

Abstract
The mammalian ortholog of the Drosophila MOF (males absent on the first) gene product is a histone H4 lysine 16-specific acetyltransferase. Recent studies have shown that depletion of human MOF (hMOF) in human cell lines leads to genomic instability, spontaneous chromosomal aberrations, cell cycle defects, altered nuclear morphology, reduced transcription of certain genes, and defective DNA damage response to ionizing radiation (IR). Here we show that MOF plays an essential role in mammals during embryogenesis and oncogenesis. Ablation of the mouse Mof gene (mMof) by gene targeting resulted in early embryonic lethality and cell death. Lethality correlated with the loss of H4 lysine 16 acetylation (H4K16ac) and could not be rescued by concomitant inactivation of ATM or p53. In comparison to primary cells or normal tissue, all immortalized human normal and tumor cell lines and primary tumors demonstrated similar or elevated hMOF and H4K16ac levels. Accordingly, MOF overexpression correlated with increased cellular proliferation, oncogenic transformation, and tumor growth. Thus, these data reveal that the acetylation of histone H4 at K16 by MOF is an epigenetic signature of cellular proliferation common to both embryogenesis and oncogenesis and that MOF is an essential factor for embryogenesis and oncogenesis.
MOF belongs to the MYST family of acetyltransferases, which have been associated with acute myeloid leukemia (MOZ), transcriptional silencing in Saccharomyces cerevisiae (SAS2 and YBF2/SAS3), interactions with human immunodeficiency virus Tat in humans (TIP60), and dosage compensation in Drosophila melanogaster (MOF), in addition to their role in DNA damage repair (2, 4, 12, 13, 17, 18, 43, 44, 46). Akhtar and Becker (1) demonstrated that Drosophila MOF is a histone acetyltransferase that acetylates chromatin specifically at histone H4 lysine 16 (H4K16). Depletion of human MOF (hMOF) in human cells results in the loss of acetylation at lysine 16 of histone H4 (6, 12, 41, 45, 46), strongly arguing that the highly conserved MOF protein may be the major histone acetyltransferase, which acetylates histone H4 at K16.
Acetylation at K16 of histone H4 (H4K16ac) is a prevalent and reversible posttranslational chromatin modification in eukaryotes, and recent studies have highlighted its significance. Shogren-Knaak and coworkers have found that a single histone H4K16ac modification modulates both higher-order chromatin structure and functional interactions between a nonhistone protein and the chromatin fiber (39). Shia and coworkers have demonstrated that the presence of H4K16ac and H2A.Z synergistically prevent the ectopic propagation of heterochromatin in the subtelomeric regions of yeast (36). Furthermore, it is well understood that H4K16ac disrupts higher-order chromatin structure, changes the functional interactions between chromatin-associated proteins (39), and serves as a switch for altering chromatin from a repressive to a transcriptionally active state in yeast and humans (36). Interestingly, Dou and coworkers reported that MOF-mediated histone acetyltransferase activity, specific for H4K16, is required for optimal transcription activation on a chromatin template in vitro and in vivo (6). The loss of monoacetylation at lysine 16 of histone H4 specifically at nucleosome repetitive sequences is a common hallmark of tumor cell lines and human cancer where it is progressively lost from early preneoplastic stages to the most malignant stage (8). Furthermore, inhibition of SIRT1 deacetylase in breast and colon cancer cells has been shown to cause increased acetylation of H4K16 at two specific endogenous promoters that correlated with decreased proliferation (29). In contrast, mouse embryonic fibroblasts (MEFs) deficient for SIRT1, have dramatically increased resistance to replicative senescence (5). These results demand further evaluation of H4K16ac in tumor and normal cells.
Vigorous cellular proliferation is common and essential during both embryogenesis and oncogenesis. Therefore, these two processes may potentially have common chromatin modification signatures characteristic of their proliferation status. Recent studies have demonstrated that hMOF depletion, resulting in the loss of H4K16 acetylation correlates with a decrease in DNA damage-induced activation of ATM and prevents ATM from phosphorylating downstream effectors, such as p53 and CHK2 (12, 45). hMOF physically interacts with ATM and p53 (6, 12), and in vitro, hMOF binds to and acts synergistically with p53 to increase histone H4K16ac and target gene transcription (6). While H4K16ac is known to alter higher-order chromatin structure into a relaxed conformation (39), it is important to determine whether there is a correlation between this modification and cellular growth both during embryonic development and in the process of tumorigenesis.
In this study, we determined whether MOF, which specifically acetylates H4K16, is required for cell growth and proliferation during either embryogenesis or tumorigenesis and whether proliferating tumors and tumor-derived cell lines demonstrated loss of MOF and H4K16 acetylation. Specifically, we analyzed the impact of mammalian MOF expression on embryonic development and oncogenic transformation, two processes that rely on cellular proliferation. We examined MOF and H4K16ac levels in single-cell zygotes through blastocyst-stage embryos and found that ablation of MOF correlated with loss of histone H4K16ac and paralleled embryonic lethality and cell death. Conversely, MOF overexpression increased H4K16ac levels, which correlated with oncogenic transformation and tumor growth. All tumor cell lines examined expressed hMOF and H4K16ac. Strikingly, primary human cells immortalized with the catalytic unit of telomerase (hTERT) have higher levels of hMOF and H4K16ac than primary cells do.
MATERIALS AND METHODS
Targeted mutagenesis.
Prior mapping and sequence studies indicated the mouse Mof (mMof) gene is located on chromosome 7 at the end of a subtelomeric region (see Fig. 1). An mMof gene-targeting vector was constructed from 129/SvJ mouse DNA by replacing a genomic fragment containing part of exon 2, intron 2, exon 3, and part of intron 3 with a neomycin resistance gene cassette (see Fig. 2). A diphtheria toxin A gene cassette was also included in the targeting construct as negative selection against random integration. The construct was electroporated into 129/Sv W9.5 embryonic stem (ES) cells, and G418-resistant colonies were analyzed by Southern blot analysis using a 5′ flanking probe. Three independently derived Mof+/− ES cell clones were injected into C57BL/6 blastocysts to derive male chimeras. Germ line transmission of the targeted allele was confirmed by Southern blot analysis.
FIG. 1.

mRNA expression levels, gene localization, and mMof gene product weight. (A) Northern blot of multiple normal human tissues hybridized with an hMof cDNA probe. (B) Fluorescence in situ hybridization localization of the mMof gene by in situ hybridization of biotin-labeled mMof genomic DNA to the mouse metaphase chromosome. Specific signal is observed at the subtelomeric region of the chromosome 7. Chromosome 7 was identified by the standard G-banding. (C) In vitro expression of a cloned mMof cDNA. Translation products were loaded onto a 10% sodium dodecyl sulfate-polyacrylamide gel and separated by electrophoresis. The autoradiograph shows the following: no cDNA added (lane 1), luciferase control cDNA (lane 2), and mMof cDNA (lane 3). The positions of a set of prestained protein size markers (Cambrex) (in kilodaltons) are indicated to the left of the gel. The mMof product is indicated by the arrow to the right of the gel.
FIG. 2.

mMof inactivation results in early embryonic lethality. (A) Gene targeting strategy for mMof null allele. The organization of the wild-type gene, the targeting construct, and the structure of the locus following gene targeting are shown. Exons are shown as black boxes and are numbered. The locations of PCR primers for detecting gene targeting events are shown by arrows at the bottom of targeted mMof mutant locus. The locations of the phosphoglycerate kinase gene carrying a neomycin resistance gene (PGKneopA) and the diphtheria toxin A gene (DT) are shown. The restriction sites shown are BglII (B), EcoRI (E), SphI (S), and XbaI (X). (B) Southern blot analysis of tail DNA digested with BglII and hybridized with a probe located outside the targeting construct. Tail DNA from mMof+/− (+/−) and mMof+/+ (+/+) mice was used. Note the absence of mMof−/− mice. The positions of molecular size markers in lane M (in kilobases) are indicated to the right of the gel. (C) Gross morphology of embryos. Two normal (left and right) and three mutant embryos at E7.5 (arrows). (D) All normal embryos at E8.5 were either mMof+/+ or mMof+/− as judged by CD31 staining, which detects vasculature of endothelial cells. Genotypes were confirmed by PCR analysis.
A conditional Mof allele was constructed by inserting a single loxP site together with an FRT-flanked hygromycin resistance gene cassette 1.5 kb upstream of the mMof transcription initiation site and a second loxP site within intron 3 (see Fig. 5). The construct was electroporated into W9.5 ES cells to generate Mofflox/+ cells. Using a knock-in strategy, the tamoxifen-inducible creERT2 fusion gene cassette (7) was inserted into the ubiquitously expressed ROSA26 locus by homologous recombination (11). Correctly targeted mMofflox/+/Rosa26creERT2/+ ES clones were grown in the presence of 200 nM tamoxifen to yield mMofΔflox/+/Rosa26creERT2/+ ES cells. Electroporation of these cells with the conditional targeting vector then gave rise to mMofflox/+/Rosa26creERT2/+ and mMofΔflox/flox/Rosa26creERT2/+ ES cell clones.
FIG. 5.

mMof is essential for proliferation and/or viability of ES cells. (A) Conditional inactivation of mMof. A partial restriction map of the genomic region containing mMof exons 1 to 4 (black boxes numbered 1 to 4) is shown at the top, followed by a diagram of the targeting vector used for insertion of the FRT-flanked neomycin cassette and loxP sites into the mMof gene (bottom). A diphtheria toxin A gene cassette is also included in the targeting construct as a negative selection marker. The wavy line represents plasmid sequence. The restriction sites shown are BglII (B), EcoRI (E), SalI (S), and SphI (Sp). (B) Southern blot analysis showing cre-mediated recombination of the floxed allele in mMofflox/+/Rosa26creERT2/+ and mMofΔflox/flox/Rosa26creERT2/+ ES cells after tamoxifen treatment. Genomic DNA was prepared from mMofΔflox/flox/Rosa26creERT2/+ (lanes 1, 3, 4, 7, 8, 11, 12, 15, and 16) or mMofflox/+/Rosa26creERT2/+ ES cells (lanes 2, 5, 6, 9, 10, 13, 14, 17, and 18) collected at 0, 24, 36, 48, and 60 h from cells treated with 200 nM tamoxifen (Tam) or not treated with tamoxifen (−), digested with PstI, and probed with the 5′ external probe. Note that after 24 h, the conditional (Cond) Mofflox allele is fully recombined in the presence of tamoxifen. The positions of molecular size markers in lane M (in kilobases) are indicated to the left of the gel. WT, wild type. (C) Western blot analysis for MOF and H4K16ac levels in mMofΔflox/flox/Rosa26creERT2/+ ES cells harvested at 0, 24, 48, and 60 h from cells treated with tamoxifen (Tam) (+) or not treated with tamoxifen (−). (D) Proliferation of mMofflox/+/Rosa26creERT2/+ and mMofΔflox/flox/Rosa26creERT2/+ ES cells with or without tamoxifen treatment. Cell numbers were determined at different time points to determine the population doublings in cells with and without mMof.
Staging and collecting of oocytes and embryos.
Oocytes, fertilized eggs, and early embryos were collected from mice according to a previously described procedure (49). Oocytes and embryos were fixed for 20 min in 4% paraformaldehyde in phosphate-buffered saline (PBS) and then washed three times with PBS supplemented with 1% bovine serum albumin (BSA). Fixed oocytes and embryos were permeabilized in 0.5% Triton X-100 in PBS supplemented with 1% BSA for 25 min, incubated overnight at 4°C in antibody diluted in PBS supplemented with 1% BSA, then washed to remove nonspecifically bound antibody, and incubated in 2.5 μg/ml Texas Red-conjugated donkey anti-rabbit immunoglobulin G (IgG) for 1 h at room temperature. The embryos and oocytes were then placed in 0.4 mg/ml RNase in PBS supplemented with 1% BSA for 10 min and stained and mounted in 4′,6′-diamidino-2-phenylindole (DAPI) with glycerol slow-fade mounting medium. Images were obtained on a Zeiss 410 Axiovert system. For each developmental panel, contrast and brightness remained constant. Approximately 40 oocytes/embryos were analyzed for a particular developmental stage in at least three independent trials, and relevant optical sections are shown.
Generation of various mouse genotypes.
All mice in this study have SV129 background. The Atm and p53 heterozygous mice have been described previously (33, 50). Atm−/− mice are infertile. Atm−/−/Mof+/− mice were generated by crossing male and female Atm+/−/Mof+/− genotypes. Offspring were genotyped at age 21 days by PCR-based assays using mouse tail DNA (50).
Cell lines.
The cell lines used were human lymphoblastoid (GM07349, GM10860, GM10854, GM10863, GM10851, GM10850, and GM10847), human leukemia (HL60, K562, and Jurkat), mesothelial cancer lines (REN, LRK, MS1022H, H266, H2052, and H2452), lymphoma (Raji, U937, and HT), breast (MCF-7 and MCF-10A), colon (HCT-116 and RKO), lung (H1299), osteosarcoma (MG63) lymphoblastoid cell lines, and 293 and HeLa cells. All cells were maintained as described previously (26, 40). Primary fibroblast cells lines (GM5823, HFF, and BJ) and the establishment of their hTERT-derived counterparts were described previously (48). Mouse fibroblasts 435 (with ATM) and 743 (without ATM) cells have been described previously (10).
Survival assay.
Cells were plated into 60-mm dishes in 5.0 ml of medium, incubated for 6 h, and subsequently exposed to ionizing radiation (IR) as described previously (25). The number of cells plated per dish was selected to ensure that about 50 colonies would survive treatment with a particular radiation dose. Cells were exposed to IR in a dose range of 0 to 6 Gy at room temperature. Cells were incubated for 12 or more days and then fixed in methanol-acetic acid (3:1) prior to staining with crystal violet. Only colonies containing at least 50 cells were counted.
Western blot analysis.
Cell lysates for Western blot analysis were prepared as previously described (27). Antibodies used to detect MOF, histone H4, and acetylated H4K16 were previously described (12). Immunoblotting and detection of MOF, histone H4, and acetylated H4K16 were done according to previously described procedures (12, 27, 35).
Primary cell culture, establishment of cell lines, and growth assays.
MEFs from mMof heterozygous intercrosses were isolated from embryonic day 13.5 (E13.5) embryos by standard procedures (15, 25). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and frozen at different passages for storage. Continuous passaging was utilized to allow for emergence of spontaneously immortalized clones from both wild-type and mMof heterozygous MEFs. These clones were positive for telomerase activity and were passaged for at least 100 population doublings.
For growth assays, MEFs were plated in 60-mm tissue culture dishes, and four dishes were counted on the indicated days. Four different passages (passages 1, 5, 12, and 19) were utilized in order to determine the effect of passage number on growth rate. To determine plating efficiency, MEFs were seeded at various densities, and after 15 days in culture, the colonies were fixed, stained, and counted.
Analysis of micronuclei and ratio of normochromatic to polychromatic erythrocytes.
Micronucleus formation and the ratio of normochromatic to polychromatic erythrocytes were determined by previously described procedures (15, 23). Briefly, bone marrow smears from the mice were prepared, and the stained smears were examined to determine the incidence of micronucleated cells in 1,800 polychromatic erythrocytes and the ratio of normochromatic to polychromatic erythrocytes for each animal.
Transformation assay.
The transformation assay was performed as described previously (9, 15). Exponentially growing subconfluent cells were trypsinized and plated 48 h prior to exposure to IR at a dose of 1 Gy. Immediately after irradiation, the cells were trypsinized and replated into 10-cm-diameter culture dishes at cell numbers estimated to result in either 300 viable cells per dish for the assay of neoplastic transformation or 30 viable cells for the cell survival assay. For the assay of neoplastic transformation, cells were grown in Eagle's basal medium supplemented with 10% heat-inactivated FBS, and the culture medium was changed at 12-day intervals during the 6- to 8-week incubation. Cells plated for cell survival determination were incubated as described above. At the end of 12 or 42 days of incubation, cells were fixed in formalin and stained with Giemsa. Cell survival was determined by the colony assay as described above, while neoplastic transformed foci II and III were identified according to the criteria of Reznikoff et al. (30, 31).
Reverse transcriptase PCR of hMOF mRNA.
To compare hMOF mRNA levels in paired tumor and normal tissue samples, reverse transcriptase PCR was performed. mRNAs isolated from paired tumor and normal tissue samples were obtained from the Alvin J. Siteman Cancer Center of the Washington University School of Medicine, St. Louis, MO; the Sidney Kimmel Cancer Center at John Hopkins, Baltimore, MD; and the M.D. Anderson Cancer Center, Houston, TX. The isolated RNAs from tumor and normal tissues were treated with RNase-free DNase (Roche Diagnostic Corporation, Indianapolis, IN) (1 mg/ml) for 2 h at 37°C, followed by heat inactivation at 65°C for 10 min. The reverse transcriptase reaction mixture contained 1 mg/ml DNase-treated RNA, 0.25 mg/ml pdN6 random primers (Roche Diagnostic Corporation), first-strand buffer (GIBCO-BRL), 0.5 mM deoxynucleoside triphosphate (Roche Diagnostic Corporation, Indianapolis, IN), and 200 units Moloney murine leukemia virus reverse transcriptase (GIBCO-BRL), and the mixtures were incubated for 1 h at 37°C. PCR was performed by using gene-specific primers along with the primers for β-actin. cDNAs of paired samples (tumor and normal tissue) were analyzed with ABI Prism 7700 (Applied Biosystems) with SYBR green ROX mix (Abgene, United Kingdom). Each cDNA was analyzed in triplicate, and dihydrofolate reductase (DHFR) was used as an internal control for comparison of the expression status. The relative quantification of hMOF mRNA (compared to DHFR mRNA) was determined as described previously (34, 35). The primer oligonucleotide sequences used for hMOF and DHFR quantification are available upon request. The products of the reverse transcriptase PCRs were analyzed by gel electrophoresis, and they gave single bands of the appropriate amplicon size.
Mice and tumor irradiation.
Eight-week-old NMRI nu/nu male mice were maintained in a pathogen-free mouse colony for the duration of experiments. Mice were randomly distributed four per cage, and each mouse was labeled with a code on the ear. Two million exponentially growing RKO cells with and without hMOF overexpression in a volume of 20 μl were injected (intramuscularly) in the right thigh flank, and the mice were checked daily for tumor appearance as described previously (32). Animals with a tumor of 8 mm in mean diameter were locally irradiated with a single dose of 25 Gy using a 8-MeV linear particle accelerator (dose rate of 1 Gy/min). For the regrowth of tumors, the previously described protocol was followed (32).
Chemotaxis assay.
Migration assays were done in 24-well cell culture chambers with 8-μm-pore membranes. The lower surface of the membrane was precoated with fibronectin (25 μg/ml). Cells were resuspended in chemotaxis buffer (DMEM, 0.1% bovine serum albumin, 12 mM HEPES) at 2.5 × 106 cells/ml, and 150 μl of cells from each sample was loaded into the upper chamber. Chemotaxis buffer (0.6 ml) containing DMEM, 10% FBS, epidermal growth factor (EGF) (20 ng/ml), and insulin growth factor (IGF) (20 ng/ml) was dispensed into the lower chamber. The plates were incubated for 24 h at 37°C in 5% CO2. After incubation, the inserts were removed carefully, and the cells were fixed and stained using the HEMA 3 staining kit (Fisher Scientific Company, MI). Migrated cells were counted in a minimum of five high-power fields (magnification of ×100).
RESULTS
Loss of MOF and H4K16 acetylation results in early embryonic lethality.
MOF mRNA is expressed ubiquitously in all human tissues (Fig. 1A). The mouse Mof gene (mMof) localizes on chromosome 7 (Fig. 1B) and encodes a protein product of ∼58 kDa, a molecular mass similar to that of human MOF protein (Fig. 1C) (12, 42). To determine the physiological significance of mammalian MOF, we disrupted the mMof gene by homologous recombination in embryonic stem cells (Fig. 2A) and generated germ line-transmitting chimeras.
Heterozygous (mMof+/−) intercrosses yielded wild-type (mMof+/+) and heterozygous (mMof+/−) offspring pups but no homozygous mMof null mutant pups (Fig. 2B). The complete absence of homozygous Mof null mutants in all litters examined implied that Mof deficiency is lethal to embryos (Fig. 2B). We then determined the stage of lethality by analyzing embryos from mMof+/− intercrosses at different times postcoitum. In the litters of mice at E7.5 and E8.5 that we dissected, we found approximately 25% empty or necrotic deciduae (Fig. 2C), and all the morphological normal embryos were either wild type or heterozygous (Fig. 2D and data not shown). At E7.5, all the normal embryos (wild type and heterozygotes) had gastrulated, while the Mof null mutant deciduae contained clusters of cells within a small sac or some giant cells only (Fig. 2C), indicating that Mof-deficient embryos are capable of implantation but die prior to the onset of gastrulation.
The observation that no viable mMof−/− embryos are found at the E7.5 stage of development led us to investigate the expression and localization of MOF protein and H4K16ac status during early embryogenesis (single-cell zygote to preimplantation blastocyst) (Fig. 3A). PCR-based genotyping revealed that about 25% of all zygotes did not have the mMof gene (Fig. 3B). MOF protein localized throughout the entire zygotic cell (whether mMof+/+, mMof+/−, or mMof−/−), including the cytoplasmic and chromatin regions, indicating a strong maternal contribution of MOF. In the single-cell zygotes, both male and female pronuclei chromatin in the fertilized egg stained equally strongly for acetylated histone H4K16 (H4K16ac) (Fig. 3C, top panel). All zygotes were positive for MOF and H4K16ac staining; however, about 25% of the zygotes did not have the mMof gene (Fig. 3B). Through the first few cleavages, until the 16-cell stage embryo is reached, neither MOF nor H4K16ac immunofluorescence distinguishes mMof−/− embryos from the mMof+/+ or mMof+/− embryos (Fig. 3C and D). This is most likely due to the abundance of maternal MOF protein present in mMof−/− oocytes, which probably allowed for continued cleavage during early embryonic development and normal levels of H4K16ac in the chromatin. At the 32-cell stage, mMof+/+ or mMof+/− embryos have increasingly distinct nuclear signals for MOF (Fig. 3D, top panel). H4K16ac levels in Mof null mutants at this stage remained unaffected compared to the levels in heterozygous or wild-type embryos.
FIG. 3.

Dynamics of MOF and H4K14Aac during early embryogenesis. (A) Cell proliferation of fertilized zygotes. Some zygotes showed large and slow proliferation as indicated by the white arrow. (B) PCR-based genotyping of zygotes from intercrosses of mMof+/− mice. (C) Maternally expressed MOF and H4K16acetylation in one-, two-, and eight-cell embryos. (D) mMof null embryos still have maternal MOF and H4K16ac until the 32-cell stage. (E) Reduced MOF and H4K16ac in the morula stage of mMof null embryos compared to the corresponding wild-type embryos. (F) mMof null blastocysts differentiated into inner cell mass (ICM) and trophoblasts show loss of MOF and H4K16ac. Note that in the wild-type blastocysts, MOF and H4K16ac staining is more intense in ICM cells than in trophectoderm cells. The arrows point to the H4K16ac-positive metaphase cell. DIC, differential interference contrast.
Starting at the morula stage, marked differences between mMof+/+ and mMof−/− embryos become apparent. As the maternal MOF diminishes, acetylation of H4K16 becomes undetectable from this stage (Fig. 3E). MOF protein and H4K16ac are barely detectable in mMof−/− embryos at the blastocyst stage of development (Fig. 3F). Very interestingly, some cells of the mMof+/+ late morula stage express higher levels of MOF and higher levels of H4K16ac (Fig. 3E, top panel), a pattern seen among several embryos at this stage. The polarized configuration of these cells and the upcoming differentiation of trophectoderm and inner cell mass (ICM) (Fig. 3F) suggest that these cells may be the progenitors of the ICM. This is further substantiated by the appearance of differentiated trophoblasts and pluripotent undifferentiated ICM cells in the mMof +/+ blastocyst (Fig. 3F). ICM cells have convincingly higher levels of MOF protein than trophectoderm cells, and they also have higher levels of H4K16ac. These observations are consistent with previous results with regard to the higher levels of H4K16ac in ES cells compared to trophectoderm (21). The increased acetylation of H4K16 in ES cells is suggestive of the significance of this chromatin modification for rapid proliferation during early embryonic development. Eventually, complete depletion of MOF and loss of H4K16ac from a few blastocysts (following Mendelian ratios) was observed (Fig. 3F and data not shown). This correlated with the embryos exhibiting a marked delay in developmental progression, sluggish hatching and implantation, proliferation arrest, and death before gastrulation. These embryos were identified by genotyping as mMof null embryos (data not shown). Retarded developmental progression became more evident as the embryos reached the blastocyst stages (Fig. 3F). Curiously, both the interphase chromatin and metaphase/anaphase chromosomes show comparable H4K16ac levels that are confined to the DAPI-stained DNA regions (Fig. 3F). A similar phenomenon was reported for H4K16ac in Drosophila (19). The deformed nuclear morphology observed by DAPI staining in the mMof−/− blastomeres at the morula stage and also the blastocyst stage (Fig. 3F) could be a precursor of the upcoming catastrophic effects on development.
Inactivation of ATM and p53 does not rescue lethality of mMof deletion.
Prior studies have reported that depletion of hMOF resulted in a G2/M arrest (46) that could be partly rescued by inactivation of specific cell cycle checkpoint gene products. Therefore, we examined whether the delayed developmental progression and lethality of mMof−/− embryos could be rescued by inactivating genes such as ATM or p53, which are involved in cell cycle control (20, 22, 27, 38). Although knockdown of hMOF in human cells results in a G2/M checkpoint arrest (46), inactivation of ATM or p53 could not rescue or ameliorate the early embryonic lethality of mMof nullizygous embryo (data not shown). We determined the influence on growth due to mMof haploinsufficiency in Atm heterozygous and Atm null backgrounds by monitoring the health and survival of mMof+/+/Atm+/+ (n = 34), mMof+/−/Atm+/+ (n = 45), mMof+/+/Atm+/− (n = 30), mMof+/−/Atm+/− (n = 38), mMof+/+/Atm−/− (n = 24), and mMof+/−/Atm−/− (n = 19) mice over a period of 1 year (Fig. 4A). In contrast to ∼92% of mMof+/+/Atm+/+, mMof+/+/Atm+/−, or mMof+/−/Atm+/+ mice, only ∼76% mice with the mMof+/−/Atm+/− genotype remained healthy and survived the first year of life. mMof heterozygosity in Atm null background (mMof+/−/Atm−/−) resulted in a modest weight reduction and earlier death compared to Atm null mice (Fig. 4A and B). Although the differences in survival are modest, they are statistically significant (P < 0.05). Interestingly, mMof heterozygosity in a p53-deficient background (mMof+/−/p53−/−) resulted in death at an earlier age compared to p53 null mice (Fig. 4C and data not shown).
FIG. 4.
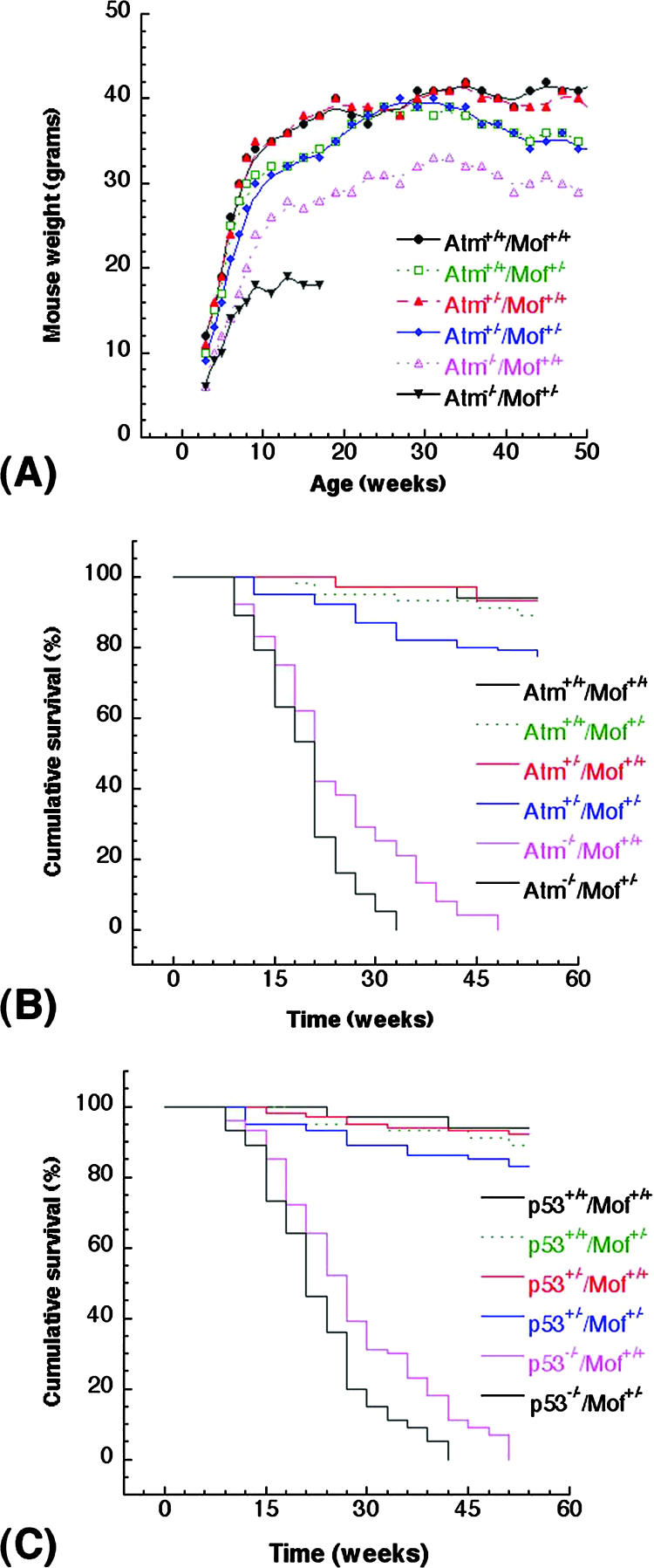
Influence of mMof heterozygosity on the survival of mice with different genotypes. (A and B) Body weight (A) and cumulative survival (B) of Atm+/+/Mof+/+, Atm+/+/Mof+/−, Atm+/−/Mof+/+, Atm+/−/Mof+/−, and Atm−/−/Mof+/− mice. The mice were weighed weekly from weaning (∼21 days) to adulthood. The differences in the weight and survival of Atm+/−/Mof+/− and Atm−/−/Mof+/− mice are modest, but the differences are statistically significant (P < 0.05 determined by the chi-square test). (C) Cumulative survival of p53+/+/Mof+/+, p53+/+/Mof+/−, p53+/−/Mof+/+, p53+/−/Mof+/−, and p53−/−/Mof+/− mice. The cumulative survival was plotted according to Kaplan-Meier analysis.
Reduced mMof levels correlate with decreased H4K16ac, cell proliferation, cell survival, and increased genomic instability.
To examine the biological function of mMof at the cellular level, we attempted to generate homozygous mutant ES cells by increasing the G418 concentration to force homogenetization and by sequential gene targeting of mMof+/− ES cells using a mMof-hygromycin resistance gene vector (data not shown). However, no homozygous mMof−/− ES cells could be obtained, suggesting that Mof function is indispensable for viability and/or proliferation of ES cells.
Thus, we designed a conditional mMof allele (Mofflox) by flanking exons 1 to 3 with loxP sites (Fig. 5A) and generated mMofflox/+ ES cells. To inactivate the conditional allele by cre-mediated recombination, we introduced the tamoxifen-inducible creERT2 fusion gene into the ROSA26 locus (ROSA26creERT2/+) using a knock-in strategy and subsequently generated mMofΔflox/+/Rosa26creERT2/+ and mMofΔflox/flox/Rosa26creERT2/+ ES cells. The phenotypic consequences of MOF deficiency were then determined by tamoxifen-induced expression of Cre recombinase and inactivation of the conditional Mofflox allele (Fig. 5A and B). Deletion of mMof resulted in the subsequent loss of MOF protein, loss of acetylation of histone H4 at K16 (Fig. 5C), and decreased cellular proliferation (Fig. 5D). Heterozygous mMof (mMof+/−) MEFs also had reduced levels of MOF (Fig. 6A) and demonstrated a modest increase in population doubling time (Fig. 6B), decreased IR-induced γ-H2AX focus formation (Fig. 6C), and a slight decrease in survival after IR exposure (Fig. 6D). The decreased survival in mMof+/− MEFs could be due to defective repair of IR-induced single- and double-strand DNA breaks. MOF, a chromatin-modifying factor, may have a critical role in the repair of DNA cross-links. We also compared the effect of mitomycin C-induced DNA cross-links on mMof +/+ and mMof +/− MEFs. Cell killing after mitomycin C treatment was higher in mMof+/− MEFs than in mMof+/+ MEFs (Fig. 6E).
FIG. 6.

Haploinsufficiency of mMof influences the DNA damage response. (A) Comparison of mMof levels in mouse embryonic fibroblasts established from wild-type (mMof+/+) and heterozygous (mMof+/−) mMof embryos. (B) Population doubling time of heterozygous mMof MEFs and wild-type MEFs. ATM-deficient MEFs are used as a positive control. (C) γ-H2AX focus formation after exposure to radiation (1 Gy). (D) Heterozygous mMof+/− cells are more sensitive to increased doses of IR exposure (>4 Gy) than wild-type mMof+/+ cells are, and the differences are statistically significant (P < 0.05 determined by chi-square test). (E) Survival after mitomycin C treatment. More mMof+/− cells are killed compared to mMof+/+ cells, and the differences are statistically significant (P < 0.001, as determined by the chi-square test).
One mechanism that could contribute to the altered growth characteristics of haploinsufficient mMof cells is increased genomic instability. This was substantiated by the observation of a modestly increased ratio of normochromatic to polychromatic erythrocytes and an increased frequency of micronucleus occurrence in erythrocytes from mMof+/− mice compared to mMof+/+ mice (Table 1). The higher frequency of spontaneous micronucleus formation observed in mMof+/− mice is consistent with the higher spontaneous chromosome aberration rate observed in cultured mMof+/− MEFs and in phytohemagglutinin-stimulated lymphocytes isolated from mMof+/− mice (Table 2 and data not shown). A reduced level of IR-induced γ-H2AX focus formation was also observed in heterozygous mMof cells compared to wild-type mMof cells. Interestingly, reduced levels of mMof increased genomic instability and decreased survival after IR or mitomycin C exposure, while total loss resulted in cell lethality.
TABLE 1.
Ratio of normochromatic erythrocytes to polychromatic erythrocytes and micronucleus formation in mMof+/− and mMof+/− micea
| Genotype | Normochromatic erythrocyte/polychromatic erythrocyte ratio
|
No. of micronucleated cells/1,800 polychromatic erythrocytes/mouse
|
||
|---|---|---|---|---|
| Mean | Range | Mean | Range | |
| mMof+/+ | 1.49 | 0.43-2.88 | 1.06 | 0-3.69 |
| mMof+/− | 1.68 | 0.51-3.19 | 1.22 | 0-3.90 |
Six age-matched sets of male and female mMof+/+ and mMof+/− mice were sacrificed, and bone marrow smears were made as described previously (24). mMof+/− mice have a modest increase in the ratio of normochromatic erythrocytes to polychromatic erythrocytes and a higher frequency of micronucleated cells than mMof+/+ mice do. The differences in the ratio of normochromatic erythrocytes to polychromatic erythrocytes and the higher frequency of micronucleated cells among Mof+/− and Mof+/+ mice are statistically significant (P < 0.05, as determined by the chi-square test).
TABLE 2.
Frequencies of chromosomal aberrations in mMof+/+ and mMof+/− MEFsa
| Genotype | No. of chromosome gaps + breaks/ 200 cells at metaphase | No. of chromatid gaps + breaks/ 200 cells at metaphase | No. of chromosome end associations/ 100 cells at metaphase | No. of bridges/ 100 cells at anaphase |
|---|---|---|---|---|
| mMof+/+ | 3 | 5 | 8 | 2 |
| mMof+/− | 10 | 15 | 19 | 8 |
Chromosome gaps and breaks, chromatid gaps and breaks, and chromosome end associations were analyzed at metaphase, and bridges were analyzed at anaphase in cells with and without haploinsufficient mMof. The aberrations in mMof+/− haploinsufficient cells are significantly different from mMof+/+ cells as assessed by chi-square analysis (P < 0.05, as determined by the chi-square test).
MOF levels correlate with the frequency of oncogenic transformation.
One of the hallmarks of malignant transformation is genomic instability, which promotes a wide range of mutations, including structural and numerical alterations of chromosomes. In order to gain further insight into the function of mammalian MOF during oncogenesis, we examined cells expressing either mutant (ΔmMof [Fig. 7A and unpublished data]) or wild-type mMof for IR-induced cell killing and oncogenic transformation. Cells expressing ectopic mMof have higher levels of mMof and H4K16ac than cells expressing ΔmMof do (Fig. 7B). Cells expressing ΔmMof had decreased survival (Fig. 7C) and reduced spontaneous and IR-induced oncogenic transformation rates compared to cells overexpressing mMof (Fig. 7D) as determined by focus formation described previously (15). We also determined the levels of mMof and H4K16ac in mouse tumors and normal tissues and found that tumor tissues have increased levels of MOF and H4K16ac in comparison to normal tissues (Fig. 7E).
FIG. 7.

The level of mMof protein correlates with cell survival and oncogenic transformation. (A) Strategy for cloning the mMof gene and its domains. mMof protein is denoted at the top with a line representing 458 amino acids. Five different fragments of mMof cDNA encoding full-length mMof protein with three domains (chromodomain region, zinc finger, and acetyl coenzyme A (acetyl Co-A) binding site) are diagrammed. The five different fragments were generated by PCR using specific primers. All clones are drawn to approximate scale. Each fragment is subcloned into pBABE retroviral or pcDNA3.1 vectors. The fragment designated ΔmMof showed dominant-negative activity for cell survival and was used in transformation assays. (B) Western blot analysis to determine the levels of MOF and H4K16ac in mouse 435 and 743 fibroblasts used in the analysis of the assays shown in panels C and D. 435 fibroblasts are mouse cells with wild-type ATM, while 743 fibroblasts are ATM null cells. Lanes 1, 3, and 4 are 435 cells transfected with the described cDNAs. HeLa cell extracts were used as a positive control for detection of MOF protein in mouse cell lines. In panels B and C, the control is 435 mouse fibroblasts. (C) Cell survival of 435 fibroblasts transfected with empty vector, 435 fibroblasts expressing wild-type mMof or ΔmMof, and 743 cells (positive control) determined by colony-forming assay. (D) Transformation incidence following exposure to 1 Gy of gamma rays in 435 (Atm+/+) mouse cells, 435 cells with empty vector, 435 cells with wild-type mMof, and 435 cells with ΔmMof expression. The spontaneous and IR-induced morphological transformation of 435 cells with and without overexpression of mMof was determined as described previously (15). (E) Levels of MOF and H4K16ac in normal thymus (N) and tumor tissues (T1 and T2) from mice. T1 and T2 are samples of two different thymic lymphomas.
Cells with prolonged life span have higher hMOF and H4K16ac levels.
A prerequisite for oncogenic transformation is the acquisition of prolonged life span. Since most immortalized cells and tumors have telomerase activity (16) and expression of the catalytic unit of telomerase (hTERT) prolongs the life span of primary fibroblasts (3, 47, 48), we determined whether MOF levels change in cells with a prolonged life span. hMOF levels were compared in isogenic cells with and without hTERT expression. Comparison of hMOF and H4K16ac levels in these isogenic cell lines revealed that hMOF and H4K16ac levels are higher in cells where life span has been prolonged by ectopic expression of hTERT (Fig. 8A).
FIG. 8.

hMOF and H4K16ac levels in primary cells and tumor cell lines. (A) hMOF and H4K16ac levels in human fibroblasts with and without ectopic expression of hTERT and in tumor cell lines. (B) Comparison of hMOF RNA levels in different tumors and normal tissues: breast (a), lung (b), and prostate (c). (C and D) hMOF and H4K16ac levels in different tumors (C) and immortalized cell lines (D).
Tumors and tumor-derived cell lines have not lost MOF expression and H4K16ac levels.
As hMOF is required for acetylation of H4K16 and cellular proliferation, we compared hMOF expression in various tumors and normal tissues. Consistent with the higher levels of hMOF seen in rapidly proliferating cells, hMOF mRNA expression was detected in all of the 300 different breast (n = 132), lung (n = 74), and prostate (n = 94) tumors examined (Fig. 8B and data not shown). Irrespective of tumor type, approximately 25 percent of normal tissue samples had reduced levels of hMOF mRNA expression than the corresponding tumor tissue sample did (Fig. 8B and data not shown). However, hMOF protein and H4K16ac were found in the entire tumor and immortalized cell lines examined (Fig. 8C and D and data not shown). Tumor cells consistently demonstrated higher levels of mMof and H4K16ac, further supporting the notion about the relationship between the status of H4K16ac status and cellular proliferation.
MOF levels correlate with tumor growth.
To determine whether hMOF levels influence tumor growth, we used a standard nude mouse xenograft assay to examine tumor growth and its response to IR exposure. Knockdown of hMOF in colorectal carcinoma (RKO) cells (Fig. 9A) inhibited cell growth. Conversely, overexpression of hMOF increased H4K16ac (Fig. 9B) and accelerated proliferation (Fig. 9C). RKO cells, with or without overexpression of hMOF, were injected into mice, and when the tumors reached 8 mm in size, the tumors were locally irradiated with a single 25-Gy dose. Untreated tumors arising from injected RKO cells overexpressing hMOF grew rapidly compared to control cells (Fig. 9D). Tumors from RKO cells, with and without hMOF overexpression, grew at relatively constant rates, whereas radiation treatment (single dose of 25 Gy) caused temporary shrinkage of the tumors, followed by regrowth in most of the tumors. Interestingly, tumors from RKO cells with hMOF overexpression regrew faster than control cells. Two months after irradiation, ∼20% of the tumors from control cells were below detectable levels compared to only ∼8% tumors derived from cells overexpressing hMOF. An immediate relapse of tumor growth was much more prominent in cells expressing hMOF. These results suggest that hMOF promotes tumor growth.
FIG. 9.

MOF influences cell population doubling and tumor growth. (A) Small interfering RNA (siRNA)-mediated reduction of hMOF protein in RKO cells. hMOF siRNA and control Luc siRNA have been described previously (12). (B) RKO cells transfected with green fluorescent protein-tagged hMOF (GFP-hMOF) have higher levels of H4K16 acetylation. (C) Population doubling of RKO cells with overexpression of hMOF and knockdown of hMOF. Knockdown of hMOF was done as described previously (12). (D) The effect of gamma irradiation treatment on tumor growth in mice. Eight-week-old NMRI nu/nu male mice were injected with 2 million exponentially growing RKO cells with and without overexpression of hMOF in the right thigh. The data points are the averages of eight mice for untreated tumors, six mice for uncured treated tumors, and three mice for the locally controlled tumors. Animals with a tumor size 8 mm in diameter were locally irradiated with a single dose of 25 Gy and examined for tumor size. (E) For chemotaxis assay, cells were seeded in the presence of EGF and IGF (20 ng/ml) by using 24-well plates containing 8.0-μm-pore inserts (Costar Corporation, Kennebunk, ME). FBS (10%) was used as a positive control. The number of cells in five high-power fields (5 HPF) (magnification of ×100) was counted. The experiments were done three times, and the data are presented as the means ± standard errors (error bars) (P < 0.05 determined by the chi-square test).
We also tested whether hMOF could influence cell migration. RKO cells with and without hMOF overexpression were subjected to a chemotaxis assay conducted in the presence and absence of EGF and IGF, factors shown to induce chemotaxis in various cell types. FBS (10%) was utilized as a positive control. Both cell types migrated towards 10% FBS, but there was a modest enhancement of migration capability in cells overexpressing hMOF compared to control cells (Fig. 9E). Cells overexpressing hMOF also showed significantly increased migration towards both EGF and IGF compared to control cells. These results are consistent with a role for MOF in oncogenesis.
DISCUSSION
There are two known histone acetyltransferase complexes that modify histone H4 at K16; the MSL (male-specific silencing) complex in humans and flies, and the SAS (something about silencing) complex in yeast cells (1, 13, 41). The MSL complex specifically acetylates H4K16 in Drosophila and humans. In human cells, knockdown of hMOF results in the loss of H4K16ac (12, 41, 46). H4K16 acetylation has been shown to destabilize nucleosomes and correlates with regions of chromatin decondensation (39). Here, we report the first extensive study showing that MOF is required for proliferation during embryogenesis and in oncogenesis. However, the exact role of MOF in these processes remains to be determined. An intriguing possibility is that MOF acetylation of histone H4 at K16 may have a regulatory role during transcription (1), which is poorly understood in mammals, but well documented in yeast and Drosophila (36, 37). The significance of H4K16 acetylation lies in the fact that in budding yeast, most of the genome exists in a decondensed state with more than three fourths of histone H4 acetylated at lysine 16 (28, 37). In Drosophila, the transcriptionally enhanced X chromosome of male flies, a site of ubiquitous H4K16ac, is also decondensed (1). In human cell lines, decreased levels of hMOF correlate with loss of H4K16ac and is associated with defective DNA repair (12). In this study, we found that the loss of H4K16ac correlated with the progressive depletion of maternal MOF during embryogenesis in mMof null embryos. In wild-type mMof embryos, the rapidly proliferating ICM cells have convincingly higher levels of MOF protein than trophectoderm cells, which is consistent with previous results showing higher levels of acetylated H4K16 in ES cells than in trophectoderm cells. Thus, increased acetylation of H4K16 in ES cells is indicative of the significance of this chromatin modification for proliferation during development. Complete ablation of MOF and loss of H4K16ac results in a marked delay in developmental progression, sluggish hatching and implantation, proliferation arrest, and death.
The loss of cell proliferation could be due to cell cycle arrest specifically induced by p53 and by ATM as suggested by a previous report of G2/M arrest in hMOF knockdown cells (46). In addition, hMOF has been reported to acetylate p53 at lysine 120, and this acetylation may help distinguish between the cell cycle arrest and apoptotic functions of p53 (45). If the mMof depletion-dependent cell cycle arrest were the cause of embryonic lethality or cellular death, then depletion of p53 or ATM should have rescued or at least partially ameliorated the observed embryonic lethality. However, our results do not support this hypothesis.
Inactivation of the mMof gene in ES cells caused depletion of Mof protein accompanied with loss of acetylated H4K16 and resulted in reduced proliferation and ultimately cell death. Taken together with the lack of success in deriving homozygous Mof mutant ES cells, these experiments strongly suggest that Mof function is indispensable for cellular proliferation and/or viability. These results also raised the question of whether tumor cells can survive in the absence of MOF and the associated loss of H4K16 acetylation.
Multiple assays were utilized previously to quantitate H4K16 monoacetylation at the global level and genome region/locus-specific level, and the data suggested that loss of monoacetylation at H4K16 occurs in tumors and tumor-derived cell lines (8). Our results, however, demonstrate that tumors and tumor-derived cell lines have similar or higher levels of hMOF or H4K16ac compared to those of normal control cells (Fig. 7E and 8 and data not shown). Similar results were found in multiple different transformed cell lines (Fig. 8 and data not shown). Moreover, we found that primary cells have lower levels of hMOF and H4K16ac than the corresponding isogenic hTERT immortalized cells do (Fig. 8A). These in vitro results were also supported by the in vivo hMOF expression analysis of 300 different tumors, which demonstrated no loss of hMOF expression. In addition, tumor cells with higher levels of hMOF had faster initial growth and immediate regrowth (recurrence) of tumors after irradiation exposure (Fig. 9D), suggesting that hMOF promotes cellular proliferation. Therefore, after examination of a large number of tumor-derived cell lines and tumor samples, we propose that hMOF and H4K16ac may be required for the process of transformation, either in vitro or in vivo.
Intriguingly, MOF overexpression was associated with both increased K16H4 acetylation and increased cell proliferation (12, 46; also the present study), while cells treated with SIRT1 inhibitors had increased K16H4 acetylation levels but a reduction in cell proliferation (29). Thus, although MOF seems to be closely associated with cell proliferation, the relationship between the H4K16 acetylation status and cell proliferation requires further study.
The loss of MOF and H4K16ac correlates with increased genomic instability (Tables 1 and 2 and Fig. 6) (12, 46), which is considered an important step in cancer development because it can greatly accelerate the genetic changes leading to tumor cell progression (14). However, induction of genomic instability and increased radiosensitivity caused by depletion of MOF result in decreased oncogenic transformation, whereas overexpression of MOF resulted in increased oncogenic transformation (Fig. 7C and D); this suggests that the loss of MOF is probably not a marker of oncogenesis.
Collectively, our data suggest that the requirement for MOF during tumor cell proliferation is similar to that during embryogenesis prior to tissue differentiation. The role of MOF in cellular proliferation is supported by the following. (i) There is a strong correlation observed between the ectopic expression of the catalytic unit of telomerase (hTERT) and the levels of hMOF and H4K16ac seen in isogenic cell systems (Fig. 8A). (ii) Depletion of MOF results in cellular lethality. (iii) Overexpression of MOF results in enhanced oncogenic transformation and faster relapse of tumor growth after radiation treatment (Fig. 7D and 9D and E). Thus, the results suggest that expression of hMOF and acetylation of H4K16 are linked with proliferation status and is supported by the fact that all the tumor-derived cell lines and immortalized cell lines examined have both hMOF and acetylated H4K16. These novel observations presented here provide a foundation for future molecular studies on the significance of epigenetic modifications in varied contexts of embryonic development, tumorigenesis, and DNA metabolism.
Acknowledgments
We thank J. Michael White and Raju Kucherlapati for their advice and support. Thanks are due to Manel Esteller and Clayton Hunt for their suggestions and comments on the manuscript and to L. Mao for providing us with the samples of mRNA from tumor and normal tissue samples.
This investigation was supported by funds from NIH (CA123232 and CA10445), Department of Defense, and the Department of Radiation Oncology of the Washington University School of Medicine to T.K.P., and NCI grant PO1 CA97403 (Project 5) to T.L.
Footnotes
Published ahead of print on 29 October 2007.
REFERENCES
- 1.Akhtar, A., and P. B. Becker. 2000. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 5367-375. [DOI] [PubMed] [Google Scholar]
- 2.Bird, A. W., D. Y. Yu, M. G. Pray-Grant, Q. Qiu, K. E. Harmon, P. C. Megee, P. A. Grant, M. M. Smith, and M. F. Christman. 2002. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419411-415. [DOI] [PubMed] [Google Scholar]
- 3.Bodnar, A. G., M. Ouellette, M. Frolkis, S. E. Holt, C. P. Chiu, G. B. Morin, C. B. Harley, J. W. Shay, S. Lichtsteiner, and W. E. Wright. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279349-352. [DOI] [PubMed] [Google Scholar]
- 4.Bone, J. R., J. Lavender, R. Richman, M. J. Palmer, B. M. Turner, and M. I. Kuroda. 1994. Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev. 896-104. [DOI] [PubMed] [Google Scholar]
- 5.Chua, K. F., R. Mostoslavsky, D. B. Lombard, W. W. Pang, S. Saito, S. Franco, D. Kaushal, H. L. Cheng, M. R. Fischer, N. Stokes, M. M. Murphy, E. Appella, and F. W. Alt. 2005. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 267-76. [DOI] [PubMed] [Google Scholar]
- 6.Dou, Y., T. A. Milne, A. J. Tackett, E. R. Smith, A. Fukuda, J. Wysocka, C. D. Allis, B. T. Chait, J. L. Hess, and R. G. Roeder. 2005. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121873-885. [DOI] [PubMed] [Google Scholar]
- 7.Feil, R., J. Wagner, D. Metzger, and P. Chambon. 1997. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun. 237752-757. [DOI] [PubMed] [Google Scholar]
- 8.Fraga, M. F., E. Ballestar, A. Villar-Garea, M. Boix-Chornet, J. Espada, G. Schotta, T. Bonaldi, C. Haydon, S. Ropero, K. Petrie, N. G. Iyer, A. Perez-Rosado, E. Calvo, J. A. Lopez, A. Cano, M. J. Calasanz, D. Colomer, M. A. Piris, N. Ahn, A. Imhof, C. Caldas, T. Jenuwein, and M. Esteller. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37391-400. [DOI] [PubMed] [Google Scholar]
- 9.Freyer, G. A., D. A. Palmer, Y. Yu, R. C. Miller, and T. K. Pandita. 1996. Neoplastic transformation of mouse C3H10T1/2 cells following exposure to neutrons does not involve mutation of ras gene as analyzed by SSCP and cycle sequencing. Mutat. Res. 357237-244. [DOI] [PubMed] [Google Scholar]
- 10.Gage, B. M., D. Alroy, C. Y. Shin, O. N. Ponomareva, S. Dhar, G. G. Sharma, T. K. Pandita, M. J. Thayer, and M. S. Turker. 2001. Spontaneously immortalized cell lines obtained from adult Atm null mice retain sensitivity to ionizing radiation and exhibit a mutational pattern suggestive of oxidative stress. Oncogene 204291-4297. [DOI] [PubMed] [Google Scholar]
- 11.Guo, K., J. E. McMinn, T. Ludwig, Y. H. Yu, G. Yang, L. Chen, D. Loh, C. Li, S. Chua, Jr., and Y. Zhang. 2007. Disruption of peripheral leptin signaling in mice results in hyperleptinemia without associated metabolic abnormalities. Endocrinology 1483987-3997. [DOI] [PubMed] [Google Scholar]
- 12.Gupta, A., G. G. Sharma, C. S. H. Young, M. Agarwal, E. R. Smith, T. T. Paull, J. C. Lucchesi, K. K. Khanna, T. Ludwig, and T. K. Pandita. 2005. Involvement of human MOF in ATM function. Mol. Cell. Biol. 255292-5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilfiker, A., D. Hilfiker-Kleiner, A. Pannuti, and J. C. Lucchesi. 1997. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 162054-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoeijmakers, J. H. 2001. Genome maintenance mechanisms for preventing cancer. Nature 411366-374. [DOI] [PubMed] [Google Scholar]
- 15.Hunt, C. R., D. J. Dix, G. G. Sharma, R. K. Pandita, A. Gupta, M. Funk, and T. K. Pandita. 2004. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol. Cell. Biol. 24899-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim, N. W., M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich, and J. W. Shay. 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 2662011-2015. [DOI] [PubMed] [Google Scholar]
- 17.Kimura, A., K. Matsubara, and M. Horikoshi. 2005. A decade of histone acetylation: marking eukaryotic chromosomes with specific codes. J. Biochem. (Tokyo) 138647-662. [DOI] [PubMed] [Google Scholar]
- 18.Kusch, T., L. Florens, W. H. Macdonald, S. K. Swanson, R. L. Glaser, J. R. Yates III, S. M. Abmayr, M. P. Washburn, and J. L. Workman. 2004. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 3062084-2087. [DOI] [PubMed] [Google Scholar]
- 19.Lavender, J. S., A. J. Birley, M. J. Palmer, M. I. Kuroda, and B. M. Turner. 1994. Histone H4 acetylated at lysine 16 and proteins of the Drosophila dosage compensation pathway co-localize on the male X chromosome through mitosis. Chromosome Res. 2398-404. [DOI] [PubMed] [Google Scholar]
- 20.Morgan, S. E., and M. B. Kastan. 1997. p53 and ATM: cell cycle, cell death, and cancer. Adv. Cancer Res. 711-25. [DOI] [PubMed] [Google Scholar]
- 21.O'Neill, L. P., and B. M. Turner. 1995. Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J. 143946-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandita, T. K. 2003. A multifaceted role for ATM in genome maintenance. Expert Rev. Mol. Med. 51-21. [DOI] [PubMed] [Google Scholar]
- 23.Pandita, T. K. 1988. Assessment of the mutagenic potential of a fungicide Bavistin using multiple assays. Mutat. Res. 204627-643. [DOI] [PubMed] [Google Scholar]
- 24.Pandita, T. K. 1983. Mutagenic studies on the insecticide Metasystox-R with different genetic systems. Mutat. Res. 12497-102. [DOI] [PubMed] [Google Scholar]
- 25.Pandita, T. K. 2006. Role of mammalian Rad9 in genomic stability and ionizing radiation response. Cell Cycle 51289-1291. [DOI] [PubMed] [Google Scholar]
- 26.Pandita, T. K., and W. N. Hittelman. 1992. Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat. Res. 13094-103. [PubMed] [Google Scholar]
- 27.Pandita, T. K., H. B. Lieberman, D. S. Lim, S. Dhar, W. Zheng, Y. Taya, and M. B. Kastan. 2000. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 191386-1391. [DOI] [PubMed] [Google Scholar]
- 28.Peterson, C. L., and M. A. Laniel. 2004. Histones and histone modifications. Curr. Biol. 14R546-R551. [DOI] [PubMed] [Google Scholar]
- 29.Pruitt, K., R. L. Zinn, J. E. Ohm, K. M. McGarvey, S. H. Kang, D. N. Watkins, J. G. Herman, and S. B. Baylin. 2006. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reznikoff, C. A., J. S. Bertram, D. W. Brankow, and C. Heidelberger. 1973. Quantitative and qualitative studies of chemical transformation of cloned C3H mouse embryo cells sensitive to postconfluence inhibition of cell division. Cancer Res. 333239-3249. [PubMed] [Google Scholar]
- 31.Reznikoff, C. A., D. W. Brankow, and C. Heidelberger. 1973. Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Res. 333231-3238. [PubMed] [Google Scholar]
- 32.Sawant, S. G., V. Gregoire, S. Dhar, C. B. Umbricht, S. Cvilic, S. Sukumar, and T. K. Pandita. 1999. Telomerase activity as a measure for monitoring radiocurability of tumor cells. FASEB J. 131047-1054. [DOI] [PubMed] [Google Scholar]
- 33.Scherthan, H., M. Jerratsch, S. Dhar, Y. A. Wang, S. P. Goff, and T. K. Pandita. 2000. Meiotic telomere distribution and Sertoli cell nuclear architecture are altered in Atm- and Atm-p53-deficient mice. Mol. Cell. Biol. 207773-7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma, G. G., A. Gupta, H. Wang, H. Scherthan, S. Dhar, V. Gandhi, G. Iliakis, J. W. Shay, C. S. Young, and T. K. Pandita. 2003. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 22131-146. [DOI] [PubMed] [Google Scholar]
- 35.Sharma, G. G., K. K. Hwang, R. K. Pandita, A. Gupta, S. Dhar, J. Parenteau, M. Agarwal, H. J. Worman, R. J. Wellinger, and T. K. Pandita. 2003. Human heterochromatin protein 1 isoforms HP1Hsα and HP1Hsβ interfere with hTERT-telomere interactions and correlate with changes in cell growth and response to ionizing radiation. Mol. Cell. Biol. 238363-8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shia, W. J., B. Li, and J. L. Workman. 2006. SAS-mediated acetylation of histone H4 Lys 16 is required for H2A.Z incorporation at subtelomeric regions in Saccharomyces cerevisiae. Genes Dev. 202507-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shia, W. J., S. G. Pattenden, and J. L. Workman. 2006. Histone H4 lysine 16 acetylation breaks the genome's silence. Genome Biol. 7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3155-168. [DOI] [PubMed] [Google Scholar]
- 39.Shogren-Knaak, M., H. Ishii, J. M. Sun, M. J. Pazin, J. R. Davie, and C. L. Peterson. 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311844-847. [DOI] [PubMed] [Google Scholar]
- 40.Smilenov, L. B., S. E. Morgan, W. Mellado, S. G. Sawant, M. B. Kastan, and T. K. Pandita. 1997. Influence of ATM function on telomere metabolism. Oncogene 152659-2665. [DOI] [PubMed] [Google Scholar]
- 41.Smith, E. R., C. Cayrou, R. Huang, W. S. Lane, J. Cote, and J. C. Lucchesi. 2005. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 259175-9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith, E. R., A. Pannuti, W. Gu, A. Steurnagel, R. G. Cook, C. D. Allis, and J. C. Lucchesi. 2000. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol. 20312-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sterner, D. E., and S. L. Berger. 2000. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64435-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suka, N., K. Luo, and M. Grunstein. 2002. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine 16 and spreading of heterochromatin. Nat. Genet. 32378-383. [DOI] [PubMed] [Google Scholar]
- 45.Sykes, S. M., H. S. Mellert, M. A. Holbert, K. Li, R. Marmorstein, W. S. Lane, and S. B. McMahon. 2006. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24841-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taipale, M., S. Rea, K. Richter, A. Vilar, P. Lichter, A. Imhof, and A. Akhtar. 2005. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol. Cell. Biol. 256798-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaziri, H., and S. Benchimol. 1998. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol. 8279-282. [DOI] [PubMed] [Google Scholar]
- 48.Wood, L. D., T. L. Halvorsen, S. Dhar, J. A. Baur, R. K. Pandita, W. E. Wright, M. P. Hande, G. Calaf, T. K. Hei, F. Levine, J. W. Shay, J. J. Wang, and T. K. Pandita. 2001. Characterization of ataxia telangiectasia fibroblasts with extended life-span through telomerase expression. Oncogene 20278-288. [DOI] [PubMed] [Google Scholar]
- 49.Wright, P. W., L. C. Bolling, M. E. Calvert, O. F. Sarmento, E. V. Berkeley, M. C. Shea, Z. Hao, F. C. Jayes, L. A. Bush, J. Shetty, A. N. Shore, P. P. Reddi, K. S. Tung, E. Samy, M. M. Allietta, N. E. Sherman, J. C. Herr, and S. A. Coonrod. 2003. ePAD, an oocyte and early embryo-abundant peptidylarginine deiminase-like protein that localizes to egg cytoplasmic sheets. Dev. Biol. 25673-88. [DOI] [PubMed] [Google Scholar]
- 50.Ziv, S., O. Brenner, N. Amariglio, N. I. Smorodinsky, R. Galron, D. V. Carrion, W. Zhang, G. G. Sharma, R. K. Pandita, M. Agarwal, R. Elkon, N. Katzin, I. Bar-Am, T. K. Pandita, R. Kucherlapati, G. Rechavi, Y. Shiloh, and A. Barzilai. 2005. Impaired genomic stability and increased oxidative stress exacerbate different features of ataxia-telangiectasia. Hum. Mol. Genet. 142929-2943. [DOI] [PubMed] [Google Scholar]