

Abstract
The transforming growth factor β (TGFβ) family has critical roles in the regulation of fertility. In addition, the pathogenesis of some human cancers is attributed to misregulation of TGFβ function and SMAD2 or SMAD4 mutations. There are limited mouse models for the BMP signaling SMADs (BR-SMADs) 1, 5, and 8 because of embryonic lethality and suspected genetic redundancy. Using tissue-specific ablation in mice, we deleted the BR-SMADs from somatic cells of ovaries and testes. Single conditional knockouts for Smad1 or Smad5 or mice homozygous null for Smad8 are viable and fertile. Female double Smad1 Smad5 and triple Smad1 Smad5 Smad8 conditional knockout mice become infertile and develop metastatic granulosa cell tumors. Male double Smad1 Smad5 conditional knockout mice are fertile but demonstrate metastatic testicular tumor development. Microarray analysis indicated significant alterations in expression of genes related to the TGFβ pathway, as well as genes involved in infertility and extracellular matrix production. These data strongly implicate the BR-SMADs as part of a critical developmental pathway in ovaries and testis that, when disrupted, leads to malignant transformation.
The transforming growth factor β (TGFβ) family regulates cell proliferation and differentiation in many developmental and physiological processes, including reproduction (39). When TGFβ family function is disrupted, a number of reproductive phenotypes result. These include infertility, germ cell loss, or, less commonly, tumor formation. For example, genetic deletion of the inhibin α subunit in mice results in sex cord stromal tumors and death through a cancer cachexia-like disease (10, 31). Deletion of follistatin (a negative regulatory protein of activin) or anti-Müllerian hormone (AMH/MIS) results in premature ovarian failure (14, 25), and loss of ovarian activin leads to infertility (37). Less is known about the in vivo contribution of the BMP subgroup in adult reproductive physiology because mutations in most BMP signaling components result in embryonic lethality or primordial germ cell loss (7).
The TGFβ family signals through cell surface serine-threonine kinase receptor complexes that phosphorylate intracellular SMAD transcription factors (30). TGFβ, activin, and nodal receptors signal through SMAD2 or SMAD3 (termed AR-SMADs), while BMP receptors signal through SMAD1, SMAD5, or SMAD8 (termed BR-SMADs) (15). Once phosphorylated, R-SMADs form complexes with SMAD4 and activate or repress target gene transcription in association with a number of cofactors (30). Loss-of-function mutations in TGFβ receptors, SMAD2, or SMAD4 are found in a minority of cancer types, such as human pancreatic and gastrointestinal tumors (13, 29). Other cancers exploit the TGFβ signaling pathway to promote tissue invasion and metastasis (44). TGFβ function is cell type dependent, and the mechanism by which TGFβ functions as both a tumor suppressor and an oncogene in carcinogenesis is unclear (40). Whether signaling by members of the BMP subfamily also displays this duality (i.e., loss of function in some instances and gain of function in others) in tumor development is unknown.
Some studies have suggested a role for the BMP pathway in human cancer. Mutations in the BMP type I receptor BMPR1A are implicated in the pathogenesis of juvenile polyposis syndrome and lead to an increase in malignant potential (20, 23). Mice with tissue-specific inactivation of Bmpr1a also develop intestinal polyposis (18). Additionally, in vitro, BMPs have growth-suppressive effects on normal and some cancer cell lines that have intact BMP signaling components (2, 5, 33, 42), and the BMP antagonist gremlin has been shown to promote tumor cell proliferation (50). Recently, the BMP signaling pathway was demonstrated to be a key inhibitory regulator of tumor-initiating cells of human glioblastomas (41). However, definitive in vivo evidence for a tumor-suppressive role of the BR-SMAD pathway tissue is lacking (40).
Functional differences between some of the BR-SMADs have not been established (34). Null mutations in Smad1 or Smad5 give embryonic lethal phenotypes in mice, but Smad8 homozygous null mice are viable and fertile (1; Z. Huang, D. Wang, K. Ihida-Stansbury, P. L. Jones, and J. F. Martin, submitted for publication). Embryonic lethality also occurs in Smad1+/− Smad5+/− double heterozygous embryos (1). The study herein was designed to ablate BR-SMADs in gonadal somatic cells to examine the function of the BMP pathway in reproduction. Here we show that Cre-mediated genetic ablation of either Smad1 or Smad5 in ovarian granulosa cells results in normal reproductive function but that combined loss of Smad1 and Smad5 results in fertility defects and granulosa cell tumors. Unlike other mouse models that develop sex cord stromal tumors, female Smad1 Smad5 double conditional knockout (dKO) mice develop peritoneal and lymphatic metastases, uncovering a novel regulatory role in this process for the BR-SMADs. Male Smad1 Smad5 dKO mice also develop metastatic testicular tumors. These data strongly implicate SMAD1 and SMAD5 as critical tumor suppressors with redundant functions.
MATERIALS AND METHODS
Mouse strains and breeding.
The Smad1 conditional allele (Smad1RobPC) (designated Smad1flox throughout) and the Smad1 null allele (Smad1Robcn) (designated Smad1− throughout) have been described elsewhere (53) (Fig. 1A). The Smad5 null allele (Smad5tm1Zuk) (designated Smad5− throughout) was generated previously (8). The Smad5 conditional allele (Smad5fle2) (designated Smad5flox throughout) has been described previously (54). The Smad8 null allele was generated by deletion of exons 4 and 5, which code for most of the MH2 domain that functions in receptor binding and contains the SSXS phosphorylation site (Huang et al., submitted). Mice in this study were maintained on a C57BL/6J;129S5/SvEvBrd mixed hybrid background. For readability, genotypes are not written in standard order (i.e., by chromosome number); the correct order is Smad8 (chromosome 3), Smad1 (chromosome 8), Amhr2 (chromosome 12), and Smad5 (chromosome 13). Mutant mice and their genotypes that were analyzed in the manuscript are listed in Table 1. When possible, mice were generated with three floxed alleles and one null allele (i.e., Smad1flox/− Smad5flox/flox and Smad1flox/flox Smad5flox/−) for Smad1 and Smad5 to increase the likelihood of complete recombination. However, Smad1+/− Smad5+/− and Smad1flox/− Smad5flox/− mice cannot be generated, due to embryonic lethality (1). Mice were genotyped from genomic tail DNA by use of PCR primers as described previously (8, 53, 54; Huang et al., submitted) and by Southern blot analysis (22). For analysis of recombination in granulosa cells, genomic DNA was prepared by standard protocols from preovulatory stage granulosa cells as described previously (38). Expression of cre from the Amhr2 locus in the ovary and testis has been validated previously (24, 25). All experimental animals were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
FIG. 1.

Generation of single, double, and triple conditional knockouts. (A) Schematic representation depicting floxed and null alleles for Smad1 (Smad1RobPC and Smad1Robcn, respectively) (53), floxed and null alleles for Smad5 (Smad5fle2 and Smad5tm1Zuk, respectively) (8, 54), and a null allele for Smad8 (Huang et al., submitted). Only a portion of each genomic locus is shown. In both Smad1 and Smad5, loxP sites (open triangles) flank exon 2 (boxed), the first coding exon, which when recombined produces a null allele. The Smad8 null allele deletes exons 4 and 5, which encode the majority of the MH2 domain. PGK phosphoglycerate kinase promoter; hprt, hypoxanthine-guanine phosphoribosyl transferase minigene. (B) Genotyping of single, double, and triple control and conditional knockouts by PCR. Each lane represents one mouse of eight possible genotypes. Representative genotyping of genomic tail DNA is shown, with expected sizes of PCR products as indicated. (C) Recombination of floxed (fl) alleles in genomic DNA from isolated granulosa cells of cre-negative (neg) and cre-positive (pos) Smad1flox/flox Smad5flox/flox 21-day-old mice. When crossed to Amhr2cre/+ mice, the deleted allele (Δ) is detected in granulosa cells. Because four independent recombinations have to occur in dKO (Smad1flox/flox Smad5flox/flox Amhr2cre/+) mice, granulosa cells isolated from young mice are mosaic for the deletions and show variable levels of recombination between mice. Each lane represents one mouse.
TABLE 1.
List of mutant mouse lines analyzed for Smad1, Smad5, and Smad8
| Designation | Genotype | Control strain | Fertility defecta | Tumor developmentb |
|---|---|---|---|---|
| Smad1 cKO | Smad1flox/−Amhr2cre/+ | Smad1flox/− | No | No |
| Smad5 cKO | Smad5flox/−Amhr2cre/+ | Smad5flox/− | No | No |
| Smad8 null | Smad8−/− | WT or Smad8+/− | No | No |
| Smad5 Smad8 dKO | Smad5flox/−Smad8−/−Amhr2cre/+ | Smad5+/−Smad8−/− | No | No |
| Smad1 Smad5 dKO | Smad1flox/−Smad5flox/floxAmhr2cre/+ | Smad1flox/−Smad5flox/flox | Yes | Yes |
| Smad1flox/floxSmad5flox/−Amhr2cre/+ | Smad1flox/floxSmad5flox/− | Yes | Yes | |
| Smad1flox/floxSmad5flox/floxAmhr2cre/+ | Smad1flox/floxSmad5flox/flox | Yes | Yes | |
| Smad1 Smad5 Smad8 tKO | Smad1flox/−Smad5flox/floxSmad8−/−Amhr2cre/+ | Smad1flox/−Smad5flox/floxSmad8−/− | Yes | Yes |
| Smad1flox/floxSmad5flox/−Smad8−/−Amhr2cre/+ | Smad1flox/floxSmad5flox/−Smad8−/− | Yes | Yes | |
| Smad1flox/floxSmad5flox/floxSmad8−/−Amhr2cre/+ | Smad1flox/floxSmad5flox/floxSmad8−/− | Yes | Yes |
Fertility defect of the mutant strain is given for female mice.
Gonadal tumor development is indicated for both male and female mutant mice.
Fertility analysis.
Fertility analysis was performed as described previously (38). Mutant mice were compared to control littermates of the same genotype that were negative for the cre allele. For example, Smad1flox/flox Smad5flox/flox served as the controls for Smad1flox/flox Smad5flox/flox Amhr2cre/+, etc. (Table 1). Each mutant or control genotype was monitored over a 6-month period while mated to known fertile wild-type (WT) mice. All breeding cages were set up when the mice reached sexual maturity (6 weeks of age for females; 8 weeks of age for males), and numbers of pups per litter were recorded. Mice were sacrificed when they showed signs of infertility (missed three litters) or developed visible tumors. Mice were monitored weekly for tumor development and were sacrificed if they showed signs of distress.
Tissue collection.
For necropsies, mice were anesthetized by isoflurane inhalation and then euthanized by cervical dislocation. Organs were dissected and fixed in 10% neutral-buffered formalin for histology or stored in RNAlater (Ambion) at −80°C until processed for RNA.
Histology and immunohistochemistry.
Tissue embedding, sectioning, and staining were performed by the Department of Pathology Core Facility (Baylor College of Medicine) using standard techniques. Histological sections were cut at 4 to 6 μm and stained with hematoxylin and eosin. Immunohistochemistry was performed as previously described (38), using formalin-fixed, paraffin-embedded sections. Immunohistochemistry was performed using the Vectastain ABC method (Vector Laboratories). Rabbit polyclonal anti-phospho-Smad2/3 (1:300; Cell Signaling Technology) and rabbit polyclonal anti-inhibin α (1:500; a gift of W. Vale, Salk Institute) antibodies were used. Troma-I (anti-cytokeratin 8) (1:50) and Troma-III (anti-cytokeratin 19) (1:10) rat monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences. Immunohistochemistry was performed on at least five samples in duplicate. Immunoreactivity was visualized by diaminobenzidine tetrahydrochloride staining, and the tissue was counterstained in hematoxylin.
Granulosa cell cultures and ligand treatment.
Collection and treatment of WT granulosa cells were performed as previously described (36). CD1 female mice 19 to 21 days old were injected intraperitoneally with 5 IU pregnant mare serum gonadotropins (Calbiochem), and granulosa cells were collected 44 to 46 h later by puncturing antral follicles, plated at a density of 5.5 × 105 cells/ml, and treated immediately upon harvest in Dulbecco's modified Eagle's medium-F-12 medium supplemented with 0.5% heat-inactivated fetal bovine serum and 10 U/ml penicillin and streptomycin. Cells were treated for 5 h and then harvested for RNA. BMP4 was a gift from Wyeth.
RNA analysis.
RNA was extracted using a Qiagen RNeasy micro kit. cDNA was prepared from 200 ng of total RNA by use of a SuperScript III first-strand synthesis kit (Invitrogen) in a 50-μl reaction. Real-time quantitative PCR (qPCR) was performed as previously described (38) by using an Applied Biosystems (ABI) Prism 7500 sequence detection system, TaqMan master mix, and predesigned gene expression assays. Data were analyzed by the ΔΔCT method (where CT is cycle threshold) using ABI 7500 system software (v1.2.3) as described by ABI and normalized to the endogenous reference (Gapd). The mean ΔCT of the control samples was used as the calibrator sample. The averages and standard errors of the means were calculated, and the relative amount of target gene expression for each sample compared to the control was plotted in Excel (Microsoft).
Microarray analysis.
RNA was extracted by use of a Qiagen RNAeasy kit, and RNA integrity, concentration, and quality were checked by the Baylor College of Medicine Microarray Core Facility. Labeling, hybridization, washing, scanning, and initial analysis were performed by the Baylor College of Medicine Microarray Core Facility using standard Affymetrix protocols and an Affymetrix mouse genome 430 2.0 array. Six chips were analyzed from three independent pools of WT granulosa cell samples (each pool was collected from two to three females) and three independent tumor samples (two samples of Smad1flox/flox Smad5flox/flox Smad8+/− Amhr2cre/+ and one sample of Smad1flox/flox Smad5flox/flox Smad8−/− Amhr2cre/+). Affymetrix CEL files were imported into GeneSpring GX (Agilent), and GC robust multiarray average was used to perform background correction and normalization. Differential gene expression was filtered using the volcano plot filter in GeneSpring, with a P value of <0.001 and a minimum twofold change. Forty-three genes would have been expected to change by chance alone. Probe sets that were considered to be “absent” in four of six samples or that had a signal intensity of less than 150 under both conditions, as determined by Affymetrix GCOS software, were removed from the gene lists. This resulted in 354 differentially expressed probe sets, which, by taking into account multiple probe sets for the same gene, corresponded to 153 genes upregulated at least twofold and 171 genes downregulated at least twofold. Gene lists were imported into DAVID bioinformatic software (http://david.abcc.ncifcrf.gov/home.jsp) for KEGG pathway analysis (12).
GSEA.
Gene set enrichment analysis (GSEA) (52) was performed using GSEA-P v2 with the default parameters (http://www.broad.mit.edu/gsea/), except that the median intensity of probes corresponding to the same gene was used to collapse probe sets. Gene sets were exported from database C2 of MSigDB v2.1 (http://www.broad.mit.edu/gsea/msigdb/index.jsp) (52), and only canonical pathways from BioCarta, Gene Ontology, GenMAPP (www.genmapp.org), Signaling Transduction KE, Signaling Alliance, and Sigma Aldrich were used. Of the 456 canonical gene sets, only 337 passed the selection criteria of a minimum size of 10 and maximum size of 500. This resulted in one gene set that was enriched in WT compared to tumor cells and one gene set enriched in tumor cells compared to WT cells by use of a false discovery rate (q < 0.25, as recommended).
Statistical analysis.
Statistical analysis was performed with JMP v5.1 (SAS Institute, Inc.) or MATLAB (Mathworks) statistical packages. Fertility data were tested using the Kruskal-Wallis analysis of ranks test for multiple comparisons and Tukey's least significant difference test for post hoc multiple comparisons to identify statistically different groups. Parametric data (qPCR data) were analyzed by Student's t test (for single comparisons) or one-way analysis of variance (for multiple comparisons) followed by a Tukey-Kramer honestly significant difference post hoc test. Parametric data are shown as means, with standard errors of the means as error bars. Statistical significance was set at α = 0.05. A minimum of three independent experiments was used for statistical comparisons.
Nucleotide sequence accession number.
The microarray data set has been deposited at NIH Gene Expression Omnibus under the series accession number GSE8156 (http://www.ncbi.nlm.nih.gov/projects/geo).
RESULTS
Absence of Smad1, Smad5, or Smad8 in somatic cells of the gonads does not affect fertility.
We generated single, gonad-specific knockouts of Smad1 and Smad5 by crossing Smad1flox/flox or Smad5flox/flox mice to mice heterozygous for each respective null allele (Smad1+/− or Smad5+/−) (8, 53, 54) (Fig. 1 and Table 1) that also expressed cre recombinase from the anti-Müllerian hormone receptor type II (Amhr2) locus (22). Amhr2cre/+ mice express cre recombinase predominantly in granulosa cells of developing ovarian follicles (4, 25, 38) and Sertoli and Leydig cells of the testis in males (24). To control for potential genetic background effects, comparisons were made between siblings of the same genotype, except for the presence or absence of cre (e.g., between Smad1flox/− and Smad1flox/− Amhr2cre/+ littermates), as well as between lines (e.g., between Smad1flox/− Amhr2cre/+ and Smad5flox/− Amhr2cre/+ mice) (Fig. 2; Table 1). Heterozygous mice (Smad1+/− or Smad5+/−), Smad8−/− mice, and Smad1 or Smad5 conditional knockout (Smad1 cKO or Smad5 cKO) mice were viable and fertile (Fig. 2A and Table 1).
FIG. 2.
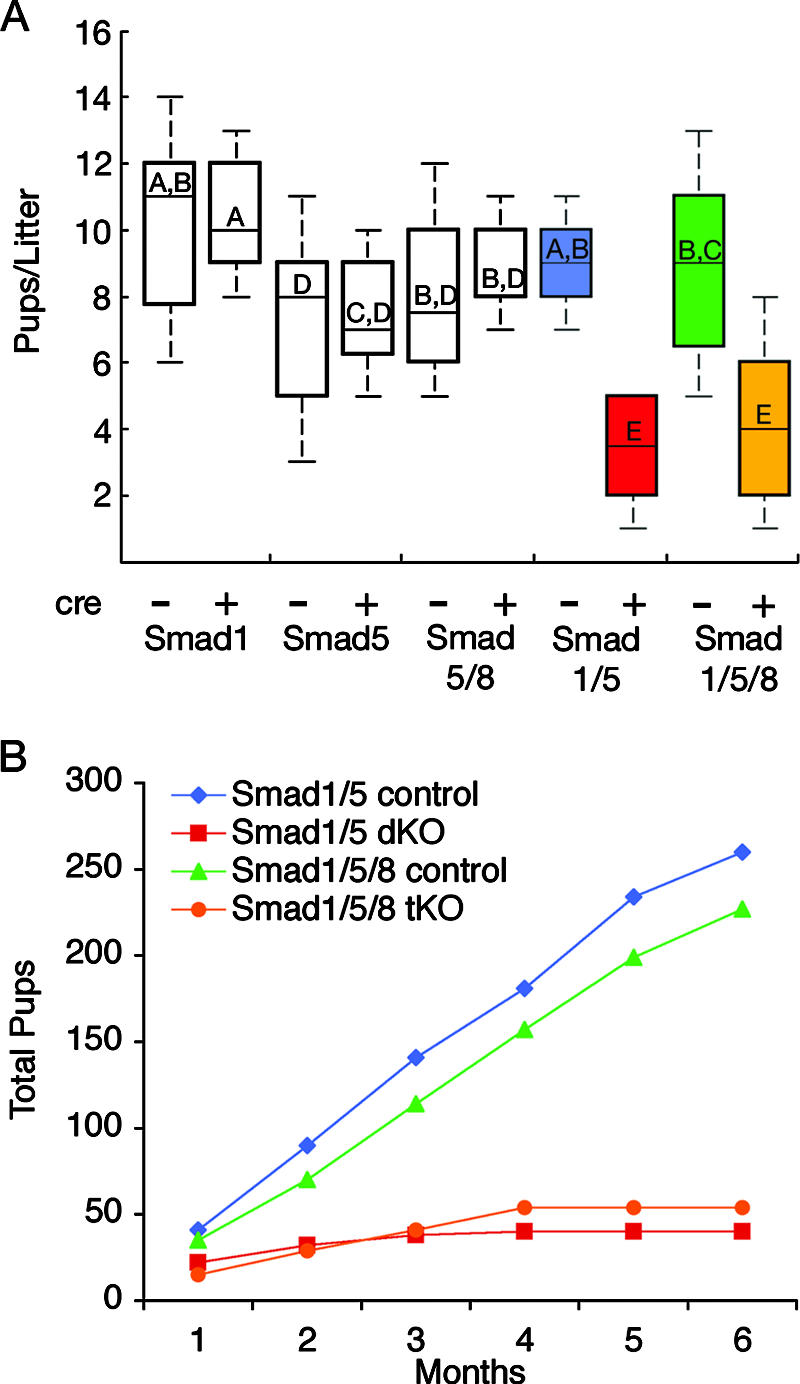
Fertility analysis of Smad1, Smad5, and Smad8 female cKO mice. (A) Adult female mice of the indicated genotypes (cre negative [−] and cre positive [+]) at 6 weeks of age were bred to WT males, and the numbers of pups were recorded for 6 months. Data are shown as median pups per litter, with the upper and lower edges of the boxes equal to the 25% and 75% quartiles and whiskers the 5% and 95% quartiles. Mice with single conditional knockouts for Smad1 (cre negative, n = 5; cre positive, n = 6) or Smad5 (cre negative, n = 10; cre positive, n = 4) or dKO for Smad5 Smad8 (controls are Smad5+/− Smad8−/−, n = 4, compared to Smad5flox/− Smad8−/− Amhr2cre/+, n = 3) have litter sizes and reproductive life spans similar to those for their control littermates. Female Smad1 Smad5 dKO (cre positive, n = 6) and Smad1 Smad5 Smad8 tKO (cre positive, n = 6) mice are significantly different from their control littermates (P < 0.001) (cre negative, n = 5 and n = 6 for dKO and tKO mice, respectively). There is no statistical difference between the numbers of Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO median pups/litter, indicating that loss of Smad8 has no additional effect on fertility. Letters written above the median line for each group indicate statistical significance by Kruskal-Wallis analysis of ranks test, followed by Tukey's post hoc test. Boxes not connected by the same letter are statistically different; for example, “A” is statistically different from “E” but not “A,B.” (B) Female Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice become infertile with age. Total accumulated pups were counted from five breeding pairs for each genotype over 6 months. The dKO and tKO mice are similar in their breeding profiles. Infertility was evident in dKO and tKO mice after 3 to 4 months of breeding.
Female Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice develop fertility defects and metastatic granulosa cell tumors.
To test for redundancy between the different BR-SMADs, we generated Smad1 Smad5 dKO mice, Smad5 Smad8 dKO mice, and a Smad1 Smad5 Smad8 triple conditional knockout (tKO) mouse. Smad5 Smad8 dKO mice were viable and fertile (Fig. 2). In contrast, Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO female mice became infertile after 3 to 4 months of breeding (Fig. 2B). Four out of 10 Smad1 Smad5 dKO female mice in our initial breeding study died before 24 weeks, while all of the control mice were viable. Upon necropsy, Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO females demonstrated unilateral or bilateral ovarian tumors (Fig. 3). Before 8 weeks, the ovaries, uteri, and oviducts were grossly normal. After 3 months, 100% of the Smad1 Smad5 dKO (15/15) and Smad1 Smad5 Smad8 tKO (15/15) female mice showed evidence of tumors. Histological analysis revealed that the tumors were poorly differentiated granulosa cell tumors, which were positive for inhibin α (Fig. 3) and negative for cytokeratins (data not shown). Aging Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice also demonstrated peritoneal metastases (Fig. 4 and Table 2) that were histologically identical to the primary granulosa cell tumor, with strong inhibin α immunoreactivity (Fig. 4E). Tumors spread to the lymph nodes, and 6 of 12 knockout females >9 months old developed hemorrhagic ascites. Smad1 and Smad5 recombined alleles were detected exclusively in the metastases, demonstrating that extraovarian tumors derived from null cells (Fig. 4F). No tumors were found in control mice or in 1-year-old Smad1 Smad5 dKO ovariectomized female mice (n = 8). Female Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice had similar fertility defects and tumor developments, supporting studies with mouse embryos that demonstrate a strong genetic interaction between Smad1 and Smad5 but not with Smad8−/− mice (1). Therefore, subsequent studies were carried out with only Smad1 Smad5 dKO mice.
FIG. 3.

Tumor development in Smad1 Smad5 and Smad1 Smad5 Smad8 female cKO mice. (A) Reproductive tract from an infertile 5.5-month-old Smad1flox/flox Smad5flox/− Amhr2cre/+ dKO mouse. The left ovary (ov) and oviduct (ovi) and both uterine (ut) horns have normal morphology. A large tumor is seen on the right. Bar, 1 cm. (B) Bilateral tumor development in an infertile 8-month-old Smad1flox/flox Smad5flox/− Smad8+/− Amhr2cre/+ dKO female mouse. Bar, 1 cm. (C) Ovary from a 32-week-old Smad1flox/flox Smad5flox/flox Smad8−/− Amhr2cre/+ tKO mouse. Most of the ovary is replaced by tumor cells, with few remaining follicles. A higher magnification is shown as panel D, demonstrating tumor (tu) cells and a single secondary follicle (sf). Bar, 100 μm. (E) Inhibin α immunoreactivity in an ovary from a 17-week-old control (Smad1flox/flox Smad5flox/flox Smad8−/−) mouse. Positive staining (brown) is limited predominantly to follicular granulosa cells, and the oviduct is negative. af, antral follicle; bu, ovarian bursa. Bar, 50 μm. (F) Ovary from a 17-week-old tKO (Smad1flox/flox Smad5flox/flox Smad8−/− Amhr2cre/+) mouse shows extensive inhibin α immunoreactivity in the granulosa cells, ovarian stroma, tumor tissue from the ovary (tu), and small patches in the oviduct (arrowheads), which likely represent growth of the tumor from the ovary into the oviduct. gr, granulosa cells. Bar, 50 μm.
FIG. 4.

Intraperitoneal metastases in female cKO mice. (A) The percentage of knockout female mice with visible metastases in the peritoneum increases with age. The number of mice in each group is as follows: 0 to 12 weeks, n = 11; 13 to 24 weeks, n = 9; 25 to 36 weeks, n = 15; and 37 to 50 weeks, n = 4. Data are from both dKO and tKO mice. (B) Three metastases to the diaphragm of an 8-month-old Smad1 Smad5 Smad8 tKO female. Bar, 5 mm. The larger growth was dissected and is shown by hematoxylin and eosin histology in panel D. Bar in panel D, 80 μm. (C) Metastases (arrows) to the spleen in an 8-month-old Smad1flox/flox Smad5flox/flox Smad8+/− Amhr2cre/+ dKO female. Bar, 5 mm. (E) Eight-month-old Smad1 Smad5 Smad8 tKO mouse with a metastatic lesion invading the liver. The tumor shows strong inhibin α-positive staining (brown staining, indicated by arrows), while the liver is negative. Bar, 200 μm. (F) PCR analysis of Smad1 and Smad5 alleles in genomic DNA from isolated metastases shows complete recombination (Δ) of the floxed alleles. Two different mice are represented, and genomic tail DNA (tail) is shown as unrecombined floxed allele controls. dia, diaphragm; om, omentum.
TABLE 2.
Locations of visible metastases in Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO female mice
| Tissue(s) | No. of mice showing metastasisa |
|---|---|
| Diaphragm | 4 |
| Liver | 3 |
| Mesentery, intestine | 3 |
| Pancreas | 3 |
| Spleen | 3 |
| Lymph nodes | 3 |
| Unknown peritoneal | 2 |
| Stomach (fascia) | 2 |
| Uterus (serosa) | 2 |
| Kidney | 1 |
| Small intestine | 1 |
| Total | 13 |
The number of mice with visible metastases in each organ is shown, of 13 mice examined (“Total”) (7 Smad1 Smad5 dKO mice and 6 Smad1 Smad5 Smad8 tKO mice) demonstrating metastatic tumor growth. Typically, in mice with metastases, more than one organ in each mouse is involved.
Male Smad1 Smad5 dKO mice are fertile but develop metastatic Sertoli-Leydig tumors.
Male Smad1 Smad5 dKO mice were fertile and, when bred to WT females, produced litters that were similar in size to those from control males (8.6 ± 0.7 [n = 8] versus 8.9 ± 0.9 [n = 4], respectively). Signs of a mild fertility defect, detected as irregularly spaced litters in 3/8 breeding pairs, appeared after 3 months of age, but this defect resulted in only a 16% decrease in litters per month (0.76 ± 0.45 litters per month for Smad1 Smad5 dKO mice, compared to an average of 0.9 ± 2.3 typically obtained in the mixed hybrid background of our mouse colonies). Thus, unlike results for female Smad1 Smad5 dKO mice, the overall ability of male mice to produce normal-sized litters was not disrupted (i.e., they were not initially subfertile). However, three males in the Smad1 Smad5 breeding pairs died of unknown causes before 6 months of age, and two of these had bilateral testicular tumors. After 28 weeks of age, all male Smad1 Smad5 dKO mice (14/14) showed gross tumor development in the testes. Tumor development occurred mostly focally (Fig. 5B and C), invading the tunica albuginea and implanting outside the testis. Less commonly, tumor cells were isolated within seminiferous tubules (data not shown). None of the control male mice developed tumors. Early-stage tumors from Smad1 Smad5 dKO males appeared to be sex cord stromal tumors with Sertoli and Leydig cell differentiation (Fig. 5C and D).
FIG. 5.

Gross morphology and histology of testicular tumors in male Smad1 Smad5 dKO mice. (A) Normal testis morphology in a 3-month-old Smad1 Smad5 dKO male. Before 12 weeks of age, only 50% of mutant males have visible tumors. epi, head of epididymis; tail, tail of epididymis; te, testis; vd, vas deferens. Bar, 2 mm. (B) Tumor development in a 14-week-old Smad1 Smad5 dKO male. The tumor is located at one pole of the testis and extends outward, implanting on the capsule. Bar, 2 mm. (C) Hematoxylin and eosin-stained cross-section of lobular tumor in the testis shown in panel B. Bar, 1 mm. (D) Inhibin (Inh) α immunoreactivity (brown staining, arrows) is focally positive in a tumor from an 11-week-old Smad1 Smad5 dKO male. Bar, 20 μm. (E) Hemorrhagic ascites formation in a 42-week-old Smad1 Smad5 dKO male compared to a control male of the same age. (F) The percentage of male Smad1 Smad5 dKO mice that develop hemorrhagic ascites increases with age.
In addition, with age, male Smad1 Smad5 dKO mice developed distended abdomens that contained hemorrhagic ascites (Fig. 5E and F). All of the male mice with ascites showed extensive testis tumor development in addition to lymphatic and peritoneal metastases. Sertoli-Leydig cell tumors from male Smad1 Smad5 dKO mice contained patchy inhibin immunoreactivity (Fig. 5D) and were negative for cytokeratins 8 and 19 (data not shown). Therefore, similarly to results for females, loss of Smad1 Smad5 in gonadal somatic cells resulted in 100% penetrance and metastases. The higher frequency of ascites development in males also suggests that the modes of metastasis differ between the sexes (i.e., primarily lymphatic spread in males versus peritoneal spread in females).
Gene expression changes in Smad1 Smad5 dKO mice.
To determine gene expression changes in the tumors, we performed microarray and qPCR analyses on dissected Smad1 Smad5 and Smad1 Smad5 Smad8 mutant granulosa cell tumors and compared these to isolated WT granulosa cells. Microarrays were performed with females because granulosa cells, unlike Sertoli cells, are readily collectable in sufficient quantities without culturing and can be used for comparison. Microarray analysis identified 171 genes significantly downregulated (P < 0.001) twofold or more, while 153 genes were significantly upregulated (P < 0.001) twofold or more (see Tables S1 and S2 in the supplemental material). Microarray results were analyzed by GSEA (52) to identify functional gene sets that are altered in tumor cells. Of the 337 gene sets examined, one set, which was associated with ovarian infertility, was significantly underrepresented in BR-SMAD knockout tumor cells compared with WT cells (false discovery rate q value of <0.25) (see Table S3 in the supplemental material). In addition, one gene set representing extracellular matrix pathway genes was overrepresented in BR-SMAD knockout tumor cells compared to WT cells (q < 0.25) (see Table S4 in the supplemental material).
By KEGG pathway analysis, only the TGFβ family signaling pathway was significantly overrepresented (P = 0.02) in the differentially expressed gene lists. This list included genes encoding ligands (Bmp7 and inhibin βB [Inhbb]), receptors (BMP receptors type II [Bmpr2] and type IB [Bmpr1b]), negative regulatory proteins (gremlin [Grem1]), and potential downstream target genes (cartilage oligomeric protein [Comp] and inhibitor of differentiation-1 [Id1]). Of the downregulated genes, five of six genes (Comp, Grem1, Inhbb, Bmpr2, and Id1) were reconfirmed by qPCR (Fig. 6A). Grem1 is a known target of BMP signaling in granulosa cells (36), but few other gene targets are known for this cell type. To determine which genes are regulated by BMPs in granulosa cells and thus those genes that may be directly affected by loss of SMAD1/5 in Smad1 Smad5 dKO tumors, primary cultures of WT granulosa cells were treated with recombinant BMP4. Only Id1 and Grem1 were upregulated within 5 h of treatment (Fig. 6B). Comp, Inhbb, or Bmpr2 gene expression did not change (Fig. 6B). This suggests that Comp, Inhbb, and Bmpr2 are altered in Smad1 Smad5 dKO tumors as an indirect consequence of loss of BR-SMAD signaling.
FIG. 6.

Gene expression changes and TGFβ activity in tumors from female Smad1 Smad5 dKO mice. (A) qPCR verification of genes expressed in Smad1 Smad5 dKO tumors compared to control granulosa cells (Smad1flox/flox Smad5flox/flox) for genes significantly downregulated in tumors in the microarray analysis. Four independent samples were used for each group. Values are means ± standard errors of the means, relative to values for the control group. Asterisks (*, P < 0.05; **, P < 0.01) indicate statistical significance by two-tailed t test. ns, not significant. (B) qPCR analysis of gene expression in 5-h control-treated and BMP4-treated (100 ng/ml) WT granulosa cells from four independent experiments indicates a significant upregulation of Grem1 and Id1 by BMP4. Asterisks denote statistical significance (**, P < 0.01) by two-tailed Student's t test. (C) qPCR verification of Bmp7, Hmga2, Mmp2, and Tgfbi expression in control granulosa cells compared to granulosa cell tumors of Smad1 Smad5 dKO mice, showing that all are significantly upregulated (P < 0.05) in tumors by one-way analysis of variance and Tukey-Kramer honestly significant difference. (D and E) Immunohistochemistry for phospho-SMAD2/3 in (D) a granulosa cell tumor from a 7-month-old Smad1 Smad5 Smad8 tKO female and (E) a tumor within a lymph node (lym) from an 8-month-old Smad1flox/flox Smad5flox/flox Smad8+/− Amhr2cre/+ dKO female. Positive immunoreactivity (brown staining) is seen in tumor nuclei (arrow, follicle-like structure within the tumor). The epithelium of the oviduct (ovi) is normally positive. Bar, 50 μm. (F) Hypothetical model for tumor development through loss of SMAD signaling in the gonad. Smad1 Smad5 dKO mice develop sex cord stromal tumors, as do inhibin α knockout mice (31). Loss of SMAD1/5 signaling suggests that the BMP pathway has a tumor suppressor function in the gonad. Interactions between the BMP and the activin/TGFβ pathways to control growth and differentiation of granulosa cells could occur at many different levels (dashed arrow) and remain to be determined.
In many human cancers, TGFβ stimulates invasion and metastasis. Canonical signaling by TGFβ results in phosphorylation and nuclear accumulation of SMAD2 and SMAD3. By immunohistochemistry, all Smad1 Smad5 dKO granulosa cell tumors and metastases contained high levels of phospho-SMAD2/3 immunoreactivity in cell nuclei (Fig. 6D and E). Phospho-SMAD2/3 was also detected in all male Smad1 Smad5 dKO tumors (n = 6) (data not shown). The nuclear phospho-SMAD2/3 immunoreactivity in Smad1 Smad5 dKO tumors supports a hypothesis that TGFβ and/or activin is functional in these tumors. Because the activin antagonist inhibin is highly expressed in Smad1 Smad5 dKO tumors and metastases (Fig. 3E and F and 4E), it is unlikely that dimeric activin is either produced or able to signal; therefore, phospho-SMAD2/3 is likely attributable to TGFβ activity. Moreover, Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO tumors express TGFβ1, TGFβ3, and the TGFβ type I receptor (Tgfbr1, or Alk5), type II receptor (Tgfbr2), and type III receptors (data not shown). An examination of the list of genes significantly upregulated in female Smad1 Smad5 tumors (see Table S2 in the supplemental material) provided further evidence for activation of the TGFβ/SMAD2/3 pathway. At least eight genes with at least one literature citation identifying the gene as a downstream target of TGFβ or SMAD2/3 were identified. This list includes Hmga2, Mmp2, and Tgfbi, and expression changes in these genes in Smad1 Smad5 tumors were verified by qPCR (Fig. 6C). Taken together, these data indicate that TGFβ signaling is active in Smad1 Smad5 dKO tumor cells when the Smad1 Smad5 pathway is disrupted in vivo and suggest a potential role for the BR-SMAD pathway in modulating TGFβ function.
DISCUSSION
Our results uncover a profound role for the BMP pathway in vivo for regulating tumorigenesis in somatic gonadal cells. In vivo evidence, including genetic models, for a tumor suppressor role for the BR-SMAD pathway has been lacking, except for the BMP type I receptor BMPR1A. However, many in vitro cell culture studies have suggested a role for the BMP family in regulating cellular differentiation and growth control. During our experiments to analyze the in vivo reproductive role of the BR-SMADs in ovarian somatic cells, we discovered that along with fertility defects, 100% of the female mice conditionally null for Smad1 Smad5 or Smad1 Smad5 Smad8 develop granulosa cell tumors. In BR-SMAD knockout males, tumors develop more slowly but also are 100% penetrant. Additionally, both male and female cKO mice demonstrate a high degree of metastases. There are few signaling pathway mutations associated with human sex cord stromal tumor development (i.e., granulosa, Sertoli and Leydig tumors), and based on the highly penetrant metastatic tumor phenotype of the Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice, the BR-SMAD pathway and the genes that it regulates become important candidate pathways for further disease research for these and other cancers.
Even though the BMP type I receptors have been shown to phosphorylate SMAD1, SMAD5, or SMAD8, the degree of redundancy between these SMADs is unknown. Smad8 null mice are viable and fertile (1), while Smad1 and Smad5 homozygous and heterozygous null embryos die at midgestation (8, 53). Several conditional alleles of Smad1 and Smad5 have been generated to circumvent the lethality and allow for the analysis of BR-SMADs in later-stage embryonic and adult tissues (1, 21, 54). However, few adult tissue-specific knockout mice have yet to be reported. Recently, it was shown that conditional deletion of Smad5 in adult hematopoietic stem cell lineages has no effect on adult hematopoiesis, and this was attributed to potential functional compensation by SMAD1, which is equivalently expressed in WT and Smad5 null hematopoietic stem cell lineages (49). Similarly, none of our single conditional knockouts for Smad1 or Smad5 or a knockout of Smad8 demonstrates significant fertility defects for the parameters that we tested. It was only through combined loss of Smad1 and Smad5 that we demonstrated the metastatic tumor phenotype in mouse gonadal cells. These data support studies with mouse embryos that indicate a strong genetic interaction between Smad1 and Smad5 (1). Thus, our data on Smad1 Smad5 dKO mice demonstrate that there is significant functional redundancy between SMAD1 and SMAD5. Therefore, genetic models for the BR-SMAD pathway in other cell and tissue types will likely have to take this into account and require modification of both of these genes to fully uncover the function of the BR-SMAD pathway.
While our studies demonstrate redundancy between SMAD1 and SMAD5, our data do not support additional redundancy with SMAD8, even though Smad8 is coexpressed with Smad1 and Smad5 in granulosa cells (data not shown). At the amino acid level, SMAD1 and SMAD5 are more similar to each other than to SMAD8, and this divergence of SMAD1/5 and SMAD8 occurred early during vertebrate evolution (1). Studies of genetic mutations in mouse embryos support the hypothesis that SMAD8 is not redundant with SMAD1 and SMAD5 because deletion of Smad8 has no effect on the phenotype of Smad1 or Smad5 null embryos and removal of one copy of Smad1 or Smad5 has no effect on the Smad8 null phenotype (1). In our studies described here, no fertility or tumor phenotype was seen in Smad8 null female mice or Smad5 Smad8 dKO mice. Furthermore, there was no additional effect of Smad8 loss in female fertility or tumor formation when Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice were compared. The lack of genetic interaction between the SMAD1/5 and SMAD8 pathways may indicate that SMAD8 functions in another non-BMP pathway in the ovary, such as for anti-Müllerian hormone/Müllerian-inhibiting substance, which has recently been shown to use different BR-SMADs sequentially during Müllerian duct regression (57). Alternatively, SMAD8, but not SMAD1/5, may interact with a SMAD-interacting protein, and loss of this interaction through loss of Smad8 is not critical during the development of the tumorigenesis phenotype.
In many human cancers, TGFβ is overexpressed and stimulates tumor invasion and metastasis. The SMAD2/3 pathway is consistently activated in tumors and metastases of both male and female Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO mice, as indicated by nuclear phospho-SMAD2/3 immunohistochemistry. It is unclear what the contribution of TGFβ activity is to tumor development in granulosa cells. We analyzed several candidate genes that are known to be directly downstream of TGFβ signaling and implicated in tumor development and metastasis. We found that several of these genes, including Hmga2, Mmp2, and Tgfbi, are significantly upregulated. Upregulation of Mmp2 expression has been linked to metastatic potential in some cancers (32). Overexpression of Tgfbi and Hmga2 has been demonstrated in a number of human cancers (6, 46, 47), and mouse models have shown that Hmga2 overexpression is sufficient to induce mesenchymal tumor development (55). How the suite of genes expressed in Smad1 Smad5 dKO sex cord stromal tumors contribute to malignant transformation remains to be determined.
In some cell types (e.g., endothelial cells), TGFβ also signals through SMAD1/5 when the type I receptor activin receptor-like kinase 1 (Acvrl1, also called Alk1) is expressed (17). Therefore, an additional possibility is that loss of TGFβ signaling through SMAD1/5 in the Smad1 Smad5 dKO mouse triggers the tumor phenotype. However, this does not appear to be a likely explanation for tumor development in the Smad1 Smad5 and Smad1 Smad5 Smad8 cKO mice, because Alk1 is not expressed in granulosa cells and we did not detect phosphorylation of SMAD1 or SMAD5 in TGFβ-treated WT granulosa cells by immunoanalysis (data not shown). In addition, a recent study has demonstrated that ALK1 activates BR-SMADS in endothelial cells after binding BMP9 and BMP10, and thus these ligands, and not TGFβ, may represent the physiological ligands for this receptor (11).
It is unknown whether activation of the TGFβ pathway in the Smad1 Smad5 dKO and Smad1 Smad5 Smad8 tKO tumors is a primary event caused by loss of SMAD1/5 or occurs secondarily during gonadal tumor development. However, it has been shown in some cell types that the BR-SMAD pathway directly counteracts TGFβ activity through opposing regulation of the same gene promoter by SMAD1/5 and SMAD2/3. For instance, BMP7 reverses the TGFβ1-induced epithelial-mesenchymal transition through a SMAD-dependent pathway in renal distal tubular and mammary ductal epithelial cells in part by antagonistic regulation of the E-cadherin promoter: TGFβ signaling through SMAD3 downregulates transcription through the E-cadherin promoter, while BMP7 signaling through SMAD5 upregulates it (56). BMP7 restores E-cadherin expression to normal values even when both the SMAD2/3 and SMAD1/5 pathways are activated concurrently, suggesting that the BR-SMAD pathway can override the AR-SMAD pathway. While E-cadherin is not expressed in granulosa cells or Smad1 Smad5 dKO tumors, a similar mechanism of SMAD promoter antagonism on granulosa cell target genes may operate in granulosa cells during folliculogenesis. Approximately 25 other genes have been shown to be coregulated by the two SMAD pathways in epithelial cells, including the Id genes (26). However, few direct downstream target genes of BMP or TGFβ have been reported for granulosa cells. In this study, we found that BMP4 regulates Id1 in WT granulosa cells and that, furthermore, the expression of Id1 is downregulated significantly in Smad1 Smad5 tumor cells, although the functional consequence of this is not known. The identification of additional Smad1 Smad5 target genes during folliculogenesis and granulosa cell differentiation will be critical for understanding tumor development in the Smad1 Smad5 dKO mouse model.
Death in most cancer patients is primarily the result of metastatic disease (51), and granulosa cell tumors metastasize in an estimated 5 to 25% of cases. Different types of ovarian cancer (i.e., epithelial and sex cord stromal cancers) have similar sites of metastasis (45) and frequently spread directly into the peritoneal cavity by growth onto adjacent organs, seeding of tumor cells present in peritoneal fluid, and dissemination via the lymphatic system (35). Very few mouse knockouts develop metastatic tumors, and among mouse models of granulosa cell tumors, the Smad1 Smad5 dKO model is unique because previous models (4, 31, 43) do not demonstrate tumor spread outside the gonad. The pattern of peritoneal spread of the female Smad1 Smad5 dKO tumors closely mimics the human dissemination pattern, and both male and female Smad1 Smad5 dKO mouse models demonstrate evidence of tumor spread through the lymphatic system. Thus, the Smad1 Smad5 dKO mouse model is an important step in uncovering novel redundant roles of SMAD1 and SMAD5 in controlling tumor cell migration to distant sites.
However, there are phenotypic dissimilarities of Smad1 Smad5 dKO mice to other mouse models of granulosa tumors, which suggest functional differences. Smad1 Smad5 dKO granulosa cell tumors lack tubule-like structures that typify inhibin α (Inha) null sex cord stromal tumors (31) and also do not develop the cancer cachexia-like wasting syndrome seen in Inha knockout mice. Granulosa cell tumors also develop in female mice engineered to express a stable form of β-catenin (4). We examined β-catenin immunoreactivity in mouse Smad1 Smad5 dKO tumors, but in the majority of Smad1 Smad5 dKO tumor cells, β-catenin is not nuclear but membrane bound (data not shown). In contrast to Smad1 Smad5 dKO females, β-catenin females show a low penetrance (57% in females by 7.5 months, versus 100% penetrance at 3 months in Smad1 Smad5 dKO female mice) and do not develop metastases (4) and males do not develop tumors (D. Boerboom, personal communication). Additional studies will be necessary to understand the pathologies leading to tumor development in these mouse models and the ways they can contribute to our understanding of sex cord stromal cell tumor formation.
Additional genetic mouse models have defined the roles of TGFβ family ligands or SMAD proteins in granulosa cell physiology and provide a foundation for understanding the role for the TGFβ family in granulosa cell tumor development (Fig. 6F). The data in this study demonstrate that when the BR-SMADs are deleted, tumors develop. When loss-of-function mutations in the activin/TGFβ pathway are made, inappropriate granulosa cell differentiation occurs and no tumors develop (37, 38). Activin gain of function (via deletion of the inhibin α subunit leading to activin overexpression) results in gonadal sex cord stromal tumors, mediated in part by the AR-SMAD SMAD3 (27, 28, 31). Combined, these phenotypes suggest a possible interplay between the BR-SMAD and AR-SMAD signaling pathways, which when unbalanced may lead to tumor development (Fig. 6F).
It is unknown if tumor suppression by SMAD1 and SMAD5 is a mechanism found in nongonadal cell types. No germ line mutations have been identified in the limited numbers of studies of humans that examined BR-SMADs (3, 16). However, if BR-SMADS are as redundant in humans, as we and others (1) suspect, then a combination of loss of function in both SMAD1 and SMAD5 would be required for developing human cancers, and this may be an unlikely event. However, because haploinsufficiency in many transcription factors causes human diseases, a significant distortion in SMAD1 SMAD5 gene expression or activity in humans may have the same phenotypic effect as the Smad1 Smad5 double null mutation and result in tumorigenesis. Thus, there are many possible scenarios that would result in downregulated SMAD1 SMAD5 expression or activity, including mutations in common upstream SMAD1 SMAD5 regulatory pathways, mutations in a SMAD1/5-interacting protein, or mutations in a common downstream target gene. Evidence for downregulation in human cancers includes loss of SMAD1 expression in cervical carcinomas (48) and loss of SMAD8 in breast, colon, and prostate cancer (9, 19). An important consideration for future cancer research will be to determine whether loss of Smad1 Smad5 Smad8 and their downstream targets in gonadal and nongonadal cells also results in tumor formation and metastatic disease in humans.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants HD32067 (to M.M.M.), HD44156 (to E.J.R.), HD12324 (to J.F.M.), and HD30284 (to R.R.B.), a European Union grant (T-Angiovasc QLG1-CT-2001-01032) and Interuniversity Attraction Poles Programme-Belgian Science Policy project IUAP5/35 (to L.U., A.Z., and D.H.), and a Ruth L. Kirschstein NRSA fellowship (5F32HD46335) and Burroughs Wellcome Career Award in the Biomedical Sciences grant (to S.A.P.).
We thank Wylie Vale (Salk Institute, La Jolla, CA) for the gift of the inhibin α polyclonal antibody, Milton Finegold and Alan Herron (Baylor College of Medicine) for pathological assessment of male tumors, Mai Tran (Baylor College of Medicine) for technical assistance, Lori-Ann Mistretta for assistance with GSEA, and Herman Dierick and Aleksandar Rajkovic for discussions and critical reading of the manuscript.
Footnotes
Published ahead of print on 29 October 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Arnold, S. J., S. Maretto, A. Islam, E. K. Bikoff, and E. J. Robertson. 2006. Dose-dependent Smad1, Smad5 and Smad8 signaling in the early mouse embryo. Dev. Biol. 296104-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck, S. E., B. H. Jung, A. Fiorino, J. Gomez, E. D. Rosario, B. L. Cabrera, S. C. Huang, J. Y. Chow, and J. M. Carethers. 2006. Bone morphogenetic protein signaling and growth suppression in colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 291G135-G145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bevan, S., K. Woodford-Richens, P. Rozen, C. Eng, J. Young, M. Dunlop, K. Neale, R. Phillips, D. Markie, M. Rodriguez-Bigas, B. Leggett, E. Sheridan, S. Hodgson, T. Iwama, D. Eccles, W. Bodmer, R. Houlston, and I. Tomlinson. 1999. Screening SMAD1, SMAD2, SMAD3, and SMAD5 for germline mutations in juvenile polyposis syndrome. Gut 45406-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boerboom, D., M. Paquet, M. Hsieh, J. Liu, S. P. Jamin, R. R. Behringer, J. Sirois, M. M. Taketo, and J. S. Richards. 2005. Misregulated Wnt/beta-catenin signaling leads to ovarian granulosa cell tumor development. Cancer Res. 659206-9215. [DOI] [PubMed] [Google Scholar]
- 5.Brubaker, K. D., E. Corey, L. G. Brown, and R. L. Vessella. 2004. Bone morphogenetic protein signaling in prostate cancer cell lines. J. Cell. Biochem. 91151-160. [DOI] [PubMed] [Google Scholar]
- 6.Buckhaults, P., C. Rago, B. St. Croix, K. E. Romans, S. Saha, L. Zhang, B. Vogelstein, and K. W. Kinzler. 2001. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 616996-7001. [PubMed] [Google Scholar]
- 7.Chang, H., C. W. Brown, and M. M. Matzuk. 2002. Genetic analysis of the mammalian TGF-β superfamily. Endocr. Rev. 23787-823. [DOI] [PubMed] [Google Scholar]
- 8.Chang, H., D. Huylebroeck, K. Verschueren, Q. Guo, M. M. Matzuk, and A. Zwijsen. 1999. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 1261631-1642. [DOI] [PubMed] [Google Scholar]
- 9.Cheng, K. H., J. F. Ponte, and S. Thiagalingam. 2004. Elucidation of epigenetic inactivation of SMAD8 in cancer using targeted expressed gene display. Cancer Res. 641639-1646. [DOI] [PubMed] [Google Scholar]
- 10.Coerver, K. A., T. K. Woodruff, M. J. Finegold, J. Mather, A. Bradley, and M. M. Matzuk. 1996. Activin signaling through activin receptor type II causes the cachexia-like symptoms in inhibin-deficient mice. Mol. Endocrinol. 10534-543. [DOI] [PubMed] [Google Scholar]
- 11.David, L., C. Mallet, S. Mazerbourg, J. J. Feige, and S. Bailly. 2007. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase (ALK1) in endothelial cells. Blood 1091953-1961. [DOI] [PubMed] [Google Scholar]
- 12.Dennis, G., Jr., B. T. Sherman, D. A. Hosack, J. Yang, W. Gao, H. C. Lane, and R. A. Lempicki. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4P3. [PubMed] [Google Scholar]
- 13.Derynck, R., R. J. Akhurst, and A. Balmain. 2001. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29117-129. [DOI] [PubMed] [Google Scholar]
- 14.Durlinger, A. L., P. Kramer, B. Karels, F. H. de Jong, J. T. Uilenbroek, J. A. Grootegoed, and A. P. Themmen. 1999. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 1405789-5796. [DOI] [PubMed] [Google Scholar]
- 15.Feng, X. H., and R. Derynck. 2005. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 21659-693. [DOI] [PubMed] [Google Scholar]
- 16.Gemma, A., K. Hagiwara, F. Vincent, Y. Ke, A. R. Hancock, M. Nagashima, W. P. Bennett, and C. C. Harris. 1998. hSmad5 gene, a human hSmad family member: its full length cDNA, genomic structure, promoter region and mutation analysis in human tumors. Oncogene 16951-956. [DOI] [PubMed] [Google Scholar]
- 17.Goumans, M. J., G. Valdimarsdottir, S. Itoh, A. Rosendahl, P. Sideras, and P. ten Dijke. 2002. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 211743-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He, X. C., J. Zhang, W. G. Tong, O. Tawfik, J. Ross, D. H. Scoville, Q. Tian, X. Zeng, X. He, L. M. Wiedemann, Y. Mishina, and L. Li. 2004. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet. 361117-1121. [DOI] [PubMed] [Google Scholar]
- 19.Horvath, L. G., S. M. Henshall, J. G. Kench, J. J. Turner, D. Golovsky, P. C. Brenner, G. F. O'Neill, R. Kooner, P. D. Stricker, J. J. Grygiel, and R. L. Sutherland. 2004. Loss of BMP2, Smad8, and Smad4 expression in prostate cancer progression. Prostate 59234-242. [DOI] [PubMed] [Google Scholar]
- 20.Howe, J. R., J. L. Bair, M. G. Sayed, M. E. Anderson, F. A. Mitros, G. M. Petersen, V. E. Velculescu, G. Traverso, and B. Vogelstein. 2001. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 28184-187. [DOI] [PubMed] [Google Scholar]
- 21.Huang, S., B. Tang, D. Usoskin, R. J. Lechleider, S. P. Jamin, C. Li, M. A. Anzano, T. Ebendal, C. Deng, and A. B. Roberts. 2002. Conditional knockout of the Smad1 gene. Genesis 3276-79. [DOI] [PubMed] [Google Scholar]
- 22.Jamin, S. P., N. A. Arango, Y. Mishina, M. C. Hanks, and R. R. Behringer. 2002. Requirement of Bmpr1a for Mullerian duct regression during male sexual development. Nat. Genet. 77. [DOI] [PubMed] [Google Scholar]
- 23.Jarvinen, H., and K. O. Franssila. 1984. Familial juvenile polyposis coli; increased risk of colorectal cancer. Gut 25792-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeyasuria, P., Y. Ikeda, S. P. Jamin, L. Zhao, D. G. De Rooij, A. P. Themmen, R. R. Behringer, and K. L. Parker. 2004. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol. Endocrinol. 181610-1619. [DOI] [PubMed] [Google Scholar]
- 25.Jorgez, C. J., M. Klysik, S. P. Jamin, R. R. Behringer, and M. M. Matzuk. 2004. Granulosa cell-specific inactivation of follistatin causes female fertility defects. Mol. Endocrinol. 18953-967. [DOI] [PubMed] [Google Scholar]
- 26.Kowanetz, M., U. Valcourt, R. Bergstrom, C. H. Heldin, and A. Moustakas. 2004. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol. Cell. Biol. 244241-4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, Q., J. M. Graff, A. E. O'Connor, K. L. Loveland, and M. M. Matzuk. 2007. SMAD3 regulates gonadal tumorigenesis. Mol. Endocrinol. 212472-2486. [DOI] [PubMed] [Google Scholar]
- 28.Looyenga, B. D., and G. D. Hammer. 2007. Genetic removal of smad3 from inhibin-null mice attenuates tumor progression by uncoupling extracellular mitogenic signals from the cell cycle machinery. Mol. Endocrinol. 212440-2457. [DOI] [PubMed] [Google Scholar]
- 29.Massague, J., S. W. Blain, and R. S. Lo. 2000. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103295-309. [DOI] [PubMed] [Google Scholar]
- 30.Massague, J., J. Seoane, and D. Wotton. 2005. Smad transcription factors. Genes Dev. 192783-2810. [DOI] [PubMed] [Google Scholar]
- 31.Matzuk, M., M. Finegold, J. Su, A. Hsueh, and A. Bradley. 1992. α-Inhibin is a tumor-suppressor gene with gonadal specificity in mice. Nature 360313-319. [DOI] [PubMed] [Google Scholar]
- 32.Minn, A. J., G. P. Gupta, P. M. Siegel, P. D. Bos, W. Shu, D. D. Giri, A. Viale, A. B. Olshen, W. L. Gerald, and J. Massague. 2005. Genes that mediate breast cancer metastasis to lung. Nature 436518-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazaki, H., T. Watabe, T. Kitamura, and K. Miyazono. 2004. BMP signals inhibit proliferation and in vivo tumor growth of androgen-insensitive prostate carcinoma cells. Oncogene 239326-9335. [DOI] [PubMed] [Google Scholar]
- 34.Miyazono, K., S. Maeda, and T. Imamura. 2005. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 16251-263. [DOI] [PubMed] [Google Scholar]
- 35.Naora, H., and D. J. Montell. 2005. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat. Rev. Cancer 5355-366. [DOI] [PubMed] [Google Scholar]
- 36.Pangas, S. A., C. J. Jorgez, and M. M. Matzuk. 2004. Growth differentiation factor 9 regulates expression of the bone morphogenetic protein antagonist, gremlin. J. Biol. Chem. 27932281-32286. [DOI] [PubMed] [Google Scholar]
- 37.Pangas, S. A., C. J. Jorgez, M. Tran, J. Agno, X. Li, C. W. Brown, T. R. Kumar, and M. M. Matzuk. 2007. Intraovarian activins are required for female fertility. Mol. Endocrinol. 212458-2471. [DOI] [PubMed] [Google Scholar]
- 38.Pangas, S. A., X. Li, E. J. Robertson, and M. M. Matzuk. 2006. Premature luteinization and cumulus cell defects in ovarian-specific Smad4 knockout mice. Mol. Endocrinol. 201406-1422. [DOI] [PubMed] [Google Scholar]
- 39.Pangas, S. A., and M. M. Matzuk. 2007. The TGFB family in the reproductive tract. In R. Derynck and K. Miyazono (ed.), The TGFB family. Cold Spring Harbor Press, Cold Spring Harbor, NY. [DOI] [PMC free article] [PubMed]
- 40.Pardali, K., and A. Moustakas. 2006. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta 177521-62. [DOI] [PubMed] [Google Scholar]
- 41.Piccirillo, S., B. Reynolds, N. Zanetti, G. Lamorte, E. Binda, G. Broggi, H. Brem, A. Olivi, F. Dimeco, and A. Vescovi. 2006. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain-tumour-initiating cells. Nature 444761-765. [DOI] [PubMed] [Google Scholar]
- 42.Pouliot, F., and C. Labrie. 2002. Role of Smad1 and Smad4 proteins in the induction of p21WAF1,Cip1 during bone morphogenetic protein-induced growth arrest in human breast cancer cells. J. Endocrinol. 172187-198. [DOI] [PubMed] [Google Scholar]
- 43.Risma, K., C. Clay, T. Nett, T. Wagner, J. Yun, and J. Nilson. 1995. Targeted overexpression of luteinizing hormone in transgenic mice leads to infertility, polycystic ovaries, and ovarian tumors. Proc. Natl. Acad. Sci. USA 921322-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts, A. B., and L. M. Wakefield. 2003. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 1008621-8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rose, P. G., M. S. Piver, Y. Tsukada, and T. Lau. 1989. Metastatic patterns in histolologic variants of ovarian cancer. Cancer 641508-1513. [DOI] [PubMed] [Google Scholar]
- 46.Schneider, D., J. Kleeff, P. O. Berberat, Z. Zhu, M. Korc, H. Friess, and M. W. Buchler. 2002. Induction and expression of betaig-h3 in pancreatic cancer cells. Biochim. Biophys. Acta 15881-6. [DOI] [PubMed] [Google Scholar]
- 47.Sgarra, R., A. Rustighi, M. A. Tessari, J. Di Bernardo, S. Altamura, A. Fusco, G. Manfioletti, and V. Giancotti. 2004. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 5741-8. [DOI] [PubMed] [Google Scholar]
- 48.Shim, C., W. Zhang, C. H. Rhee, and J. H. Lee. 1998. Profiling of differentially expressed genes in human primary cervical cancer by complementary DNA expression array. Clin. Cancer Res. 43045-3050. [PubMed] [Google Scholar]
- 49.Singbrant, S., J. L. Moody, U. Blank, G. Karlsson, L. Umans, A. Zwijsen, and S. Karlsson. 2006. Smad5 is dispensable for adult murine hematopoiesis. Blood 1083707-3712. [DOI] [PubMed] [Google Scholar]
- 50.Sneddon, J. B., H. H. Zhen, K. Montgomery, M. van de Rijn, A. D. Tward, R. West, H. Gladstone, H. Y. Chang, G. S. Morganroth, A. E. Oro, and P. O. Brown. 2006. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc. Natl. Acad. Sci. USA 10314842-14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steeg, P. S. 2006. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 12895-904. [DOI] [PubMed] [Google Scholar]
- 52.Subramanian, A., P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander, and J. P. Mesirov. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 10215545-15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tremblay, K. D., N. R. Dunn, and E. J. Robertson. 2001. Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development 1283609-3621. [DOI] [PubMed] [Google Scholar]
- 54.Umans, L., L. Vermeire, A. Francis, H. Chang, D. Huylebroeck, and A. Zwijsen. 2003. Generation of a floxed allele of Smad5 for cre-mediated conditional knockout in the mouse. Genesis 375-11. [DOI] [PubMed] [Google Scholar]
- 55.Zaidi, M. R., Y. Okada, and K. K. Chada. 2006. Misexpression of full-length HMGA2 induces benign mesenchymal tumors in mice. Cancer Res. 667453-7459. [DOI] [PubMed] [Google Scholar]
- 56.Zeisberg, M., J. Hanai, H. Sugimoto, T. Mammoto, D. Charytan, F. Strutz, and R. Kalluri. 2003. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9964-968. [DOI] [PubMed] [Google Scholar]
- 57.Zhan, Y., A. Fujino, D. T. MacLaughlin, T. F. Manganaro, P. P. Szotek, N. A. Arango, J. Teixeira, and P. K. Donahoe. 2006. Mullerian inhibiting substance regulates its receptor/SMAD signaling and causes mesenchymal transition of the coelomic epithelial cells early in Mullerian duct regression. Development 1332359-2369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.