV and the corresponding allele of the founder.
TABLE 4.
Ratio of recombinational to mutational events and the per site ratio of recombination to mutation parameter
| Clonal complex | No. of SLVs arising by:
|
r/mb | No. of polymorphisms arising by:
|
Per site r/m parameterc | ||
|---|---|---|---|---|---|---|
| Recombination (r)a | Point mutation (m) | Recombination | Point mutation | |||
| ST143 | 11 (5) | 0 | 11/0 | 41 | 0 | 41/0 |
| ST233 | 8 (4) | 0 | 8/0 | 20 | 0 | 20/0 |
| ST112 | 7 (2) | 0 | 7/0 | 22 | 0 | 22/0 |
| ST86 | 7 (2) | 0 | 7/0 | 38 | 0 | 38/0 |
| ST6 | 7 (2) | 0 | 7/0 | 17 | 0 | 17/0 |
| ST241 | 5 (1) | 0 | 5/0 | 41 | 0 | 41/0 |
| ST161 | 4 (1) | 1 | 4/1 | 18 | 1 | 18/1 |
| ST89 | 5 (1) | 0 | 5/0 | 13 | 0 | 13/0 |
| ST69 | 5 (2) | 0 | 5/0 | 16 | 0 | 16/0 |
| Total | 59 (20) | 1 | 59/1 | 226 | 1 | 226/1 |
The number of SLVs that arose by recombination with a single-nucleotide change in a variant allele is shown in parentheses.
Ratio of recombinational to mutational events = total number of SLVs that arose by recombination (r)/total number of SLVs that arose by point mutation (m).
Per site r/m parameter = total number of polymorphisms that arose by recombination/total number of polymorphisms that arose by point mutation.
In the 60 SLVs, a total of 226 nucleotide sequence differences arose by recombination with only one base pair change arising by point mutation. The per site ratio of recombination to mutation parameter was 226:1.
Investigation of yqiL-negative isolates.
All of the 46 Australian isolates yielded a PCR product for six of the seven loci following PCR (6). Four isolates (686-5, 686-10, 3064-1, and 3217-3) failed to amplify the yqiL gene with the original primers (6) and with the new primers designed for this study. The new yqiL primers successfully amplified yqiL from the control S. uberis. Southern blot analysis confirmed that these isolates lacked the yqiL gene (data not shown). BLASTN and BLASTX analyses of the yqiL gene against the S. uberis genome sequence in the Sanger Institute database identified a single yqiL homologue that exhibited 38% amino acid (57/148) and 57% nucleotide sequence identity (171/299) with the S. uberis yqiL allele 1. This open reading frame (ORF) was located in the region from 1795067 bp to 1796317 bp of the genome of S. uberis strain 0140J, 400 kb distant from yqiL. On further analysis using BLASTP, the yqiL ORF homologue exhibited extensive identity with both the thiolase B gene from Oceanobacillus iheyensis (46% amino acid identity [186/404]) (52) and the acetyl coenzyme A (acetyl-CoA) acetyltransferase gene from Clostridium difficile (46% amino acid identity [186/402]) (46). A search of the Conserved Domain Database at NCBI (35) showed that the S. uberis yqiL ORF homologue and both the thiolase B and the acetyl-CoA acetyltransferase genes contained several conserved domains characteristic of enzymes involved in the synthesis of acetoacyl-CoA (cd00751) (25, 60).
Using ORF Finder (NCBI, http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and BLASTX (NCBI, http://www.ncbi.nlm.nih.gov/BLAST), a penicillin binding protein was found 15 kb upstream of the yqiL gene.
hasA and hasC PCR.
While all of the 46 Australian S. uberis isolates possessed the hasC gene, only 39 (85%) of the isolates possessed the hasA gene (Table 2). Three out of 10 hasA PCR-negative isolates and the three hasA PCR-positive control isolates were positive by Southern blotting using the hasA probe. Chi-square analysis of the association between disease status and the presence of hasA was statistically significant (P = 0.014) (Table 5). In addition, chi-square analysis of the association between GCCs and the presence of hasA was statistically significant (P = 0.005) (Table 6).
TABLE 5.
Association between disease status and the presence of hasAa
| hasA | No. (%) of isolates and disease statusb
|
Total no. (%) of isolates | ||
|---|---|---|---|---|
| C | SC | L | ||
| Present | 20 (100) | 13 (81) | 6 (60) | 39 (85) |
| Absent | 0 | 3 (19) | 4 (40) | 7 (15) |
| Total no. of isolates | 20 | 16 | 10 | 46 |
Analysis with the chi-square test showed statistical significance (P = 0.014) in the association between disease status and the presence of hasA.
C, clinical mastitis; SC, subclinical mastitis; L, low somatic cell count.
TABLE 6.
Association between the global clonal complexes and the presence of hasAa
| hasA | No. (%) of isolates in GCC
|
No. (%) of isolates
|
|||
|---|---|---|---|---|---|
| ST5 | ST143 | ST86 | Otherb | Total | |
| Present | 6 (100) | 11 (92) | 1 (25) | 21 (87.5) | 39 (85) |
| Absent | 0 | 1 (8) | 3 (75) | 3 (12.5) | 7 (15) |
| Total no. of isolates | 6 | 12 | 4 | 24 | 46 |
Analysis with the chi-square test showed statistical significance (P = 0.005) in the association between GCC and the presence of hasA.
Isolates not belonging to any GCC.
DISCUSSION
Analyses of the Australian collection of S. uberis isolates by both MLST and PFGE clearly demonstrated a genetically diverse population. While there was no particular clone of S. uberis that predominated in the Australian data set, there were a few cases of identical clones being isolated from different cows, suggesting that possible transmission between cows or acquisition from a common source did occur, but these were uncommon events. These findings are in agreement with other S. uberis epidemiological studies (1, 40, 57, 61).
The data presented in this paper strongly suggest that the isolates in cluster I, in particular those belonging to GCC ST5 and GCC ST143, were highly associated with clinical and subclinical mastitis. The dendrogram derived following UPGMA for lineage analysis (Fig. 1) clearly distinguished a cluster of isolates associated with clinical and subclinical mastitis (cluster I) from a cluster of isolates from cows with low somatic cell counts in their milk (cluster III). Ninety percent of isolates belonging to cluster I were from cows with clinical and subclinical mastitis. The vast majority (94%) of the Australian isolates belonging to the GCC ST5 and GCC ST143 were distributed in cluster I, and there was a statistically significant association between GCC type and disease status (P = 0.006) (Table 3). In addition, eBURST analysis (Fig. 2) identified two possible LCCs in cluster I, suggesting the presence of genetically closely related strains in the cluster, supporting a previous study that demonstrated low levels of heterogeneity in isolates from clinical mastitis (27). The isolates belonging to GCC ST5 and GCC ST143 may possess virulence factors promoting invasion of host tissue, survival in the host environment or evasion of the host immune response, and internalization in the mammary gland cells (53). This hypothesis was further supported by hasA analysis. In this study, 95% of the isolates in GCC ST5 and GCC ST143 possessed hasA, whereas only 25% of the isolates in GCC ST86 possessed hasA gene with chi-square analysis demonstrating a significant link between the possession of hasA and the GCC type (P = 0.005) (Table 6). These results are in agreement with previous studies (6, 42), which suggested a link between both GCC ST5s and GCC ST143s with hasA. Although hasA is not thought to be directly involved in the pathogenicity of S. uberis (17), it nevertheless appears to be a virulence marker gene.
In contrast, cluster III, which was genetically distinct from cluster I (Fig. 1), consisted of isolates mainly from cows with low somatic cell counts and comprised all of the isolates belonging to GCC ST86. This finding was further corroborated following eBURST analysis (Fig. 3). These observations suggest that isolates in GCC ST86, although forming a clonal complex in the global S. uberis population, were not associated with mastitis. Accordingly, it may be possible that these isolates may have the ability to colonize in a host environment and evade the host immune system but lack pathogenicity, behaving like a commensal species. This hypothesis is further supported by the results of a recent study which demonstrated that most of the isolates in GCC ST86 were less associated with clinical mastitis (42).
There was an inconsistency in cluster III where one isolate (ST264) belonging to GCC ST143 grouped with GCC ST86s (Fig. 1). This could be due to the assigning system used by the MLST database, which differs from UPGMA which was used in Fig. 1 (6). ST264 shares four out of seven allelic profiles with both ST143 and ST86 and therefore could be assigned either to GCC ST143 or to GCC ST86 in the database; however, ST264 also shares five of seven profiles with its nearest neighbor (ST262) and therefore should possibly be assigned to GCC ST86 by the MLST database (Fig. 1).
Australian LCCs were distributed between GCC ST5 and GCC ST143 in the global population of S. uberis (Fig. 3), suggesting that these isolates were closely associated with both GCC ST5 and ST143. This is in contrast to a previous study which demonstrated that STs identified in United Kingdom and New Zealand were clearly separated into two GCC types, GCC ST5 and ST143 (42).
ST60, the founder of LCC ST60, was initially identified in the United Kingdom and has not been found in any other country. In a similar manner, ST184 was originally identified in New Zealand, and neither of these STs has been demonstrated outside their country of origin. Accordingly, the detection of both ST60 and ST184 in our Australian data set represents the first report of the occurrence of identical STs in different countries (New Zealand) and continents (Europe). These findings support the hypothesis that ancestors of Australian S. uberis are related to isolates in the United Kingdom and New Zealand. Specifically, the eBURST results (Fig. 3) suggest ST60 or ST184 could represent a recent ancestor of Australian isolates highly associated with mastitis and may represent key pathogenic lineages in the Australian S. uberis population.
It should be noted that not all STs belonging to the three major GCCs (Table 2) are represented in Fig. 3. Five STs (ST153, ST217, ST259, ST264, and ST272) do not appear in Fig. 3. These apparent inconsistencies were due to the assigning system used by the MLST database which differs from the eBURST analysis (6, 42). To avoid any possible confusion, the classification of GCC for each ST by the MLST database was used for all analyses.
The ISA analyses for both of the Australian data set and all isolates in the MLST database demonstrated linkage disequilibrium, suggesting a clonal population structure, but the low ISA also implied that substantial recombination had occurred in the S. uberis population. A comparison of the ISA between other species demonstrated that the ISA of S. uberis was lower than that observed in Neisseria meningitidis (ISA = 0.14) and Campylobacter jejuni (ISA = 0.256) which are known for their high rate of inter- and intraspecies recombination (13, 15, 30, 50). These results are in agreement with earlier studies using maximum likelihood tree analysis which demonstrated substantial recombination in the evolutionary history (6) and significant reticulate evolution detected between the loci used for MLST (62).
In addition, empirical recombination analysis demonstrated a substantial recombination rate among the entire MLST database including our Australian data set. The ratio of recombinational to mutational events (59:1) was at least five times higher than the rate reported for N. meningitidis and Streptococcus pneumoniae (16). This finding further supports the occurrence of substantial recombination in the S. uberis population.
In a previous study, S. uberis was shown to possess phages (22), and although transduction could be responsible for recombination, the exact mechanisms of recombination in S. uberis are still unknown. However, given the substantial recombination observed in both the Australian and global S. uberis populations, it is possible that frequent recombination of genomic DNA between isolates could lead to the genetic diversity as demonstrated by the MLST and PFGE results in this and previous studies (9, 40, 57). Consequently, frequent recombination would render phylogenetic analysis using PFGE data of very little significance unless the samples were very closely related. In addition, it has been demonstrated that frequent recombination within a bacterial population decreased the accuracy of eBURST analysis for identification of links between ancestors and descendants in a population, suggesting that additional analyses should be used in conjunction with eBURST to confirm the validity of the inferred relationships (56).
The yqiL locus is commonly used in MLST assays for various bacteria including Staphylococcus aureus (10), Staphylococcus epidermidis (55), Streptococcus pyogenes (11), Enterococcus faecalis (45), and Streptococcus uberis (6). Of the four isolates in our Australian collection shown to lack yqiL, two had identical 16S rRNA gene sequences to the reference sequence, whereas the other two yqiL-negative isolates differed only at 1 or 2 nt. Phylogenetic analysis demonstrated that they were only distantly related to Streptococcus parauberis and Streptococcus iniae with S. uberis being the closest species (data not shown). It is therefore highly unlikely that the Australian yqiL-negative isolates represent different species. This was further substantiated insofar as all of the other species identification tests, such as PCRs (20), biochemical tests, and API Strep 20 performed on the yqiL-negative isolates were consistent with S. uberis.
The investigation of the flanking region of the S. uberis yqiL gene revealed a penicillin binding protein 15 kb upstream of the yqiL locus. Since this possible surface protein was more than 6 kb distant from the yqiL gene, it was clear of the standard of locus selection (7) and would not be expected to affect the selection of the yqiL locus. While the mechanisms of yqiL deletions are unknown, it is possible that given the high rate of recombination observed in the S. uberis population in both this and previous studies (6), the deletion of the yqiL gene may have occurred by recombination. This is further substantiated by the occurrence of nonstandard yqiL alleles (yqiL alleles 4, 11, and 23 which possess a 1-nt deletion within the yqiL locus) in the S. uberis MLST database. Furthermore, strains of S. pyogenes lacking yqiL have been reported (38, 43) and recorded in the S. pyogenes MLST database (http://spyogenes.mlst.net/misc/info.asp) as nonstandard alleles.
The occurrence of yqiL-negative isolates remains enigmatic, and it is still unknown how the four yqiL-negative Australian strains could survive without what is considered to be a housekeeping gene. The identification of a yqiL homologue with homology to similar genes also involved in the synthesis of acetyl-CoA from diverse species of bacteria (25, 60) suggests that our four yqiL-negative isolates might utilize such a homologue to replace yqiL function. Since yqiL is absent from some strains of S. uberis, its status as a housekeeping gene is now in doubt.
In summary, MLST analysis of Australian isolates of S. uberis has clearly identified a cluster of specific STs highly associated with clinical and subclinical mastitis and a cluster of specific STs associated with cows with low somatic cell counts. Specifically, particular groups of STs, GCC ST5 and GCC ST143, were highly associated with clinical and subclinical mastitis. It is therefore postulated that such STs may represent a lineage of virulent S. uberis. In contrast, all of the isolates belonging to GCC ST86 were associated with low-cell-count cows and may represent a less virulent ST lineage.
The use of MLST has greatly facilitated interlaboratory comparison and has identified specific STs that were initially characterized in different countries and continents but are currently present in the Australian S. uberis population. Although four of the Australian isolates were shown to lack yqiL, the current MLST assay has been of significant utility and has identified possible highly virulent STs among the genetically diverse population of S. uberis both within Australia and in the global population as a whole.
The use of MLST will greatly facilitate the identification and characterization of pathogenic S. uberis isolates and specific STs that can be exploited for further research and vaccine development.
Acknowledgments
This work was supported by the Australian Research Council (LP349000) and Dairy Australia (project RMIT 11757).
We thank the staff at the Maffra Veterinary Centre; the staff at Gippsland Vetnostics, Traralgon, Victoria, Australia; veterinarian W. F. Morgan; the dairy farmers in Timboon and Newry in Australia for the provision of milk samples; and Geoff Hogg and Angelo Zaia, Microbiological Diagnostic Unit Public Health Laboratory, University of Melbourne, for access to sequencing facilities. We acknowledge the technical assistance of Wei Gao and Elizabeth Grabsch, Microbiology Department, Austin Hospital, and we thank Ben Fry, Flinders University, for consultation.
Footnotes
Published ahead of print on 16 November 2007.
REFERENCES
- 1.Baseggio, N., P. D. Mansell, J. W. Browning, and G. F. Browning. 1997. Strain differentiation of isolates of streptococci from bovine mastitis by pulsed-field gel electrophoresis. Mol. Cell. Probes 11:349-354. [DOI] [PubMed] [Google Scholar]
- 2.Beck, H. S., W. S. Wise, and F. H. Dodd. 1992. Cost benefit analysis of bovine mastitis in the UK. J. Dairy Res. 59:449-460. [DOI] [PubMed] [Google Scholar]
- 3.Bradley, A. J. 2002. Bovine mastitis: an evolving disease. Vet. J. 164:116-128. [DOI] [PubMed] [Google Scholar]
- 4.Bramley, A. J. 1984. Streptococcus uberis udder infection—a major barrier to reducing mastitis incidence. Br. Vet. J. 140:328-335. [DOI] [PubMed] [Google Scholar]
- 5.Brightling, P., G. A. Mein, J. Malmo, and D. P. Ryan. 1998. Countdown Downunder: farm guidelines for mastitis control. Dairy Research and Development Corporation, Melbourne, Victoria, Australia. http://www.countdown.org.au/farm.htm.
- 6.Coffey, T. J., G. D. Pullinger, R. Urwin, K. A. Jolley, S. M. Wilson, M. C. Maiden, and J. A. Leigh. 2006. First insights into the evolution of Streptococcus uberis: a multilocus sequence typing scheme that enables investigation of its population biology. Appl. Environ. Microbiol. 72:1420-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curran, B., D. Jonas, H. Grundmann, T. Pitt, and C. G. Dowson. 2004. Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 42:5644-5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dairy Australia. 2006. Markets and trade. Dairy Australia, Southbank, Victoria, Australia. http://www.dairyaustralia.com.au/index.php?option=com_content&task=view&id=31&Itemid=46.
- 9.Douglas, V. L., S. G. Fenwick, D. U. Pfeiffer, N. B. Williamson, and C. W. Holmes. 2000. Genomic typing of Streptococcus uberis isolates from cases of mastitis, in New Zealand dairy cows, using pulsed-field gel electrophoresis. Vet. Microbiol. 75:27-41. [DOI] [PubMed] [Google Scholar]
- 10.Enright, M. C., N. P. J. Day, C. E. Davies, S. J. Peacock, and B. G. Spratt. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38:1008-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enright, M. C., B. G. Spratt, A. Kalia, J. H. Cross, and D. E. Bessen. 2001. Multilocus sequence typing of Streptococcus pyogenes and the relationships between emm type and clone. Infect. Immun. 69:2416-2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feil, E. J., and M. C. Enright. 2004. Analyses of clonality and the evolution of bacterial pathogens. Curr. Opin. Microbiol. 7:308-313. [DOI] [PubMed] [Google Scholar]
- 13.Feil, E. J., M. C. Enright, and B. G. Spratt. 2000. Estimating the relative contributions of mutation and recombination to clonal diversification: a comparison between Neisseria meningitidis and Streptococcus pneumoniae. Res. Microbiol. 151:465-469. [DOI] [PubMed] [Google Scholar]
- 14.Feil, E. J., B. C. Li, D. M. Aanensen, W. P. Hanage, and B. G. Spratt. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feil, E. J., M. C. J. Maiden, M. Achtman, and B. G. Spratt. 1999. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol. Biol. Evol. 16:1496-1502. [DOI] [PubMed] [Google Scholar]
- 16.Feil, E. J., J. M. Smith, M. C. Enright, and B. G. Spratt. 2000. Estimating recombinational parameters in Streptococcus pneumoniae from multilocus sequence typing data. Genetics 154:1439-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Field, T. R., P. N. Ward, L. H. Pedersen, and J. A. Leigh. 2003. The hyaluronic acid capsule of Streptococcus uberis is not required for the development of infection and clinical mastitis. Infect. Immun. 71:132-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontaine, M. C., J. Perez-Casal, X. M. Song, J. Shelford, P. J. Willson, and A. A. Potter. 2002. Immunisation of dairy cattle with recombinant Streptococcus uberis GapC or a chimeric CAMP antigen confers protection against heterologous bacterial challenge. Vaccine 20:2278-2286. [DOI] [PubMed] [Google Scholar]
- 19.Grundmann, H., S. Hori, and G. Tanner. 2001. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganisms. J. Clin. Microbiol. 39:4190-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hassan, A. A., I. U. Khan, A. Abdulmawjood, and C. Lammler. 2001. Evaluation of PCR methods for rapid identification and differentiation of Streptococcus uberis and Streptococcus parauberis. J. Clin. Microbiol. 39:1618-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haubold, B., M. Travisano, P. B. Rainey, and R. R. Hudson. 1998. Detecting linkage disequilibrium in bacterial populations. Genetics 150:1341-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill, A. W., and C. A. Brady. 1989. A note on the isolation and propagation of lytic phages from Streptococcus uberis and their potential for strain typing. J. Appl. Bacteriol. 67:425-431. [DOI] [PubMed] [Google Scholar]
- 23.Hill, A. W., and J. A. Leigh. 1989. DNA fingerprinting of Streptococcus uberis: a useful tool for epidemiology of bovine mastitis. Epidemiol. Infect. 103:165-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter, P. R., and M. A. Gaston. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 26:2465-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Igual, J. C., C. González-Bosch, J. Dopazo, and J. E. Pérez-Ortin. 1992. Phylogenetic analysis of the thiolase family. Implications for the evolutionary origin of peroxisomes. J. Mol. Evol. 35:147-155. [DOI] [PubMed] [Google Scholar]
- 26.Jayarao, B. M., B. E. Gillespie, M. J. Lewis, H. H. Dowlen, and S. P. Oliver. 1999. Epidemiology of Streptococcus uberis intramammary infections in a dairy herd. J. Vet. Med. B Infect. Dis. Vet. Public Health 46:433-442. [DOI] [PubMed] [Google Scholar]
- 27.Jayarao, B. M., E. E. Schilling, and S. P. Oliver. 1993. Genomic deoxyribonucleic acid restriction fragment length polymorphism of Streptococcus uberis: evidence of clonal diversity. J. Dairy Sci. 76:468-474. [DOI] [PubMed] [Google Scholar]
- 28.Jolley, K. A., E. J. Feil, M. S. Chan, and M. C. J. Maiden. 2001. Sequence type analysis and recombinational tests (START). Bioinformatics 17:1230-1231. [DOI] [PubMed] [Google Scholar]
- 29.Khan, I. U., A. A. Hassan, A. Abdulmawjood, C. Lammler, W. Wolter, and M. Zschock. 2003. Identification and epidemiological characterization of Streptococcus uberis isolated from bovine mastitis using conventional and molecular methods. J. Vet. Sci. 4:213-223. [PubMed] [Google Scholar]
- 30.Koehler, A., H. Karch, T. Beikler, T. F. Flemmig, S. Suerbaum, and H. Schmidt. 2003. Multilocus sequence analysis of Porphyromonas gingivalis indicates frequent recombination. Microbiology 149:2407-2415. [DOI] [PubMed] [Google Scholar]
- 31.Kossaibati, M. A., and R. J. Esslemont. 1997. The costs of production diseases in dairy herds in England. Vet. J. 154:41-51. [DOI] [PubMed] [Google Scholar]
- 32.Leigh, J. A. 1999. Streptococcus uberis: a permanent barrier to the control of bovine mastitis? Vet. J. 157:225-238. [DOI] [PubMed] [Google Scholar]
- 33.Lemee, L., I. Bourgeois, E. Ruffin, A. Collignon, J. F. Lemeland, and J. L. Pons. 2005. Multilocus sequence analysis and comparative evolution of virulence-associated genes and housekeeping genes of Clostridium difficile. Microbiology 151:3171-3180. [DOI] [PubMed] [Google Scholar]
- 34.Lemee, L., A. Dhalluin, M. Pestel-Caron, J. F. Lemeland, and J. L. Pons. 2004. Multilocus sequence typing analysis of human and animal Clostridium difficile isolates of various toxigenic types. J. Clin. Microbiol. 42:2609-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchler-Bauer, A., J. B. Anderson, P. F. Cherukuri, C. DeWeese-Scott, L. Y. Geer, M. Gwadz, S. Q. He, D. I. Hurwitz, J. D. Jackson, Z. X. Ke, C. J. Lanczycki, C. A. Liebert, C. L. Liu, F. Lu, G. H. Marchler, M. Mullokandov, B. A. Shoemaker, V. Simonyan, J. S. Song, P. A. Thiessen, R. A. Yamashita, J. J. Yin, D. C. Zhang, and S. H. Bryant. 2005. CDD: a conserved domain database for protein classification. Nucleic Acids Res. 33:D192-D196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDougall, S., T. J. Parkinson, M. Leyland, F. M. Anniss, and S. G. Fenwick. 2004. Duration of infection and strain variation in Streptococcus uberis isolated from cows' milk. J. Dairy Sci. 87:2062-2072. [DOI] [PubMed] [Google Scholar]
- 37.Mein, G., and F. Smolenaars. 2000. Making the most of the milk harvest—a prospectus for the Milk Quality and Harvesting Subprogram of the National Dairy Alliance. Dairy Research and Development Corporation, Melbourne, Victoria, Australia.
- 38.Pérez-Trallero, E., M. Montes, B. Orden, E. Tamayo, J. M. Garcia-Arenzana, and J. M. Marimón. 2007. Phenotypic and genotypic characterization of Streptococcus pyogenes isolates displaying the MLSB phenotype of macrolide resistance in Spain, 1999 to 2005. Antimicrob. Agents Chemother. 51:1228-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petrovski, K. R., M. Trajcev, and G. Buneski. 2006. A review of the factors affecting the costs of bovine mastitis. J. S. Afr. Vet. Assoc. 77:52-60. [DOI] [PubMed] [Google Scholar]
- 40.Phuektes, P., P. D. Mansell, R. S. Dyson, N. D. Hooper, J. S. Dick, and G. F. Browning. 2001. Molecular epidemiology of Streptococcus uberis isolates from dairy cows with mastitis. J. Clin. Microbiol. 39:1460-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pullinger, G. D., T. J. Coffey, M. C. Maiden, and J. A. Leigh. 2007. Multilocus-sequence typing analysis reveals similar populations of Streptococcus uberis are responsible for bovine intramammary infections of short and long duration. Vet. Microbiol. 119:194-204. [DOI] [PubMed] [Google Scholar]
- 42.Pullinger, G. D., M. Lopez-Benavides, T. J. Coffey, J. H. Williamson, R. T. Cursons, E. Summers, J. Lacy-Hulbert, M. C. Maiden, and J. A. Leigh. 2006. Application of Streptococcus uberis multilocus sequence typing: analysis of the population structure detected among environmental and bovine isolates from New Zealand and the United Kingdom. Appl. Environ. Microbiol. 72:1429-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson, D. A., J. A. Sutcliffe, W. Tewodros, A. Manoharan, and D. E. Bessen. 2006. Evolution and global dissemination of macrolide-resistant group A streptococci. Antimicrob. Agents Chemother. 50:2903-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rozen, S., and H. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365-386. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz-Garbajosa, P., M. J. M. Bonten, D. A. Robinson, J. Top, S. R. Nallapareddy, C. Torres, T. M. Coque, R. Canton, F. Baquero, B. E. Murray, R. del Campo, and R. J. L. Willems. 2006. Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J. Clin. Microbiol. 44:2220-2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sebaihia, M., B. W. Wren, P. Mullany, N. F. Fairweather, N. Minton, R. Stabler, N. R. Thomson, A. P. Roberts, A. M. Cerdeno-Tarrraga, H. W. Wang, M. T. G. Holden, A. Wright, C. Churcher, M. A. Quail, S. Baker, N. Bason, K. Brooks, T. Chillingworth, A. Cronin, P. Davis, L. Dowd, A. Fraser, T. Feltwell, Z. Hance, S. Holroyd, K. Jagels, S. Moule, K. Mungall, C. Price, E. Rabbinowitsch, S. Sharp, M. Simmonds, K. Stevens, L. Unwin, S. Whithead, B. Dupuy, G. Dougan, B. Barrell, and J. Parkhill. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38:779-786. [DOI] [PubMed] [Google Scholar]
- 47.Smith, C. L., and C. R. Cantor. 1987. Purification, specific fragmentation, and separation of large DNA molecules. Methods Enzymol. 155:449-467. [DOI] [PubMed] [Google Scholar]
- 48.Smith, J. M., N. H. Smith, M. Orourke, and B. G. Spratt. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. USA 90:4384-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spratt, B. G., W. P. Hanage, B. Li, D. M. Aanensen, and E. J. Feil. 2004. Displaying the relatedness among isolates of bacterial species—the eBURST approach. FEMS Microbiol. Lett. 241:129-134. [DOI] [PubMed] [Google Scholar]
- 50.Suerbaum, S., M. Lohrengel, A. Sonnevend, F. Ruberg, and M. Kist. 2001. Allelic diversity and recombination in Campylobacter jejuni. J. Bacteriol. 183:2553-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swinkels, J. M., J. G. Rooijendijk, R. N. Zadoks, and H. Hogeveen. 2005. Use of partial budgeting to determine the economic benefits of antibiotic treatment of chronic subclinical mastitis caused by Streptococcus uberis or Streptococcus dysgalactiae. J. Dairy Res. 72:75-85. [DOI] [PubMed] [Google Scholar]
- 52.Takami, H., Y. Takaki, and I. Uchiyama. 2002. Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res. 30:3927-3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamilselvam, B., R. A. Almeida, J. R. Dunlap, and S. P. Oliver. 2006. Streptococcus uberis internalizes and persists in bovine mammary epithelial cells. Microb. Pathog. 40:279-285. [DOI] [PubMed] [Google Scholar]
- 54.Tankouo-Sandjong, B., A. Sessitsch, E. Liebana, C. Kornschober, F. Allerberger, H. Hachler, and L. Bodrossy. 2007. MLST-v, multilocus sequence typing based on virulence genes, for molecular typing of Salmonella enterica subsp. enterica serovars. J. Microbiol. Methods 69:23-36. [DOI] [PubMed] [Google Scholar]
- 55.Thomas, J. C., M. R. Vargas, M. Miragaia, S. J. Peacock, G. L. Archer, and M. C. Enright. 2007. Improved multilocus sequence typing scheme for Staphylococcus epidermidis. J. Clin. Microbiol. 45:616-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turner, K. M., W. P. Hanage, C. Fraser, T. R. Connor, and B. G. Spratt. 2007. Assessing the reliability of eBURST using simulated populations with known ancestry. BMC Microbiol. 7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang, S. M., M. A. Deighton, J. A. Capstick, and N. Gerraty. 1999. Epidemiological typing of bovine streptococci by pulsed-field gel electrophoresis. Epidemiol. Infect. 123:317-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watson, D. L., M. L. McColl, and H. I. Davies. 1996. Field trial of a staphylococcal mastitis vaccine in dairy herds: clinical, subclinical and microbiological assessments. Aust. Vet. J. 74:447-450. [DOI] [PubMed] [Google Scholar]
- 59.Wells, S. J., S. L. Ott, and A. H. Seitzinger. 1998. Key health issues for dairy cattle—new and old. J. Dairy Sci. 81:3029-3035. [DOI] [PubMed] [Google Scholar]
- 60.Yang, S. Y., X. Y. Yang, G. Healy-Louie, H. Schulz, and M. Elzinga. 1990. Nucleotide sequence of the fadA gene. Primary structure of 3-ketoacyl-coenzyme A thiolase from Escherichia coli and the structural organization of the fadAB operon. J. Biol. Chem. 265:10424-10429. [PubMed] [Google Scholar]
- 61.Zadoks, R. N., B. E. Gillespie, H. W. Barkema, O. C. Sampinion, S. P. Oliver, and Y. H. Schukken. 2003. Clinical, epidemiological and molecular characteristics of Streptococcus uberis infections in dairy herds. Epidemiol. Infect. 130:335-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zadoks, R. N., Y. H. Schukken, and M. Wiedmann. 2005. Multilocus sequence typing of Streptococcus uberis provides sensitive and epidemiologically relevant subtype information and reveals positive selection in the virulence gene pauA. J. Clin. Microbiol. 43:2407-2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zadoks, R. N., L. L. Tikofsky, and K. J. Boor. 2005. Ribotyping of Streptococcus uberis from a dairy's environment, bovine feces and milk. Vet. Microbiol. 109:257-265. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, W., B. M. Jayarao, and S. J. Knabel. 2004. Multi-virulence-locus sequence typing of Listeria monocytogenes. Appl. Environ. Microbiol. 70:913-920. [DOI] [PMC free article] [PubMed] [Google Scholar]