

Abstract
The relationship between Ca2+ release (“Ca2+ sparks”) through ryanodine-sensitive Ca2+ release channels in the sarcoplasmic reticulum and KCa channels was examined in smooth muscle cells from rat cerebral arteries. Whole cell potassium currents at physiological membrane potentials (−40 mV) and intracellular Ca2+ were measured simultaneously, using the perforated patch clamp technique and a laser two-dimensional (x–y) scanning confocal microscope and the fluorescent Ca2+ indicator, fluo-3. Virtually all (96%) detectable Ca2+ sparks were associated with the activation of a spontaneous transient outward current (STOC) through KCa channels. A small number of sparks (5 of 128) were associated with currents smaller than 6 pA (mean amplitude, 4.7 pA, at −40 mV). Approximately 41% of STOCs occurred without a detectable Ca2+ spark. The amplitudes of the Ca2+ sparks correlated with the amplitudes of the STOCs (regression coefficient 0.8; P < 0.05). The half time of decay of Ca2+ sparks (56 ms) was longer than the associated STOCs (9 ms). The mean amplitude of the STOCs, which were associated with Ca2+ sparks, was 33 pA at −40 mV. The mean amplitude of the “sparkless” STOCs was smaller, 16 pA. The very significant increase in KCa channel open probability (>104-fold) during a Ca2+ spark is consistent with local Ca2+ during a spark being in the order of 1–100 μM. Therefore, the increase in fractional fluorescence (F/Fo) measured during a Ca2+ spark (mean 2.04 F/Fo or ∼310 nM Ca2+) appears to significantly underestimate the local Ca2+ that activates KCa channels. These results indicate that the majority of ryanodine receptors that cause Ca2+ sparks are functionally coupled to KCa channels in the surface membrane, providing direct support for the idea that Ca2+ sparks cause STOCs.
Keywords: Ca2+ sparks, ryanodine receptor, sarcoplasmic reticulum, potassium currents, smooth muscle
introduction
Cytoplasmic Ca2+ ions, which enter the cell's cytoplasm through L-type voltage-dependent channels in the plasma membrane and through Ca2+ release channels in the sarcoplasmic reticulum (SR),1 regulate contraction of arterial smooth muscle by activating myosin light chain kinase. Under most circumstances, this activating Ca2+ will be broadly (“globally”) distributed throughout the cytoplasm and be sufficiently long lasting for Ca2+ to approach equilibrium with myosin light chain kinase and major calcium buffers. Thus, global Ca2+ regulates contraction of arterial smooth muscle. However, in the immediate vicinity of open Ca2+ channels, “local” Ca2+ concentrations are not in equilibrium with Ca2+ buffers, and therefore could reach much higher levels than global concentrations (Neher, 1998). Local Ca2+ transients (“Ca2+ sparks”) caused by opening of ryanodine-sensitive Ca2+ release channels (RyR) in the SR have been detected in arterial smooth muscle, using a laser scanning confocal microscope and the fluorescent indicator fluo-3 (Nelson et al., 1995; see also Bonev et al., 1997; Jaggar et al., 1998; Porter et al., 1998). Calcium sparks have been subsequently measured in myocytes from portal vein (Mironneau et al., 1996), airway (Sieck et al., 1997), esophagus (Kirber et al., 1997), and ileum (Gordienko et al., 1998).
We proposed that these calcium sparks serve as local calcium signals to activate KCa channels in the surface membrane (Nelson et al., 1995; see also Fay, 1995), based on a number of lines of evidence. (a) SR can make intimate contacts with the plasma membrane (Devine, et al., 1972; Somlyo, 1985). (b) Immunostaining indicating that RyRs are distributed along the cell membrane (Gollasch et al., 1998). (c) Calcium sparks occurred close to the cell membrane (Nelson et al., 1995). (d) Calcium sparks had similar kinetics and frequencies as transient KCa channel currents (Nelson et al., 1995; Bonev et al., 1997; Jaggar et al., 1998; Porter et al., 1998). These latter KCa currents were originally described by Benham and Bolton (1986) as “spontaneous transient outward currents” (STOCs). (e) Inhibitors of sarcoplasmic reticulum release (e.g., ryanodine) prevented both calcium sparks and STOCs (Nelson et al., 1995).
Activation of KCa channels (“a STOC”) by a local calcium release event (“a calcium spark”) would cause a global hyperpolarization of the membrane potential, closing voltage-dependent calcium channels, and, therefore, feed back to decrease global Ca2+ and contraction (see e.g., Brayden and Nelson, 1992; Knot and Nelson, 1998; Knot et al., 1998). A single STOC can cause a very significant membrane potential hyperpolarization (>20 mV; Ganitkevich and Isenberg, 1990). In contrast, a single Ca2+ spark, based on its spatial spread, would activate <1% of a cell's myosin light chain kinase. Therefore, the location (particularly relative to KCa channels), timing, and extent of such Ca2+-release events will determine whether they contribute significantly to global Ca2+, leading to contraction, or to local stimulation of KCa channels, leading to a reduction of that global Ca2+. In support of the latter mechanism in arterial smooth muscle, inhibitors of Ca2+ sparks or KCa channels increase in a nonadditive manner global Ca2+ of pressurized cerebral arteries by 50 nM, which leads to a 30% vasoconstriction (Knot and Nelson, 1998; Knot et al., 1998).
The goal of this study was to determine the quantitative relationship between Ca2+ sparks and transient potassium currents in arterial smooth muscle cells. Local calcium release transients are monitored in two distinct ways: (a) as an optical change in fluorescence as calcium binds to dye molecules in the surrounding cytoplasm, and (b) as KCa channel currents activated by Ca2+. Much like the light and thunder that arise from the electrical discharge of lightning, these two observable events would have individual characteristics, reflecting the nature of the originating transient event. Definitive evidence for causality of local calcium release events activating KCa channels would come from a quantitative analysis of simultaneous measurements of fluorescence and potassium currents in single myocytes. Furthermore, the previous nonsimultaneous measurements do not address the issue of whether all Ca2+ sparks activate nearby KCa channels, or whether some sparks fail to activate STOCs. The stoichiometry of Ca2+ spark events to STOCs should provide key information about the relationship between Ca2+ spark sites and KCa channels; i.e., the fraction of Ca2+ spark sites that are close enough to KCa channels in the sarcolemmal membrane to cause a STOC. The relationship between the measured change in fluorescence and the change in KCa channel open probability during a Ca2+ spark would give information about the fidelity of the fluorescence measurements as well as the local Ca2+ sensed by the KCa channels.
We provide here the first quantitative evidence of functional coupling between local Ca2+ release events and KCa channel activation in these cells. Whole cell KCa channel currents were measured using the perforated patch approach of the whole cell configuration, so as to minimize intracellular solute and structural changes. Ca2+ sparks were measured simultaneously with potassium currents by using the 2-dimensional (x–y) scanning mode of a laser scanning confocal microscope and the fluorescent Ca2+ indicator fluo-3. Virtually all (>96%) detectable Ca2+ sparks were associated with a STOC. A significant fraction of STOCs (41%) were not associated with Ca2+ sparks, consistent with these events being caused by small sparks or by sparks that were out of focus. The amplitudes of the sparks and STOCs were correlated. Our results strongly support our previous hypothesis that Ca2+ sparks cause STOCs (Nelson et al., 1995; Bonev et al., 1997; Porter et al., 1998). Comparison of the potassium currents and fluorescence changes caused by a calcium spark indicate that KCa channels sense much higher intracellular calcium than reported by the fluorescent indicator. The one-to-one functional coupling presented here between Ca2+ sparks and STOCs has important implications regarding the location of Ca2+ spark sites and their role in the regulation of cell function.
materials and methods
Cell Isolation
Sprague-Dawley rats (12–14 wk old) of either sex were killed by peritoneal injection of pentobarbital solution (150 mg/kg). The brain was removed and placed into ice cold oxygenated (95% O2/5% CO2) physiological salt solution containing (mM) 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.0023 EDTA, and 11 glucose, pH 7.4. Cerebral basilar arteries were carefully dissected in ice-cold physiological salt solution and cleaned of surrounding connective tissue. Arteries were then washed with a low Ca2+ enzyme solution (ES) containing (mM) 55 NaCl, 80 sodium glutamate, 5.6 KCl, 10 HEPES, 2 MgCl2, 10 glucose, pH 7.3, and then digested with papain and collagenase as follows. The artery was first incubated in ES, with the same ionic composition as described above, containing 0.3 mg/ml papain and 1 mg/ml dithioerythritol for 20 min at 37°C, and then transferred to a second incubation in ES with 100 μM CaCl2 containing 1 mg/ml collagenase (types F and H in a 70/30% mixture, respectively) and incubated for 10 min at 37°C. The enzyme digestion was stopped by placing the artery in ice cold low Ca2+ ES for 15 min. The digested tissue was triturated with a fire polished glass Pasteur pipette to yield single smooth muscle cells. After trituration the cells were kept on ice and used within the same day.
Simultaneous Patch Clamp and Fluorescence Recordings
The isolated myocytes were plated in the recording chamber and loaded with the Ca2+-indicator fluo-3 by incubation in ES containing 10 μM fluo-3 acetoxymethylester and 2.5 μg/ml pluronic acid (Molecular Probes, Inc.) in the dark for 30 min at room temperature. The incubation was followed by a 30-min wash in the same low Ca2+ buffer. Cells were then washed with physiological Ca2+ bathing solution (see below) and used for simultaneous patch clamp and confocal recordings after a stabilizing period of ∼10 min. All measurements were made 15–45 min after the stabilizing period. Synchronicity of the current and fluorescence measurements was achieved by means of a light-emitting diode placed near the recording chamber and switched on and off for 1.8 ms from a D/A output of Digidata board during the acquisition protocol, usually 10 s at −40 mV. This system allowed us to align the fluorescence and current traces with a precision only limited by the sampling frequency of the electrical recording (620 μs/point).
Confocal Microscopy
The cells were scanned with a laser scanning confocal system (OZ; Noran Instruments) hosted by an Indy workstation (Silicon Graphics, Inc.) and Intervision software package. The confocal system is mounted in an inverted Diaphot microscope with a 60× water immersion lens (NA, 1.2; Nikon Inc.). Fluo-3 fluorescence was excited with a krypton/argon laser at 488 nm and emitted light was detected by the confocal detector at wavelengths >515 nm. Images were typically of 48.4 × 50.6 μm (or 220 × 230 pixels) and acquired every 8.33 ms (120 images/s) during 10-s laser exposure triggered from the patch amplifier, and stored on writable CDs for future analysis. The confocal aperture was adjusted to expand the z axis so that recordings included a relatively large fraction of the cell. The measurement depth of our recordings was estimated to be 3.0–3.5 microns (z-axis width at half intensity, using a mirrored cover slip), corresponding to ∼50% of the cell volume. Since these isolated cells are thin, the confocal exclusion using these settings was adequate to screen for extraneous sources of light while maximizing our signal quality. This recording arrangement conveyed several significant advantages for this study. (a) The images measured were derived from a much larger fraction of each cell's volume (∼50%), permitting quantitative comparison with a majority of the STOCs. (b) Variability in spark amplitude was minimized due to variations in the relative z-axis position of the spark and the focal plane. (c) The volume within which quantitative signals were averaged for further analysis was roughly cubic; i.e., 2.2 × 2.2 × 3 μm (see below). (d) The efficiency of light collection was much higher, permitting longer recordings from the same cell with less photodamage.
Electrophysiological Recordings
Potassium currents were measured in the whole cell, perforated patch configuration of the patch clamp technique (Horn and Marty, 1988), using an Axopatch 200A amplifier (Axon Instruments). The bathing solution contained (mM): 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4. To minimize contraction, in some cases the bathing solution also contained 5 μM Wortmannin or 20 μM cytochalasin D. The pipette solution contained (mM) 110 potassium aspartate, 30 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, and 0.05 EGTA, pH 7.2, and 250 mg/ml amphotericin B. Membrane currents were recorded while holding the cells at a steady membrane potential of −40 mV. Currents were filtered at 500 Hz and digitized at 1.6 kHz (620 μs/ point). Cells were simultaneously scanned for fluorescence changes as indicated above.
Chemicals
Unless otherwise stated, all chemicals used in this study were obtained from Sigma Chemical Co. and Calbiochem-Novabiochem International. All experiments were conducted at room temperature (20°–22°C).
Data Analysis
To determine the amplitude of the STOCs, analysis was performed off line, using a custom analysis program. The threshold of STOCs was set at three times the single KCa channel amplitude at −40 mV or at 6 pA. The activity of KCa channels in the absence of Ca2+ sparks is very low at −40 mV (nPo ∼10−3; see Bonev et al., 1997), with the probability of three simultaneous openings being exceedingly low. Image analysis was performed using custom written analysis programs using Interactive Data Language software (Research Systems Inc.). Baseline fluorescence (Fo) was determined by averaging the 30 images (of 1,200) with no activity. Ratio images were then constructed and replayed for careful examination to detect active areas where sudden increases in F/Fo occurred. F/Fo vs. time traces were further analyzed in Microcal Origin (Microcal Software, Inc.), and represent the averaged F/Fo from a box region of 2.2 × 2.2 μm centered in the active area of interest to achieve the fastest and sharpest changes. This box size (4.8 μm2) was determined empirically to be the best compromise between temporal and spatial precision of Ca2+ sparks and the signal to noise ratio.
Statistical Analysis
Results are expressed as means ± SEM where applicable. All the statistical analysis was made with SigmaStat 2.03 software (Jandel Scientific Software). Spearman rank order correlation test was used for correlation analysis and Mann-Whitney rank sum test for significant differences.
results
STOCs Were Associated with Ca2+ Sparks
To minimize disruption of the cell's cytoplasm, whole cell current was measured in isolated cerebral artery myocytes, using the perforated patch approach of the patch clamp technique (Hamill et al., 1981; Horn and Marty, 1988). Outward potassium currents (“STOCs”) have previously been shown to be through iberiotoxin-sensitive, large conductance potassium (KCa) channels in cerebral arterial myocytes (Nelson et al., 1995). Intracellular Ca2+ was measured using the Ca2+-sensitive fluorescent dye (fluo-3) and a Noran Instruments laser scanning confocal microscope, with images obtained every 8.3 ms. Under such conditions, Ca2+ sparks (n = 128) and STOCs (n = 208) were measured with an average spark frequency of 0.7 ± 0.1 Hz and an average STOC frequency of 1.4 ± 0.3 Hz at −40 mV (10 cells from different arteries). Fig. 1 A illustrates the life cycle of a Ca2+ spark, which peaks in ∼20 ms and decays over 200 ms. Fig. 1 B illustrates simultaneous recordings of whole cell current (at −40 mV) (in blue) and fluorescence measurements for two spark sites that were identified in the cell (indicated by green and red boxes and F/Fo traces). Approximately 2.0 ± 0.3 (Table I) Ca2+ spark sites per cell were detected (n = 10). The Ca2+ sparks in the red site were relatively large (F/Fo, 2.8 ± 0.4), and the associated STOCs were also relatively large (57 ± 10 pA). This Ca2+ spark site generated four sparks over a 10-s recording period. The spark site indicated in green had much smaller sparks (mean 1.5 F/Fo) and much smaller associated STOCs (mean 7.9 pA).
Figure 1.
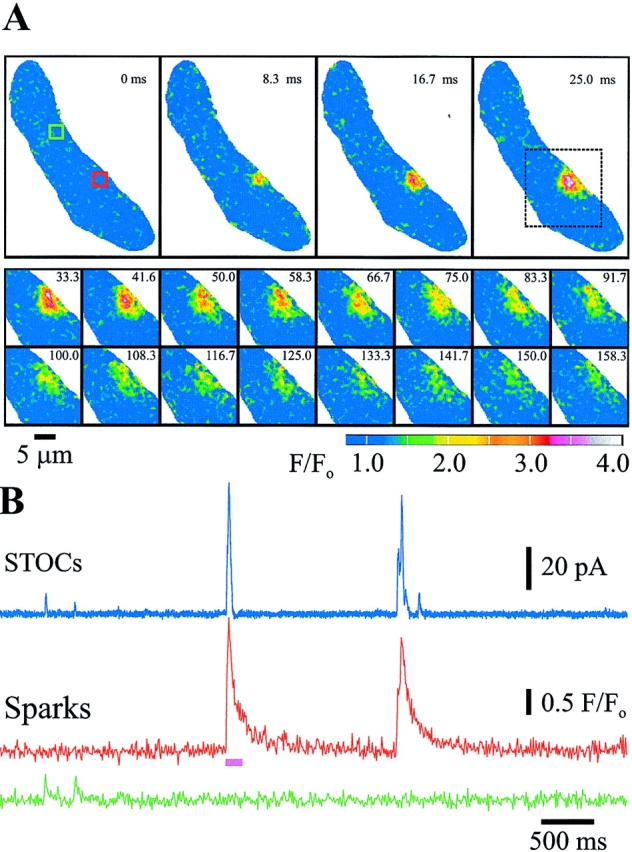
Ca2+ sparks generate STOCs in myocytes from rat basilar cerebral artery. (A) Original sequence of two-dimensional confocal images obtained every 8.33 ms of an entire smooth muscle cell (top), followed by subsequent images of the region of interest (dotted box) illustrating the time course of the fractional increase in fluorescence (F/Fo) and decay of a typical Ca2+ spark. The images are color coded as indicated by the bar. (B) Simultaneous STOC (pA) and spark (F/ Fo) measurements, at –40 mV, illustrating temporal association. (Blue) Current, (red and green) the F/Fo average of the red and green boxes (2.2 μm/side) indicated in A, respectively. The pink bar indicates the segment of the trace illustrated in A.
Table I.
Summary of Simultaneous Measurements of Ca2+ Sparks and STOCs
| STOCs | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Ca2+ sparks | Spark associated | Sparkless | |||||||||||||
| mean ± SEM | n* | mean ± SEM | n* | mean ± SEM | n* | |||||||||||
| Amplitude | 2.0 ± 0.1 | F/FO | 128 | 33.6 ± 1.9 | 123 | 16.2‡ ± 1.3 | 85 | pA | ||||||||
| Area§ | 13.6 ± 1.2 | μm2 | 119 | — | — | |||||||||||
| Rise time‖ | 22.7 ± 1.1 | ms | 114 | 16.5 ± 0.8 | 117 | 9.5‡ ± 0.6 | 81 | ms | ||||||||
| τ1 ¶ | 31.5 ± 2.5 | ms | 102 | 13.4 ± 0.6 | 115 | 9.4‡ ± 0.6 | 80 | ms | ||||||||
| τ2 ¶ | 274.8 ± 21.9 | ms | 102 | — | — | |||||||||||
| t 1/2 ** | 55.9 ± 3.5 | ms | 102 | 9.3 ± 0.4 | 115 | 6.5‡ ± 0.5 | 80 | ms | ||||||||
| Number of sites | 2.1 ± 0.3 | 10†† | — | |||||||||||||
| Δ Time peak§§ | 6.7 ± 0.9 | ms | 116 | — | — | |||||||||||
| D Time onset‖‖ | −0.3 ± 0.4 | ms | 114 | — | — | |||||||||||
| Sparks w/o STOCs | 2.0 ± 0.1 | F/FO | 5 | 4.7¶¶ ± 0.3 | 5 | pA | ||||||||||
| STOCs total | — | — | 26.5 ± 1.4 | 208 | pA | |||||||||||
Number of events from 10 cells used to calculate mean values.
Significantly different compared with corresponding spark-associated STOC parameters (P < 0.01; Mann-Whitney Rank Sum Test).
Measured at 50% amplitude of the peak.
Measured from 10 to 90% increase of the signal.
Fitted decay time constants, biexponential for Ca2+ sparks and monoexponential for STOCs.
Half time for decay.
Number of cells.
Ca2+ spark peak time − STOC peak time.
Ca2+ spark onset time − STOC onset time; onset times were determined at 10% increase of the signal.
Associated current.
Figs. 1 and 2 illustrate a close temporal association of two very low frequency (<1.5 Hz) events, a Ca2+ spark and a STOC. Virtually all (123 of 128, 96%) measured Ca2+ sparks were accompanied by a STOC with amplitude >6 pA, occurring within the time required to acquire one image (8.3 ms). Even the small number (five) of Ca2+ sparks that did not match our criteria for association with a STOC were, in fact, paired with small transient currents (mean amplitude 4.7 pA, Table I), which were below our STOC threshold of 6 pA.
Figure 2.

Almost every Ca2+ spark (96%) generates a STOC. Time course of whole cell current and F/Fo from three different cells. Blue traces represent whole cell currents at −40 mV. F/Fo traces represent the average from a square box of 2.2 μm/ side centered at identified active sites in the cell. One site is displayed in cell 1, one site in cell 2, and four sites in cell 3 (A–D; maximum distance between box centers was 7.3 μm [A and D]; minimum distance between box centers was 2.9 μm [B and C]).
The Ca2+ sparks and STOCs occurred virtually simultaneously whenever they were paired. Using the photodiode synchronization method described above, we could precisely align the electrical recordings with our optical signals. The mean time difference between the onset (10% over baseline) of a Ca2+ spark and a STOC was −0.3 ± 0.4 ms (n = 114), with the negative number indicating that the sparks preceded the STOCs (Table I). For these low frequency events, the chances that such close temporal association could have been coincidental is essentially nil. The mean time difference of the peak of an associated STOC and Ca2+ spark was 6.4 ± 1.0 ms (n = 118), with the peak of the STOC occurring earlier than the peak of the Ca2+ spark (Table I). The decay half time of STOCs (9.3 ± 0.4 ms) was shorter than the decay half time of Ca2+ sparks (55.9 ± 3.6 ms) (Table I). The Ca2+ spark decay was well fitted by two exponentials (time constants were: τ1 = 31.5 ms and τ2 = 274.8 ms, Table I, with both components contributing similarly, 49 and 51%, respectively), whereas the STOC decay was fitted by one exponential (τ1 = 13.4 ms, Table I).
These observations indicate the close relationship between Ca2+ sparks and STOCs. In addition, several previous lines of evidence suggested that Ca2+ sparks cause STOCs, including the observations that inhibitors of Ca2+ sparks (ryanodine and thapsigargin) block STOCs, and kinetic similarities between Ca2+ sparks and STOCs (Nelson et al., 1995). Furthermore, the close temporal relationship between spark and STOC onset, and the observation that STOCs subsequently peak and decline before sparks, indicate that the majority of Ca2+ spark sites are close enough to the cell membrane to activate KCa channels quickly, before the full spatial spread of the Ca2+-dye complex.
Relationship between Ca2+ Spark and STOC Amplitudes
One apparent aspect of our recordings is that both the sparks and the STOCs varied in amplitude. Fig. 2 illustrates Ca2+ sparks and STOCs from three different cells, with one cell having four spark sites. Fig. 2 as well as Fig. 1 illustrates that STOCs were associated with Ca2+ sparks, and that larger amplitude sparks were associated with larger STOCs. Fig. 4 also shows the spread in amplitudes of the observed STOCs, ranging from events just above our threshold of 6 pA to 101 pA.
Figure 4.

Many small STOCs are not associated with sparks. STOC amplitude distribution histogram at −40 mV, indicating the contribution of STOCs associated with sparks (green bars) and sparkless STOCs (yellow bars). Mean amplitude of Ca2+ spark– associated STOCs was 33.5 pA. STOCs that were not associated with a Ca2+ spark had a mean amplitude of 16 pA.
A causal relationship between the Ca2+ release and the generation of STOCs would predict that if Ca2+ spark amplitude varied due to differences in the amount of Ca2+ locally released from the SR during each spark, then the amplitude of the associated STOC would also vary in relation to the release amount. In contrast, other possible sources of variability (e.g., uneven dye distribution, spark events at different positions in the confocal plane, differences in the distances between spark sites, and KCa channels) would cause uncorrelated amplitudes between the two events. We have therefore studied the quantitative relationship between Ca2+ spark and STOC amplitudes. Fig. 3 shows a plot of Ca2+ sparks and STOC amplitudes from all the cells used in this study (n = 10). The amplitudes of Ca2+ sparks and STOCs were strongly correlated (correlation coefficient 0.8). The mean amplitudes of associated Ca2+ sparks (F/Fo) and STOCs (pA) at −40 mV were 2.04 ± 0.05 and 33.6 ± 1.9 pA, respectively (n = 123) (Table I). This correlation provides strong evidence that the common element (amount of Ca2+ released) between the two observed events is the factor causing the common variation.
Figure 3.

Ca2+ spark and STOC amplitudes are correlated. Scatter plot of Ca2+ spark amplitude vs. STOC amplitude at −40 mV. Each color represents the contribution of each particular cell (n = 10 cells). Amplitudes were taken at peaks. Correlation coefficient was 0.8 according to Spearman rank order correlation test (P < 0.05, n = 123).
STOCs that Were Not Associated with a Detectable Ca2+ Spark
Although virtually all Ca2+ sparks observed were associated with a STOC (Figs. 1 and 2), we also observed many STOCs (41%) that were not associated with a spark above the detection threshold of F/Fo > 1.2 (Fig. 4, Table I). However, as shown in Fig. 4, STOCs that were not associated with Ca2+ sparks had smaller amplitudes (mean, 16.3 ± 1.3 pA, n = 83 STOCs, see Table I) and had faster rise and decay times (Table I). If the amplitude correlation illustrated in Fig. 3 also held for these STOCs, then they would have been associated with small sparks. Since a significant fraction (∼50%) of the cell volume was scanned, it is unlikely that large calcium sparks outside the scan volume would have been missed, but it is likely that small and fast calcium sparks outside the scan volume could have been missed.
discussion
Ca2+ Sparks Cause STOCs: Implications for the Location and Nature of Ca2+ Spark Sites
This study provides the first quantitative analysis of simultaneous Ca2+ spark and potassium current measurements in smooth muscle. Mironneau et al. (1996) provided one simultaneous recording of a spark (line scan mode) and STOC in a portal vein myocyte (Mironneau et al., 1996), without quantitative information. In a preliminary report, evidence that some, but not all, Ca2+ sparks cause STOCs in esophageal myocytes was provided (Kirber et al., 1997). Our results indicate that virtually all detectable Ca2+ transients are associated with a detectable potassium current in arterial smooth muscle, that these events initiate virtually simultaneously, and that they have closely correlated amplitudes. These results therefore demonstrate a close functional coupling between Ca2+ spark sites and KCa channels in the surface membrane. In support of the close proximity of Ca2+ spark sites (i.e., RyR) to the surface membrane, SR elements are within 20 nm of the sarcolemmal membrane (Devine et al., 1972; Somlyo, 1985), and RyR have been shown to colocalize with the plasma membrane (Carrington et al., 1995; Gollasch et al., 1998). Taken together with previous pharmacological evidence, we can conclude that Ca2+ sparks cause STOCs in arterial smooth muscle.
Sparkless STOCs
Although we establish here that essentially all detectable Ca2+ sparks cause a STOC in cerebral artery myocytes, many STOCs were observed that were not associated with a measurable Ca2+ spark. These “sparkless” STOCs appeared to result from small and fast Ca2+ sparks in regions of the cell that were not scanned. The sparkless STOCs had a smaller mean amplitude (16 pA) than STOCs associated with Ca2+ sparks (33.6 pA) (Table I). Since the lower amplitude STOCs were generally caused by smaller Ca2+ sparks (Fig. 3), the missed Ca2+ sparks are likely to be small as well. Given that the scanned volume covers only about half the cell, and falls off in a graded way for events outside the optimum recording plane, small events were more likely to be missed. In addition, the optical sampling rate (8.33 ms per frame) also restricts the detection limits in the time domain. We show that the sparkless STOCs were faster (both rise and fall time, Table I) than those that were associated with sparks, indicating that the corresponding Ca2+ transients were also likely to be faster. For such small, fast events, it is increasingly likely that the peak of the transient fell between frames, and was therefore further underestimated. These combined “spatio-temporal filtering” effects of the optical recording apparatus would increase the likelihood that small, rapid spark events outside the recording plane would be missed, giving rise to a population of sparkless STOCs. Although our recording system provides significant advantages over other imaging alternatives, it seems clear that KCa channels are far superior to the fluorescent dye fluo-3 in sensing local and rapid calcium changes.
The missed smaller sparks and their associated sparkless STOCs might conceivably represent a completely different source of transient Ca2+. For example, these smaller, faster Ca2+ transients might be caused by Ca2+ entering the cell through L-type Ca2+ channels (Wang et al., 1997). We believe, however, that these events are more likely to belong to the lower end of RyR-mediated Ca2+ release events. This latter hypothesis appears more likely since thapsigargin and ryanodine abolish all the STOC activity in these cells (Nelson et al., 1995).
Ca2+ Spark and STOC Amplitudes Were Correlated
The amplitudes of both the Ca2+ sparks and the STOCs reported here are distributed from just above the detection threshold to 3.6 F/Fo and 101 pA, respectively. A number of factors contribute to distribution of Ca2+ spark amplitudes. First, spark events might vary in z-axis position with respect to the confocal recording plane. Second, the release channel(s) that give rise to the sparks might have variable open duration, thus releasing more or less Ca2+ during each spark. Third, the number and distribution of release channels that participate in each spark might differ from one event or cell region to another. Each of these factors undoubtedly plays a role in our observed distributions. Similarly, there are several reasons why STOC amplitudes might be broadly distributed. Assuming STOCs are caused by local Ca2+ release, the distance of that release from the membrane, the local density of KCa channels, the quantity of Ca2+ released, and the spatial spread of the released Ca2+ are all factors that would influence the magnitude of the STOC. We report here, however, a strong correlation between the amplitude of individual spark and STOC events (Fig. 3). This observation reduces the possible explanation to those that would be common to both processes. We therefore expect that the correlation of Ca2+ sparks and STOCs amplitudes reflects a common, underlying variability in the quantity of Ca2+ released during one event.
On the Kinetics of Ca2+ Sparks and STOCs
The rise time of a Ca2+ spark (∼20 ms) (Nelson et al., 1995; this study, Table I) may reflect in part the kinetics of fluo-3 (Escobar et al., 1997). The kinetics of a STOC did not appear to be affected by fluo-3 (time to peak in cells without fluo-3, 17 ms [Nelson et al., 1995]; time to peak in cells with fluo-3, 16 ms [this study, Table I]), suggesting that fluo-3 (∼50 μM) does not buffer the local Ca2+ that activates the KCa channels. This observation is consistent with a very short gap (20 nm) (Devine et al., 1972; see also Neher, 1998 for a discussion of Ca2+ diffusion over short distances) between the SR with Ca2+ spark sites and the surface membrane. The relatively slow rise time of a STOC (10–20 ms) may reflect the activation time of KCa channels by Ca2+, and not diffusion (Markwardt and Isenberg, 1992). The rapid decay of STOCs may reflect the diffusion and the steep Ca2+ dependence of KCa channels.
The decay of Ca2+ sparks in arterial smooth muscle appears to be biexponential (Table I), and much slower than the decay of the associated STOCs. The faster decay time constant (31 ms) is similar to that measured for Ca2+ sparks in heart muscle (Santana et al., 1997), and consistent with the diffusion of the Ca2+– fluo-3 complex. The nature of the slower time constant of decay is unknown. Although it is clear that a STOC is associated with a Ca2+ spark, the precise interpretation of timing of the onsets and peaks of STOCs and Ca2+ sparks (Table I) is much less certain. Our precision is limited by a number of factors including image sampling rate (120 Hz) and the kinetics of fluo-3.
Fidelity of Fluo-3 Ca2+ Measurements
A quantitative consideration of the Ca2+ sensitivity of KCa channels also supports the idea that Ca2+ spark sites are close to the sarcolemmal membrane containing KCa channels. The whole-cell activity (nP o) of KCa channels, in the absence of Ca2+ sparks, has been directly measured using the perforated patch, whole cell configuration of the patch clamp technique, and is ∼10−2 at 0 mV (Bonev et al., 1997; Porter et al., 1998). Therefore, taking into account the voltage dependence of KCa channels (e-fold per 10–20 mV; Carl et al., 1996), activity (n cell P oBase) of KCa channels in the absence of Ca2+ sparks would be ∼10−3 at physiological membrane potentials (−40 mV), where n cell is the total number of functional KCa channels in a single cell and P oBase is the average open probability of these n cell channels. The activity of KCa channels that were activated by a Ca2+ spark or n spark P oSpark can be determined by dividing the STOC by the unitary current (see Nelson et al., 1995), where n spark is the number of KCa channels that have been significantly activated by a Ca2+ spark and P oSpark is the mean P o of these n spark channels. n spark P oSpark was ∼16 (33/2 pA) at −40 mV for these experiments. Thus, the minimum increase of the open probability (P o) of KCa channels can be determined by dividing n cell P oSpark by n sparkPoBase (or 16/10−3).
This calculation shows that channel open probability increased at least 104 during a Ca2+ spark, even assuming that the Ca2+ transients were felt equally by all the KCa channels in the membrane (i.e., n cell = n spark). However, since a Ca2+ spark (average size, 13 μm2, Table I) covers ∼1% of the surface membrane (1,300 μm2), then only 1% of homogeneously distributed KCa channels would be activated by a Ca2+ spark. The P o of KCa channels near the Ca2+ spark would increase more than 104 and closer to 106, depending on the distribution of KCa channels. For channel P o to increase by 104–106, local Ca2+ would have to increase by at least 10–1,000-fold, assuming open probability increases to second to fourth power of [Ca2+] (for review see McManus, 1991; Carl et al., 1996), with the beta subunit of the KCa channel increasing Ca2+ sensitivity (Tanaka et al., 1997). Therefore, the local intracellular Ca2+ that activates the KCa channels during spark ranges from 1 to 100 μM (assuming 100-nM resting Ca2+). These estimates of relatively high local Ca2+ in the junctional space between the RyR in the SR and the KCa channels are in accord with simulations of local Ca2+ changes during a Ca2+ spark in the junctional space in cardiac muscle (Cannell and Soeller, 1997). In contrast, the mean increase in Ca2+ during a Ca2+ spark, as measured with fluo-3, would predict an increase in KCa channel P o of no more than 16-fold, assuming [Ca2+]4 relationship with channel P o. These estimates indicate that local Ca2+ sensed by the KCa channels is much higher (1–100 μM) than peak of the Ca2+ spark (∼200–300 nM range at peak amplitude) measured with fluorescent Ca2+ indicator fluo-3.
In conclusion, our results support the idea that Ca2+ sparks cause STOCs, and that the RyR that generate Ca2+ sparks in cerebral arterial myocytes are located in SR elements that are close enough for local Ca2+ to cause significant activation (104–106-fold increase in open probability) of KCa channels in the plasma membrane.
Acknowledgments
We thank G. Herrera, and Drs. T.J. Heppner, J.H. Jaggar, L.F. Santana, and G.C. Wellman for comments on the manuscript.
This work was supported by the National Science Foundation grant BIR-9601682, IBN-9631416, and National Institutes of Health grants HL-44455 and HL-51728. This work was done during the tenure of a fellowship award from the American Heart Association, Maine/New Hampshire/Vermont Affiliate, Inc. (Guillermo Pérez).
Abbreviations used in this paper
- ES
enzyme solution
- Fo
baseline fluorescence
- RyR
ryanodine receptors
- SR
sarcoplasmic reticulum
- STOC
spontaneous transient outward current
references
- Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. J Physiol (Camb) 1986;381:385–406. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev AD, Jaggar JH, Rubart M, Nelson MT. Activators of protein kinase C decrease Ca2+spark frequency in smooth muscle cells from cerebral arteries. Am J Physiol. 1997;273:C2090–C2095. doi: 10.1152/ajpcell.1997.273.6.C2090. [DOI] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of Ca2+-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Soeller C. Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys J. 1997;73:112–122. doi: 10.1016/S0006-3495(97)78052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl A, Lee HK, Sanders KM. Regulation of ion channels in smooth muscles by Ca2+ . Am J Physiol. 1996;271:C9–C34. doi: 10.1152/ajpcell.1996.271.1.C9. [DOI] [PubMed] [Google Scholar]
- Carrington WA, Lynch RM, Moore ED, Isenberg G, Fogarty KE, Fay FS. Superresolution three-dimensional images of fluorescence in cells with minimal light exposure. Science. 1995;268:1483–1487. doi: 10.1126/science.7770772. [DOI] [PubMed] [Google Scholar]
- Devine CE, Somlyo AV, Somlyo AP. Sarcoplasmic reticulum and excitation-contraction coupling in mammalian smooth muscles. J Cell Biol. 1972;52:690–718. doi: 10.1083/jcb.52.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar AL, Velez P, Kim AM, Cifuentes F, Fill M, Vergara JL. Kinetic properties of DM-nitrophen and Ca2+indicators: rapid transient response to flash photolysis. Pflügers Arch. 1997;434:615–631. doi: 10.1007/s004240050444. [DOI] [PubMed] [Google Scholar]
- Fay FS. Ca2+sparks in vascular smooth muscle: relaxation regulators. Science. 1995;270:588–589. doi: 10.1126/science.270.5236.588. [DOI] [PubMed] [Google Scholar]
- Ganitkevich VY, Isenberg G. Isolated guinea pig coronary smooth muscle cell. Acetylcholine induces hyperpolarization due to sarcoplasmic reticulum calcium release activating potassium channels. Circ Res. 1990;67:525–528. doi: 10.1161/01.res.67.2.525. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Wellman GC, Knot HJ, Jaggar JH, Damon DH, Bonev AD, Nelson MT. Ontogeny of local sarcoplasmic reticulum Ca2+signals in cerebral arteries. Circ Res. 1998;83:1104–1114. doi: 10.1161/01.res.83.11.1104. [DOI] [PubMed] [Google Scholar]
- Gordienko DV, Bolton TB, Cannell MB. Variability in spontaneous subcellular Ca2+release in guinea-pig ileum smooth muscle cells. J Physiol (Camb) 1998;507:707–720. doi: 10.1111/j.1469-7793.1998.707bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+sparks in intact cerebral arteries. Am J Physiol. 1998;274:C1755–C1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- Kirber MT, Etter EF, Singer JJ, Walsh JV, Jr, Fay FS. Simultaneous 3D imaging of Ca2+sparks and ionic currents in single smooth muscle cells. Biophys J. 1997;72:A295. [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol (Camb) 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+channels. J Physiol (Camb) 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt F, Isenberg G. Gating of maxi K+ channels studied by Ca2+concentration jumps in excised inside-out multi-channel patches (myocytes from guinea pig urinary bladder) J Gen Physiol. 1992;99:841–862. doi: 10.1085/jgp.99.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus OB. Ca2+-activated potassium channels: regulation by Ca2+ . J Bioenerg Biomembr. 1991;23:537–560. doi: 10.1007/BF00785810. [DOI] [PubMed] [Google Scholar]
- Mironneau J, Arnaudeau S, Macrez-Lepretre N, Boittin FX. Ca2+ sparks and Ca2+ waves activate different Ca2+-dependent ion channels in single myocytes from rat portal vein. Cell Calc. 1996;20:153–160. doi: 10.1016/s0143-4160(96)90104-9. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by Ca2+sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT. Frequency modulation of Ca2+sparks is involved in regulation of arterial diameter by cyclic nucleotides. Am J Physiol. 1998;274:C1346–C1355. doi: 10.1152/ajpcell.1998.274.5.C1346. [DOI] [PubMed] [Google Scholar]
- Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation–contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol (Camb) 1997;503:21–29. doi: 10.1111/j.1469-7793.1997.021bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck GC, Kannan MS, Prakash YS. Heterogeneity in dynamic regulation of intracellular Ca2+in airway smooth muscle cells. Can J Physiol Pharmacol. 1997;75:878–888. [PubMed] [Google Scholar]
- Somlyo AP. Excitation–contraction coupling and the ultrastructure of smooth muscle. Circ Res. 1985;57:497–507. doi: 10.1161/01.res.57.4.497. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Meera P, Song M, Knaus HG, Toro L. Molecular constituents of maxi KCachannels in human coronary smooth muscle: predominant alpha + beta subunit complexes. J Physiol (Camb) 1997;502:545–557. doi: 10.1111/j.1469-7793.1997.545bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Fleischmann BK, Kotlikoff MI. Modulation of maxi-K+ channels by voltage-dependent Ca2+channels and methacholine in single airway myocytes. Am J Physiol. 1997;272:C1151–C1159. doi: 10.1152/ajpcell.1997.272.4.C1151. [DOI] [PubMed] [Google Scholar]