

Abstract
Ions bound near the external mouth of the potassium channel pore impede the C-type inactivation conformational change (Lopez-Barneo, J., T. Hoshi, S. Heinemann, and R. Aldrich. 1993. Receptors Channels. 1:61– 71; Baukrowitz, T., and G. Yellen. 1995. Neuron. 15:951–960). In this study, we present evidence that the occupancy of the C-type inactivation modulatory site by permeant ions is not solely dependent on its intrinsic affinity, but is also a function of the relative affinities of the neighboring sites in the potassium channel pore. The A463C mutation in the S6 region of Shaker decreases the affinity of an internal ion binding site in the pore (Ogielska, E.M., and R.W. Aldrich, 1998). However, we have found that this mutation also decreases the C-type inactivation rate of the channel. Our studies indicate that the C-type inactivation effects observed with substitutions at position A463 most likely result from changes in the pore occupancy of the channel, rather than a change in the C-type inactivation conformational change. We have found that a decrease in the potassium affinity of the internal ion binding site in the pore results in lowered (electrostatic) interactions among ions in the pore and as a result prolongs the time an ion remains bound at the external C-type inactivation site. We also present evidence that the C-type inactivation constriction is quite local and does not involve a general collapse of the selectivity filter. Our data indicate that in A463C potassium can bind within the selectivity filter without interfering with the process of C-type inactivation.
Keywords: C-type inactivation, ion interactions, potassium affinity, Shaker
introduction
In response to prolonged membrane depolarization, voltage-gated potassium channels restrict the movement of ions through the ion-selective pore by the process of inactivation. The Shaker potassium channel displays two types of inactivation, N- and C-type, that are mediated by distinct regions of the channel protein. N-type inactivation occurs within a few milliseconds after channel opening in the Shaker B channel variant and results from the physical occlusion of the cytoplasmic entrance to the pore by the amino terminus of the protein (Hoshi et al., 1990; Zagotta et al., 1990; Demo and Yellen, 1991). C-type inactivation occurs over a period of seconds in the Shaker B channel and involves a structural rearrangement of the outer mouth of the pore (Yellen et al., 1994; Liu et al., 1996). The C-type inactivation rate is also dependent on the occupancy of an external ion binding site, which must be empty before the C-type conformational change can proceed (Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995). The external site is nearly saturated in physiological solutions, due to outward potassium flux through the channel that creates a local accumulation of ions (Baukrowitz and Yellen, 1995). Prevention of potassium efflux by internal blockers increases the C-type inactivation rate by preventing the external site from being refilled after the last ion has exited from the pore to the external solution (Baukrowitz and Yellen, 1995). In the absence of added potassium, the Shaker channel conducts sodium (Starkus et al., 1997; Ogielska and Aldrich, 1998); however, the rate of C-type inactivation is extremely rapid in symmetrical sodium solutions (Starkus et al., 1997), indicating that either sodium does not interfere with the C-type inactivation process or that its dwell time at the C-type inactivation controlling site is short compared with potassium. The C-type inactivation rate is also sensitive to amino acid substitutions in the pore-forming regions of the channel (Lopez-Barneo et al., 1993; Heginbotham et al., 1994; Yang et al., 1997), but how these mutations affect the inactivation rate of the channel is not well understood. Amino acid substitutions could either directly affect the energetics of the C-type inactivation conformational change, or alternatively they could affect the ion occupancy of the C-type modulatory site in the pore.
The recent determination of the crystal structure of a prokaryotic potassium channel from Streptomyces lividans (KcsA) (Doyle et al., 1998) allows for a more detailed interpretation of the molecular mechanism by which amino acid substitutions may affect the C-type inactivation rate of the channel. The structure has confirmed previous knowledge that potassium channels are tetramers (MacKinnon, 1991; Liman et al., 1992; Kavanaugh et al., 1992), with each subunit contributing equally to the formation of the pore (Doyle et al., 1998). Shaker is a member of the voltage-gated potassium channel family where each subunit has six transmembrane regions (termed S1 to S6), as well as a membrane spanning loop (P-region) between S5 and S6. Although KcsA has only two membrane spanning domains, these are analogues of the fifth and sixth transmembrane regions (S5 and S6) of voltage-gated potassium channels. The sequence homology between KcsA and Shaker is highest within the P-region and both channels possess the selectivity-determining signature sequence (TXXTXGYG). The structure of the KcsA pore is therefore most likely reflective of the general structure of the pore of all potassium selective channels. The narrow ion selective region of the pore is formed by the signature sequence, with permeant ions complexed by the backbone carbonyl groups of the signature sequence amino acids (Doyle et al., 1998). The selectivity filter of KcsA contains three permeant ion binding sites, an external site and two overlapping internal sites (Doyle et al., 1998). By calculating the differences in the electron density maps obtained from crystals in Rb+ solutions versus K+ solutions, Doyle et al. (1998) determined that the narrow region can be simultaneously occupied by two ions, one bound at the external site and one rapidly equilibrating between the two internal sites. Although all potassium channels have a signature sequence, potassium affinity varies among channel subtypes, indicating that regions outside the conserved signature sequence are important in determining the nature of ion binding sites in the pore (Korn and Ikeda, 1995; Starkus et al., 1997; Kiss and Korn, 1998; Ogielska and Aldrich, 1998).
Potassium channels select between ions by an affinity mechanism and potassium affinity determines the relative permeability of other ions (Hille and Schwarz, 1978; Yellen, 1984; Neyton and Miller, 1988a,b; Baukrowitz and Yellen, 1996; Korn and Ikeda, 1995; Kiss et al., 1998). The Shaker potassium channel has a high potassium affinity (Starkus et al., 1997; Ogielska and Aldrich, 1998), and mutation of a single residue in S6 (A463C) decreases the apparent potassium affinity from the micromolar into the millimolar range (Ogielska and Aldrich, 1998). The crystal structure of the KcsA channel suggests a possible mechanism for this effect: the mutation may alter the interaction between the side chain at position 463 in S6 (a methionine in KcsA) and a valine within the signature sequence (V443 in Shaker) (Fig. 1). The main chain carbonyl group of this signature sequence valine contributes to the formation of the most internal ion binding site at the cytoplasmic face of the narrow ion selective region (Doyle et al., 1998; Fig. 1). In accordance with the idea that the pore valine is crucial to the integrity of a high affinity potassium binding site is the observation that mutations at that position can render the Shaker channel nonselective (Heginbotham et al., 1994). Substitutions at position 463 have also previously been shown to affect the rate of C-type inactivation (Hoshi et al., 1991). The C-type inactivation rate varies by two orders of magnitude between the Shaker B and ShakerA alternative splice variants, and the difference in the time course of inactivation is dependent on whether position 463 is occupied by an alanine (ShB) or a valine (ShA) (Hoshi et al., 1991). Given that the A463C mutation affects the apparent potassium affinity of a potassium binding site and that mutations at this position alter the C-type inactivation rate of the channel, we examined the effects of the A463C mutation on C-type inactivation. We reasoned that perhaps effects of mutations at A463 on C-type inactivation are directly resultant from changes in ion occupancy at the inner potassium ion binding site. If that is the case, then the decrease in the apparent potassium affinity in A463C should result in faster C-type inactivation. We found, however, that the rate of C-type inactivation in A463C is much slower than wild type both in potassium solutions and in symmetrical sodium solutions. Our results indicate that the affinity decrease at the more internal site decreases the repulsive interactions among ions in the pore, resulting in higher occupancy of the external C-type inactivation regulatory site. Furthermore, we have found that the C-type inactivation conformational change proceeds unimpeded with a potassium ion bound at a deeper site in the pore indicating that the inactivation process involves a very localized rearrangement of the external mouth of the channel protein.
Figure 1.

A mutation at position 463 in S6 affects the affinity of an ion binding site in the pore. Crystal structure of a region of the Streptomyces lividans prokaryotic potassium channel highlighting the S6 methionine and P-region valine interaction. (A) A side view of the interacting side chains (boxed region) within a single subunit. The GYG amino acids are labeled for easier orientation. (B) Top view of the methionine:valine interaction with all four subunits shown. The backbone carbonyl of the signature sequence valine stabilizes a potassium ion at its binding site in the ion conducting pore.
materials and methods
Molecular Biology
The A463C mutation was made by generating PCR primers using noninactivating ShBΔ6-46 as a template. The PCR product was gel purified and spliced into the channel using NsiI and SpeI as the restriction sites. The mutant was sequenced through the mutated region to check that no secondary mutations occurred. Finally, the mutant cDNA was linearized with EcoR1 and RNA was synthesized using the T7 polymerase (Ambion Inc.). The transcribed RNA was injected into Xenopus laevis oocytes as previously described (Zagotta et al., 1989).
Electrophysiology
Macroscopic recordings were done in cell free patches (either inside-out or outside-out) 3–9 d after injection. Patch pipettes were made of borosilicate glass (VWR Micropipettes). Their tips were coated with wax (Kerr Sticky Wax) and fire polished before use. Pipettes had initial resistances <2 MΩ and all recordings were done at 21°C. Leak subtraction was done using the P/4 protocol and no corrections were made for series resistance. Data were acquired using an Axopatch 200-A (Axon Instruments) patch clamp amplifier. The data were digitized using a Macintosh-based computer system using Pulse acquisition software (HEKA Electronik) and the ITC-16 hardware interface (Instrutech Corp.). Data were analyzed using Igor Pro graphing and curve fitting software (Wavemetrics Inc.).
Solutions
Unless otherwise indicated, the internal solution contained (mM): 10 EGTA, 10 Hepes, and either 140 NaCl or 140 KCl. The external solution was composed of (mM): 2 CaCl2, 5 Hepes, and either 2 KCl, 140 NaCl, or 0 KCl, 140 XCl (where X denotes either K, N-methyl-d-glucamine [NMG],1 Na, or tetraethylammonium). Solutions sometimes also contained symmetrical 30 mM NMGCl, although this change had no effect on the currents. In potassium blocking experiments, the NMG was replaced by equimolar amounts of potassium to conserve osmolarity and keep the sodium concentration constant. The pH was 7.1 for all internal solutions and 7.2 for the external solutions.
In solutions without added potassium, the free potassium concentration was measured by flame photometry and was determined to be <40 μM (Scientific Environmental Laboratories Inc.). Solutions were exchanged using a sewer pipe flow system (DAD 12) purchased from ALA Scientific Instruments Inc. The system consisted of 12 syringe reservoirs each fitted with a solenoid valve and a thin tube leading to a single polyacrylamide-coated quartz pipe (100 μM i.d.) that formed the output. The patch was situated in front of the pipe and immersed in the laminar flow. Flow was aided by air pressure applied to each reservoir (200 psi), and solution exchange was computer controlled and occurred in <1 s. Sometimes the solution changes were incomplete due to air bubble formation in the tubing and/or vesicle formation. When these technical problems were encountered the experiments were terminated and discounted.
results
Ionic Dependence of the C-type Inactivation Rate in A463C
Substitutions at position A463 in the S6 of the Shaker potassium channel affect the rate of C-type inactivation, a process that is governed by ion occupancy (Hoshi et al., 1991; Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995). We examined the effects of the potassium affinity mutation A463C on C-type inactivation and its dependence on the ionic conditions. As previously described (Hoshi et al., 1990; Lopez-Barneo et al., 1993), in the wild-type Shaker B channel with N-type inactivation removed, C-type inactivates along a single exponential time course with a time constant of 1.4 s in physiological conditions (Fig. 2 A); increasing external potassium to 140 mM slows this inactivation time constant to 2.6 s (Fig. 2 B). Baukrowitz and Yellen (1995) have shown that even in the absence of external potassium, the efflux of potassium through the channel is sufficient to occupy the site that influences C-type inactivation significantly. Accordingly, decreasing the internal potassium concentration diminishes the flux through the channel and results in faster C-type inactivation rates (K d = ∼2 mM) (Baukrowitz and Yellen, 1995; Starkus et al., 1997).
Figure 2.

Properties of C-type inactivation with potassium as the conducting ion. Experiments were done in inside-out patches. Current traces were elicited by voltage steps to +50 mV. Except for F, all experiments were performed in the presence of 140 mM internal potassium, and currents were elicited by 10-s voltage steps. Data from the wild-type Shaker channel are shown in A and B, while data from A463C are shown in C–F. (A) The wild-type Shaker channel inactivates with a time constant of 1.4 s in the presence of 2 mM K and 140 mM Na in the external solution. (B) Increasing the external potassium to 140 mM slows C-type inactivation in the wild-type Shaker channel (τ = 2.6 s). (C) C-type inactivation is slow in A463C with 2 mM K and 140 mM Na in the external solutions. Neither increasing the external potassium concentration to 140 mM (D), nor replacing it with external NMG+ (E) has any effect on the time course of C-type inactivation in A463C. However, with 5 mM internal and no external potassium (NMG+), the efflux through the pore is decreased and the A463C channel readily inactivates (F).
Substituting a cysteine residue for the alanine at position 463 decreases the affinity of an internal ion binding site in the pore (Ogielska and Aldrich, 1998), and we hypothesized that perhaps the A463C channels would inactivate rapidly due to a decrease in the occupancy of that site by potassium. However, we found that the A463C mutant inactivated more slowly than wild type in low external potassium (2 mM K, Fig. 2 C). Furthermore, raising the external potassium concentration did not further slow the inactivation (140 mM K, Fig. 2 D) in A463C as it did in the wild type channel. This result is inconsistent with the simple idea that A463C decreases the occupancy of a site that directly regulates C-type inactivation. Two alternatives are possible: (a) the A463C mutation disrupts the conformational change associated with C-type inactivation and the channel cannot inactivate regardless of the potassium concentration, or (b) the C-type inactivation site is already maximally occupied with 2 mM external potassium, and therefore further increases in concentration are ineffective. To distinguish between these two possibilities, we examined the inactivation rate under conditions that should decrease potassium occupancy at the external (C-type) site. We reasoned that if inactivation is slow in A463C due to saturation of the C-type site, then decreasing the potassium occupancy at that site should result in faster inactivation. The inactivation rate did not increase when external potassium was replaced with the impermeant ion NMG (Fig. 2 E), suggesting that perhaps potassium efflux is sufficient to saturate the external site (see Baukrowitz and Yellen, 1995). Indeed, when we decreased the internal potassium concentration from 140 to 5 mM, the decrease in the potassium efflux did result in faster C-type inactivation (Fig. 2 F). This result indicates that the C-type inactivation mechanism is present in A463C, and that it can still be regulated by potassium occupancy.
The saturation of the potassium effect even in the absence of external potassium indicates that the A463C mutation appears to increase the occupancy of potassium at the site that governs C-type inactivation as compared with the wild-type channel under the same ionic conditions. Following this reasoning, we wondered whether sodium occupancy would be increased as well and therefore whether C-type inactivation of A463C measured in symmetrical sodium solutions would be slower than in the wild-type channel. The high potassium affinity of the wild-type Shaker channel makes the study of sodium conduction difficult (Starkus et al., 1997; Ogielska and Aldrich, 1998). However, Starkus et al. (1997) have found that the wild-type Shaker channel inactivates rapidly with sodium as the conducting ion (see also Fig. 3 A). Either sodium does not hinder C-type inactivation as efficiently as potassium in the wild-type channel, or the site that influences C-type inactivation is not occupied enough by sodium to significantly slow the C-type inactivation conformational change. The decrease in the apparent potassium affinity in A463C allows for large sodium fluxes in the absence of added potassium (Ogielska and Aldrich, 1998). However, no inactivation is observed during a 400-ms pulse in symmetrical sodium (Fig. 3 B), although a slow decline is evident with longer pulse duration (Fig. 3 C). This is unlike the wild-type channel, which inactivated in <20 ms in identical sodium solutions (Fig. 3 A). The high steady state current remaining after the inactivation transient has been attributed to sodium conduction through the C-type–inactivated state of the Shaker channel (Starkus et al., 1998). Accordingly, there are two possible explanations for the slow inactivation observed in the A463C mutant: (a) sodium is better able to interfere with C-type inactivation in the mutant than in the wild-type channel, or (b) A463C inactivates extremely rapidly in these solutions and the observed sodium current is due to sodium conducting through an inactivated state of the channel.
Figure 3.
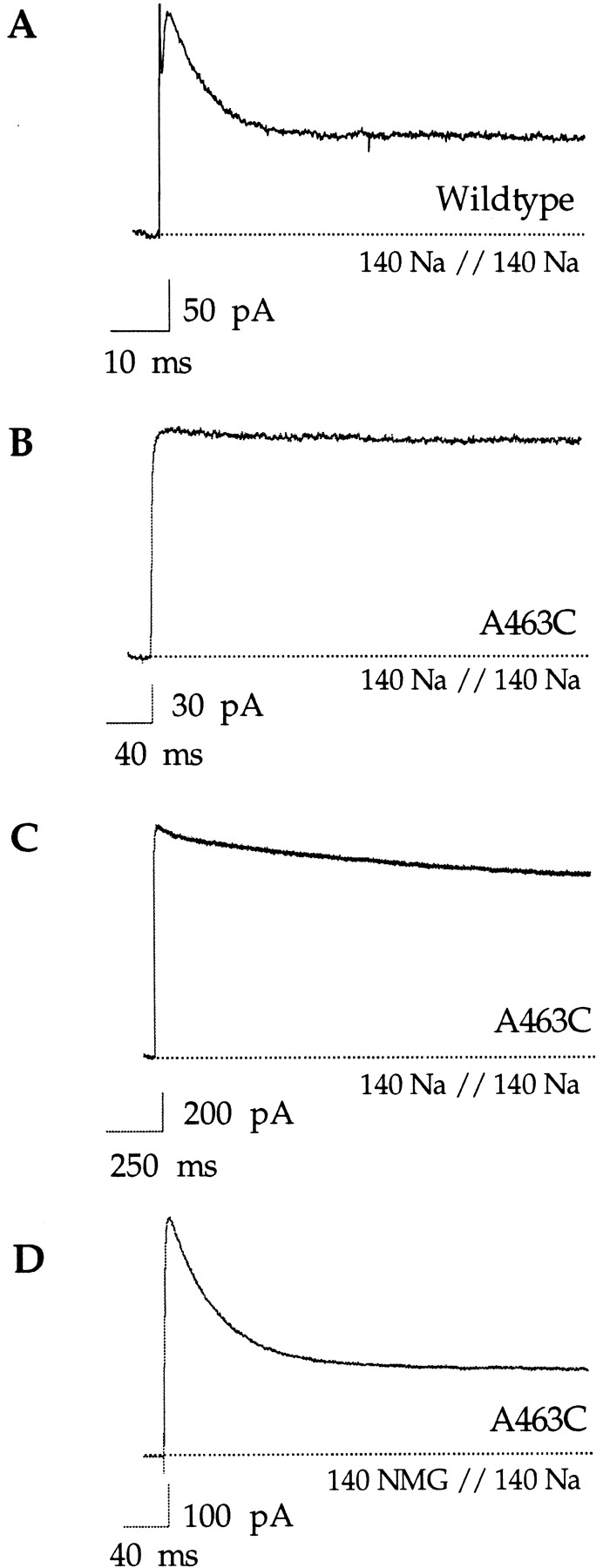
Properties of C-type inactivation with sodium as the conducting ion. All traces were elicited with voltage steps to +100 mV and in the presence of 140 mM internal sodium. Experiments were done in inside-out patches. (A) The wild-type Shaker channel inactivates rapidly (<20 ms) in the absence of added potassium and in the presence of symmetrical 140 mM sodium. Currents were elicited by a 100-ms voltage step. (B) In contrast, no inactivation is observed in the A463C mutant under the same ionic conditions. Currents were elicited by a 400-ms voltage step. (C) A slow decline in current is observed with longer pulses (2,000 ms). (D) In the presence of external NMG+, the A463C mutant rapidly inactivates to a steady state level. Pulse duration was 400 ms.
We have previously shown that in the presence of external sodium the ability of internal potassium to block sodium currents is decreased, suggesting that external sodium can enter the pore and destabilize the internal blocking potassium ion in A463C (Ogielska and Aldrich, 1998). We reasoned that perhaps external sodium could likewise interfere with C-type inactivation in the A463C mutant, predicting that in the absence of external sodium the channel would readily inactivate. If the experiment shown in Fig. 3 C is repeated with the impermeant ion NMG+ as the primary external monovalent, then the channel readily inactivates with a time constant of 43 ± 8 ms (n = 19; Fig. 3 D). With potassium as the conducting ion, the flux through the channel was sufficient to saturate the C-type inactivation site (Fig. 2 E). Although sodium interferes with C-type inactivation in A463C, sodium efflux seems to be insufficient to saturate the external site (Fig. 3 D). The observation that the inactivation rate varies depending on which ion species is permeating through the pore may be reflective of the inherent potassium selectivity of the C-type inactivation site or, alternatively, may be the result of a lower Na+ occupancy at the C-type site due to a lower Na+ flux rate through the channel. If sodium prevents C-type inactivation in the A463C mutant by binding within the pore, then low concentrations of external sodium should be able to slow the inactivation process. Indeed, when 1 mM sodium is added to the external NMG+ solution inactivation is slowed (Fig. 4 A). The observed decrease in the peak amplitude may be the result of a multi-ion interaction in the pore, but this was not investigated further. To facilitate comparison, the steady state current was subtracted and the current amplitudes were scaled (Fig. 4 A, bottom). The observation that sodium directly slows C-type inactivation argues against the possibility that the apparent lack of inactivation in symmetrical sodium solutions is due to the channels already being inactivated and conducting through the inactivated state. In the absence of external sodium, however, the A463C channels probably conduct Na+ through both the open and inactivated states as previously observed in the wild-type Shaker channel (see Starkus et al., 1998). Although the idea that the steady state current results from ions conducting through the C-type–inactivated state is intriguing, its origin in the A463C mutant is unclear and beyond the scope of this study.
Figure 4.

Both external sodium and internal potassium can slow inactivation in the A463C mutant. The control traces were elicited by a 400-ms voltage step to +100 mV in the presence of 140 mM internal sodium and external NMG+. The experiment in A was performed with an outside-out patch, while the experiment in B was with an inside-out patch. Inactivation can be slowed by 1 mM external sodium (A) and by 2.5 mM internal potassium (B). For easier comparison, the current amplitudes are scaled and the baselines subtracted in the bottom sections of both panels.
Since in the presence of external NMG+ and internal sodium, the external site is more often empty and A463C readily inactivates, we asked whether the addition of internal potassium could slow C-type inactivation in these solutions. We reasoned that by increasing internal potassium concentration we should increase the potassium occupancy at the more external (C-type) site and C-type inactivation should be concomitantly slowed. In Fig. 4 B the control trace was elicited in the presence of external NMG+ and internal Na+ and rapid inactivation was observed. Under these ionic conditions, the apparent potassium affinity of the internal site is 2.5 mM (Ogielska and Aldrich, 1998). The addition of 2.5 mM potassium to the internal solution resulted in the expected channel block (∼45%) and the remaining current inactivated with a slower time course than the control trace (Fig. 4 B). Subtracting the steady state current and scaling the amplitudes of the two traces facilitates the comparison (Fig. 4 B, bottom). The inactivation time constant increases roughly twofold from 43 ± 8 ms (n = 19) to 82 ± 11 ms (n = 5). These experiments demonstrate that as potassium appreciably occupies the channel pore it is able to interfere with C-type inactivation in A463C.
Decreased Ion–Ion Interactions Result in Increased Occupancy of the C-Type Site
Previous work has shown that the A463C mutant decreases the apparent potassium affinity and, at subsaturating concentrations, the occupancy of an internal ion binding site in the pore (Ogielska and Aldrich, 1998). Present experiments, however, suggest that A463C also increases the occupancy of an external site as reflected by the slow rates of C-type inactivation in both sodium and potassium solutions. One possibility is that the A463C mutation alters the overall structure of the pore and therefore affects the properties of several ion binding sites. Alternatively, increased occupancy at the external (C-type) site may be a secondary effect of the decreased affinity at the more internal site. If ions at the internal and external sites can mutually destabilize one another, then decreasing the occupancy of the internal site should increase the occupancy of the external site by decreasing the electrostatic interactions among ions in the pore. The observation that external sodium ions can enter the pore and decrease the apparent affinity of a blocking potassium ion at the internal site in the A463C mutant is consistent with the hypothesis that ions interact with one another in the channel pore (Ogielska and Aldrich, 1998).
In the presence of external sodium, the external (C-type) site is occupied, preventing inactivation (Fig. 3, B and C). We reasoned that by increasing ion occupancy at the internal site we should increase repulsive ion interactions in the pore and destabilize the sodium ion bound at the external (C-type) site. The emptying of the external site would be reflected in an increased rate of C-type inactivation. Potassium binds with a higher affinity than sodium and therefore blocks the sodium current at low concentrations. In symmetrical sodium solutions, the apparent internal potassium affinity of A463C is ∼6 mM (Ogielska and Aldrich, 1998). Adding low (millimolar) concentrations of internal potassium should therefore increase the occupancy of the internal site and concomitantly increase the repulsive interactions among ions in the pore. Increased repulsive interactions should destabilize the sodium ion bound at the external site and increase the C-type inactivation rate of the channel (Fig. 5 A).
Figure 5.

Increased occupancy of the external site of A463C results from decreased repulsive interactions between ions in the pore. (A) A model of the effects of increasing ion occupancy at the internal site on C-type inactivation in symmetrical sodium solutions. Potassium ions are depicted as • and sodium ions as ○. (B) The control trace was elicited by a 1,000-ms voltage step to +100 mV in an inside-out patch in symmetrical 140 mM NaCl. The addition of 1 mM internal potassium results in 13 ± 0.03% block and an increase in the C-type inactivation rate.
In symmetrical sodium solutions, the occupancy of the external (C-type) site by sodium is high, and no inactivation is observed (Fig. 5 B). When the occupancy of the internal site was increased by the addition of 1 mM potassium to the internal solution, significant inactivation was observed in addition to channel block (∼13%) (Fig. 5 B). Under these ionic conditions, A463C inactivates with a time constant of 198 ± 16 ms (n = 10), slower than in the absence of external sodium (43 ± 8 ms). These results are consistent with a decrease in the occupancy of sodium at the external site due to repulsive interactions with potassium at the internal site. Alternatively, the slow decline in current could be attributed to a slow potassium block at a second site in the pore. In the latter case, increasing the concentration of internal potassium should accelerate the blocking rate, making the current decline faster. Instead, the addition of 5 mM potassium further slowed the decline of the current (τ = 297 ± 10 ms; n = 3; data not shown). We interpret this slowing to mean that at higher concentration potassium itself occupies the external site and slows C-type inactivation.
The increased occupancy at the external (C-type) site in A463C is therefore best explained by a secondary effect of the decreased potassium affinity of an internal ion binding site. Our results suggest that ions remain bound longer at the more external site in A463C because of decreased repulsive interactions in the pore, not because the intrinsic affinity at that site has been directly increased by the A463C mutation.
External K+ Blocks Na+ Currents without Affecting C-Type Inactivation
Having found that the A463C channel readily inactivates once the external site is depleted of ions, we wanted to further examine the interactions between potassium and the C-type inactivation gate. We used sodium as the permeant ion to measure the apparent potassium affinity of the external ion binding site. We reasoned that the apparent affinity of the external (C-type) site most likely cannot be determined in blocking studies using internal potassium. Once a potassium ion traverses the pore and reaches the external (C-type) site, it presumably rapidly equilibrates with the external solution, and occupancy of that site would therefore not be perceived as current block. Based on this reasoning, we expected that low concentrations of external potassium would block the sodium current by binding to the external (C-type) site and concomitantly slow the rate of C-type inactivation.
Currents were recorded from outside-out patches in the presence of external NMG+ and internal Na+. Sweeps were 180 ms in duration to give the channels ample time to inactivate (Fig. 6 A). Micromolar concentrations of potassium were sufficient to block the observed sodium currents (Fig. 6 A). The fraction of unblocked currents (I/Imax) is plotted against external potassium concentration in Fig. 6 B (•). The data are well fitted with a single binding isotherm yielding an apparent potassium affinity of ∼100 μM. To compare the inactivation rates, the steady state current was subtracted and the control current was scaled with the current in the presence of 50 μM external potassium (Fig. 6 C). Contrary to our expectation, C-type inactivation progresses at the same rate regardless of the presence or absence of 50 μM external potassium (41 ± 7 ms, n = 4, and 43 ± 8 ms, n = 19, respectively). We interpret these data to mean that external potassium is binding to a high affinity site in the pore that is distinct from the external (C-type) site. This is unlike what was observed in a chimeric channel between Kv1.3 and Kv2.1. In that construct, external potassium both blocked the sodium currents with a high affinity and simultaneously slowed down the C-type inactivation process (Kiss and Korn, 1998).
Figure 6.

External potassium blocks sodium currents in A463C with a high affinity but does not slow C-type inactivation. Data shown in A were obtained from an outside-out patch in the presence of external NMG+ and internal Na+. Currents were elicited by voltage steps to +100 mV and externally applied potassium blocked the observed currents in a concentration-dependent manner (A). The data were quantified by measuring percent current remaining (I/Imax) at +100 mV versus the external potassium concentration (B, •). The error bars represent the SEM. A fit to the data with a single binding isotherm yields a K d value of 100 μM. The empty symbols represent the identical experiment except that it was performed in the presence of symmetrical sodium. Currents were elicited by voltage steps to +100 mV or by voltage ramps from −100 to +200 mV (data not shown). The blocking data were identical regardless of the voltage protocol used and as a result it was pooled. The error bars represent the SEM. A fit to the data with a single binding isotherm yields a K d value of 300 μM. (C) The control current and the current in the presence of 50 μM external potassium from A were scaled in amplitude and the baselines were subtracted. No change in the C-type inactivation rate is observed in the presence of 50 μM external potassium.
To examine further the properties of the high affinity binding site that is accessible from the external solution, we asked whether external potassium could still block in the presence of 140 mM external sodium. The experiments were performed in symmetrical sodium solutions and we found that external potassium could still block the sodium current with a high affinity, K d = ∼300 μM (Fig. 6 B, ○). The apparent potassium affinity is decreased in the presence of external sodium as compared with external NMG+ (∼300 vs. ∼100 μM), presumably as a result of ion–ion interactions. Potassium and sodium are either competing for entry into the channel or else cannot occupy the pore simultaneously. External potassium blocks sodium currents at submillimolar concentrations independent of the occupancy of the external (C-type) site (compare Fig. 6 B, ○ and •).
Given the sensitivity of C-type inactivation to the presence of external ions (Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995), the observation that it proceeds through a localized constriction of the outer mouth of the pore (Yellen et al., 1994; Liu et al., 1996), and the visualization of an ion bound at an external site in the crystal structure (Doyle et al., 1998), it is most likely that the site that controls C-type inactivation is the external site. Since the rate of C-type inactivation is unaffected when externally applied potassium is bound at its blocking site (Fig. 6 C), the high affinity site cannot be the external (C-type) site. External potassium presumably first binds to the C-type site but rapidly proceeds to a higher affinity blocking site, the occupancy of which does not interfere with C-type inactivation (Fig. 7, outlined scheme). The finding that external potassium still blocks sodium currents with a high affinity in the presence of external sodium indicates that the blocking ion presumably first displaces the bound sodium and subsequently binds at a higher affinity site in the pore. For example, sodium may bind and unbind rapidly at the C-type inactivation site while potassium enters deeper into the pore of the channel (Fig. 7, gray scheme). The rapid equilibration of the external (C-type) site is in agreement with the finding by Harris et al. (1998) that ions bound at the outermost site in the Shaker channel are in rapid equilibrium with the external solution.
Figure 7.

State diagram model of external potassium block of sodium currents in A463C. Sodium ions are represented by ○, while potassium ions are shown as •. (Outlined scheme) In the presence of external NMG+, external potassium first binds to the C-type inactivation site but proceeds to a deeper, higher affinity site in the pore. The occupancy of the deeper site by potassium does not interfere with the onset of C-type inactivation. (Shaded scheme) In the presence of external Na+, external potassium must first displace the bound sodium ion before it can proceed to the deeper site. After the higher affinity site is reached, the external (C-type) site is refilled by a sodium ion.
It is not clear from the data whether the high affinity site accessible from the external solution is the site affected by the A463C mutation. The crystal structure of KcsA predicts that the A463C mutation alters the interaction between the side chain at position 463 in S6 and a conserved valine in the signature sequence (V443) that participates in the formation of an internal ion binding site in the pore (Fig. 1; Doyle et al., 1998). The structural alterations result in a decrease in the measured apparent internal potassium affinity (micromolar into the millimolar range) (Ogielska and Aldrich, 1998). However, present data indicates that externally presented potassium binds to a high affinity (micromolar) site deep within the pore of the A463C mutant channel. If potassium is binding to the same site, regardless of whether it entered the pore from the internal or external solution, then the measured apparent affinity difference must somehow depend on the pathway taken to reach that site, and specifically on the relative occupancy of the neighboring sites. Since potassium is a permeant blocker, the measured apparent affinities do not reflect equilibrium binding affinities and therefore need not be pathway independent. Alternatively, it is possible that the high affinity site is one of the two overlapping internal sites predicted by the KcsA structure. Perhaps the A463C mutation alters the conformation of the V443 backbone in such a way that a potassium ion can no longer rapidly equilibrate between the two internal sites and the different apparent affinities are reflective of the two possible positions a potassium ion can assume.
Since the channel inactivates at the same rate regardless of whether the high affinity site is occupied by potassium, the C-type inactivation gating conformational change must not involve a global alteration of the selectivity filter region of the pore. Our finding is in agreement with Harris et al. (1998), who showed that the C-type inactivation conformational change trapped barium at a high affinity potassium binding site in the Shaker channel pore. We have shown that when an internal site is loaded with K+, the Na+ occupancy of the C-type site is decreased as a result of increased electrostatic interactions among ions in the pore (Fig. 5). Since a K+ ion bound at a more internal site can repel Na+ from the external (C-type) site, thus increasing the overall rate of inactivation, we wondered why the inactivation rate did not increase when externally applied K+ is bound to a high affinity site deep within the pore (Fig. 6 C). We reasoned that perhaps, under these ionic conditions (NMG+ outside and Na+ inside), Na+ occupied the C-type inactivation site so infrequently that the presence of the bound K+ did not significantly affect the occupancy of the external (C-type) site. We expected that, under conditions when the C-type site is significantly occupied by Na+ (symmetrical Na+ solutions), the rate of inactivation would be affected when an externally applied K+ ion is bound at a deeper high affinity site. However, we found that even in symmetrical sodium solutions the addition of 100 μM external K+ blocked the channel without affecting the C-type inactivation rate (data not shown). This difference between the effects of internally and externally applied potassium ions on C-type inactivation is similar to the difference in apparent blocking affinities depending upon internal and external application. This most likely results from ion interactions and the differences in occupancy of the various binding sites depending upon the directions of Na+ and K+ movement in the channel. The complex behavior of permeant blockers illustrates the interdependence of binding interactions at the sites in the pore and the fact that the measured apparent affinities do not reflect true equilibrium binding constants.
discussion
Our results underscore the importance of ion interactions in the pore of the potassium channel. We have shown that changing the affinity of one site in the pore can profoundly influence the occupancy of neighboring sites. The A463C mutation does not disturb the overall selectivity for potassium over sodium at the most internal site, but decreases ion affinity at that site. Ions spend less time there and repulsive ion–ion interactions in the pore are decreased. Functionally, occupancy of the external (C-type) site is reflected in the slow rates of C-type inactivation. Our findings are in agreement with those of Baukrowitz and Yellen (1995), who have found that an ion spends more time at the C-type inactivation site when it is not destabilized by other ions in the pore.
The recently solved potassium channel crystal structure allows for a reinterpretation of the mechanism of C-type inactivation and the molecular constituents of ion binding sites in the pore. The P-region of Shaker is highly homologous to that of KcsA, suggesting that the determinants of the ion binding sites are conserved between these channels. Independent evidence for three distinct ion binding sites in the potassium channel pore comes from elegant barium blocking studies in the calcium-activated K+ channel (Neyton and Miller, 1988a,b) and in the Shaker channel (Harris et al., 1998). Barium has the same crystal radius as potassium but, because of its divalent charge, it blocks current by binding to potassium binding sites with a high affinity. In both channel types, a high-affinity internal potassium binding site was identified. At low concentrations, external potassium was found to bind at a more external site, the “lock-in” site, and prevent the dissociation of the blocker into the external solution. The apparent potassium affinity of the external “lock-in” site in Shaker (Harris et al., 1998) correlates with the measured apparent affinity of the C-type inactivation site (Baukrowitz and Yellen, 1995). At higher concentrations, another site was filled, the “enhancement” site, and occupancy at that site promoted the exit of the blocker into the internal solution.
Recent work suggests that the C-type inactivation conformational change occurs at a position internal to residue T449 in the Shaker channel (Molina et al., 1997) and involves a local rearrangement of a few amino acids in that region (Liu et al., 1996). Further evidence for a local constriction comes from the work of Harris et al. (1998), who noted that the channel can still inactivate with a barium ion bound at a high affinity site in the pore and that the C-type conformational change prevents the dissociation of the blocker into the external solution, similar to what we find with potassium. An enticing suggestion was made by Doyle et al. (1998), who noted that the tyrosine at position 445 points away from the pore and interacts with the aromatic residues W434 and W435, creating a “cuff” of aromatic amino acids that may hold the pore open. Perhaps the aromatic cuff relaxes during C-type inactivation, preventing the passage of potassium but not of the smaller sodium ion. Such a hypothesis is supported by the observations that mutations at position W434 and Y445 greatly enhance the rate of C-type inactivation (Heginbotham et al., 1994; Yang et al., 1997; Harris et al., 1998) and that sodium readily conducts through the W434F mutant (Starkus et al., 1998). Interestingly, neither mutation (W434F or Y445F) removes the ability of external potassium to retard the onset of C-type inactivation, suggesting that the site is still present in both mutants (Heginbotham et al., 1994; Yang et al., 1997; Harris et al., 1998). In the Shaker channel, the Y445F mutation leaves the “lock-in” site unaffected but appears to disrupt the enhancement site (Harris et al., 1998). This observation suggests that the external ion binding site that governs the rate of C-type inactivation may be composed of carbonyl oxygens donated by the G446 residues.
The determination of the structure of the KcsA potassium channel (Doyle et al., 1998) and our recent findings concerning the role of ion–ion interactions in the pore allow for a reinterpretation of previous results concerning the role of residues at position 449 and 463 in the Shaker channel. Although amino acid substitutions at both positions cause significant changes in the C-type inactivation rate, we have previously found that their effects are not always energy additive (Ogielska et al., 1995). Position 449 is occupied by a threonine in the wild-type channel, and mutating position 463 from the wild-type alanine to a valine increases the rate of inactivation 100-fold (Hoshi et al., 1992). However, the inactivation rate is slow if position 449 is occupied by a valine or tyrosine, regardless of whether position 463 is an alanine or a valine. The observation that position 449 appears to play a dominant role over position 463 in the determination of the overall inactivation rate of the channel led us to speculate that residues at those positions interact either through a direct or an indirect mechanism. Since the dominant role of 449Y over 463V is preserved even when the mutations are placed in different subunits (Ogielska et al., 1995), we proposed that the effects are mediated through the ion-conducting pore rather than direct side chain or backbone interactions. Our speculation that the two side chains are most likely not interacting directly is supported by the crystal structure (Doyle et al., 1998), although we cannot definitively rule out the possibility of structural backbone alterations. We propose, however, that amino acid substitutions at both positions indirectly affect the probability of ion occupancy at the C-type inactivation site.
The residue at position 449 is exposed to solution and is localized at the external mouth of the pore (Heginbotham and MacKinnon, 1992; Lu and Miller, 1995; Kurz et al., 1995; Molina et al., 1997; Doyle et al., 1998). Although substitutions at position 449 can alter the rate of C-type inactivation in either direction, the observation that 449K and 449E both increase the rate of this process led to the speculation that mutations at this position affect the access of external ions to the C-type inactivation site (Lopez-Barneo et al., 1993). Molina et al. (1997) have shown that the T449Y substitution affects the ability of external tetraethylammonium to interfere with C-type inactivation, but does not disrupt the fidelity of the external ion binding site that governs the rate of C-type inactivation. The idea that 449 acts as a sentry for a deeper site is supported by work regarding the effects of mutations at that position on the blocking potency of external barium (Hurst et al., 1996). Following this type of reasoning, we would propose that when 449 is a tyrosine or a valine the probability of the C-type inactivation site being occupied by an ion is increased since the rate of inactivation is slow in both cases.
The A463C mutation decreases the affinity of an internal ion binding site that leads to lower occupancy of that site and decreased repulsive interactions in the pore. Ions remain bound longer at the external C-type inactivation site simply because the electrostatic interactions in the pore are diminished by the A463C mutation. By analogy, we can propose that the alanine to valine substitution at position 463 increases the affinity of the internal ion binding site and therefore increases the electrostatic interactions in the pore. The occupancy of the C-type inactivation site is accordingly decreased, resulting in a fast C-type inactivation rate.
If both mutations, T449Y and A463V, indirectly affect the probability of the C-type inactivation site being occupied, then their nonadditive contributions to the overall rate of C-type inactivation could be the result of their opposing influence on the occupancy of that site. If that is the case, their influence should be equivalent regardless of whether both substitutions are in the same or different subunits, as has been previously shown (Ogielska et al., 1995).
Acknowledgments
We thank T. Middendorf, D. Cox, and P. Zei for helpful comments on the manuscript and D. Cox for help with the analysis. We also thank R. MacKinnon for enlightening discussions and providing us with the crystal structure data.
This work was supported by a grant from the National Institutes of Health (NS23294) and a National Institutes of Health Training Grant (GM08327). R.W. Aldrich is an investigator with the Howard Hughes Medical Institute.
Abbreviation used in this paper
- NMG
N-methyl-d-glucamine
Footnotes
Portions of this work were previously published in abstract form (Ogielska, E.M., and R.W. Aldrich. 1998. Biophys. J. 74:A19).
references
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external K+: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+channel behaves like an open channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Harris RE, Larsson HP, Isacoff EY. A permeant ion binding site located between two gates of the Shaker K+channel. Biophys J. 1998;74:1808–1820. doi: 10.1016/s0006-3495(98)77891-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham L, MacKinnon R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+channel signature sequence. Biophys J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, Schwarz W. Potassium channels as multi-ion single file pores. J Gen Physiol. 1978;72:409–442. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta W, Aldrich R. Biophysical and molecular mechanisms of Shakerpotassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta W, Aldrich R. Two types of inactivation in Shaker K+channels: effects of alterations in the carboxy terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Toro L, Stefani E. Molecular determinants of external barium block in Shakerpotassium channels. FEBS Lett. 1996;388:59–65. doi: 10.1016/0014-5793(96)00516-9. [DOI] [PubMed] [Google Scholar]
- Kavanaugh MP, Hurst RS, Yakel J, Varnum MD, Adelman JP, North RA. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron. 1992;8:493–497. doi: 10.1016/0896-6273(92)90277-k. [DOI] [PubMed] [Google Scholar]
- Kiss L, Immke D, LoTurco J, Korn S. The interaction of Na+ and K+in voltage-gated potassium channels: evidence for cation binding sites of different affinity. J Gen Physiol. 1998;111:195–206. doi: 10.1085/jgp.111.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L, Korn SJ. Modulation of C-type inactivation by K+at the potassium channel selectivity filter. Biophys J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Ikeda SR. Permeation selectivity by competition in a delayed rectifier potassium. Science. 1995;269:410–412. doi: 10.1126/science.7618108. [DOI] [PubMed] [Google Scholar]
- Kurz LL, Zuhlke RD, Zhang H, Joho RH. Side-chain accessibilities in the pore of a K+channel probed by sulfhydryl-specific reagents after cysteine-scanning mutagenesis. Biophys J. 1995;68:900–905. doi: 10.1016/S0006-3495(95)80266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Lu Q, Miller C. Ag+ as a probe of pore forming residues in a K+channel. Science. 1995;268:304–307. doi: 10.1126/science.7716526. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann S, Aldrich R. Effects of external cations and mutations in the pore region on C-type inactivation of Shakerpotassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- Molina A, Castellano AG, Lopez-Barneo J. Pore mutations in Shaker K+channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J Physiol (Camb) 1997;499:3661–3667. doi: 10.1113/jphysiol.1997.sp021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton J, Miller C. Potassium blocks barium permeation through a calcium activated potassium channel. J Gen Physiol. 1988a;92:549–567. doi: 10.1085/jgp.92.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure of the high conductance Ca2+ activated K+channel. J Gen Physiol. 1988b;92:569–586. doi: 10.1085/jgp.92.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Aldrich RW. A mutation in S6 of Shaker potassium channels decreases the K+affinity of an ion binding site revealing ion–ion interactions in the pore. J Gen Physiol. 1998;112:243–257. doi: 10.1085/jgp.112.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Rayner MD, Heinemann SH. Ion conduction through C-type inactivated Shakerchannels. J Gen Physiol. 1997;110:539–550. doi: 10.1085/jgp.110.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Rayner MD, Heinemann SH. Macroscopic Na+ currents in the “nonconducting” Shakerpotassium channel mutant W434F. J Gen Physiol. 1998;112:85–93. doi: 10.1085/jgp.112.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? . J Gen Physiol. 1997;109:779–789. doi: 10.1085/jgp.109.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. Relief of Na+ block of Ca2+ activated K+channels by external cations. J Gen Physiol. 1984;84:187–199. doi: 10.1085/jgp.84.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G, Sodickson D, Chen T, Jurman ME. An engineered cysteine in the external mouth of a K channel allows inactivation to be modulated by metal binding. Biophys J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopusoocytes. Proc Natl Acad Sci USA. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shakerpotassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]