

Abstract
We report that the histone acetyltransferase Gcn5p is involved in cell cycle progression, whereas its absence induces several mitotic defects, including inefficient nuclear division, chromosome loss, delayed G2 progression, and spindle elongation. The fidelity of chromosome segregation is finely regulated by the close interplay between the centromere and the kinetochore, a protein complex hierarchically assembled in the centromeric DNA region, while disruption of GCN5 in mutants of inner components results in sick phenotype. These synthetic interactions involving the ADA complex lay the genetic basis for the critical role of Gcn5p in kinetochore assembly and function. We found that Gcn5p is, in fact, physically linked to the centromere, where it affects the structure of the variant centromeric nucleosome. Our findings offer a key insight into a Gcn5p-dependent epigenetic regulation at centromere/kinetochore in mitosis.
Epigenetic changes and chromatin signaling regulate gene expression by modifying chromatin structure. Multisubunit chromatin complexes reversibly remodel nucleosome organization and act in the catalytic modification of histone N-terminal tails, producing a combinatorial code of posttranslational modifications (25, 48, 50). Gcn5p (2, 14, 30), the ancestor of the histone acetyltransferase (HAT) family, marks histone H3 and H4 tails with an ɛ-acetyl group on specific lysines (41, 57). Gcn5 does not act on its own but rather as the catalytic subunit of two separate and conserved HAT complexes named ADA and SAGA (Spt-Ada-Gcn5-acetyltransferase). Specifically, Spt proteins are exclusively present in SAGA, while Spt20p is necessary for maintaining the integrity and function of the whole complex (58). Histone acetylation by Gcn5p is implicated in the displacement of nucleosomes from promoters during transcriptional activation and also in aiding the recruitment of TATA binding protein, RNA polymerase II, and coactivators (17). Acetylation therefore facilitates the formation of an accessible “open” chromatin structure corresponding to the transcribing genome (9, 31, 51). During cell division, chromatin remodeling expands to wide chromosomal regions, producing long waves of compaction and decondensation over the whole genome at each cell division (5, 55). Disturbing the HAT/histone deacetylase balance therefore alters protein activities on a cellular scale, which leads to various diseases, including cancer.
Several reports have highlighted the involvement of Gcn5p alone or in combination with other HATs such as Sas3 in the cell cycle (19, 22) and in the transcriptional regulation of a set of genes required at the end of telophase (28). In mammalian cells, loss of the homologue Gcn512 is lethal during embryogenesis, induces a high level of apoptosis (60), and affects G2/M transition in null mouse embryonic stem cells (32). In yeast, the specialized centromeric nucleosome, which contains the histone H3 variant Cse4p (37), is necessary for the assembly and interactions of inner kinetochore components at the centromere (7) and for the correct attachment of the chromosomes to the spindle in metaphase. Mutations in the centromeric/kinetochore components or epigentic modifications of this structure may lead to chromosome missegregation and G2/M delay. Since kinetochore assembly depends on the structure of the underlying centromere, an aberrant acetylation at this site may lead to a defective kinetochore, resulting in mitotic defects and cell death (56, 63). In fission yeast, the acetylation of histone H4 by Alp5 is also required for the correct attachment of the chromosome to the mitotic spindle (39, 46). Hyperacetylation following treatment with histone deacetylase inhibitors alters centromeric chromatin and induces high chromosome loss (10, 40, 59). Taken together, this evidence demonstrates that epigenetic regulation extends far behind histones. A growing number of nonhistone protein substrates (e.g., transcription factors) have been found to be modified at the posttranslational level and have a direct impact on key signal transduction pathways (27). Structural proteins such as the outer kinetochore component Dam1 were reported to be methylated by the histone methyltransferase Set1 (64).
These findings suggest that HATs, their substrates, and the cellular pathways that they fine-tune are still poorly understood, (65) despite the growing number of chromatin modifiers known to be involved in different cellular processes. Here we report that Gcn5p controls the metaphase-to-anaphase transition in budding yeast and is required for correct chromosome segregation and centromere/kinetochore function in mitosis. Genetic interactions between Gcn5p and DNA-bound kinetochore components together with the effect of Gcn5p on the variant centromeric nucleosome and its physical association at the centromere demonstrate that this HAT is directly implicated in the control of metaphase-to-anaphase transition.
MATERIALS AND METHODS
Strains, media, and reagents.
All yeast strains used for this work are listed in Table 1. Cells were grown in YEPD rich medium (1% yeast extract, 2% Bacto peptone, 2% glucose, and 20 μg/ml adenine) and in SD minimal medium (0.67% yeast nitrogen base and 2% glucose supplemented with required amino acids). To test benomyl sensitivity, benomyl (Sigma-Aldrich, St. Louis, MO) was added to solid medium from a 10-mg/ml stock solution (in dimethyl sulfoxide). All strains were grown at 28°C unless otherwise specified. Gene disruption and tagging were performed as previously described (21, 33) and controlled by PCR and Western blot analysis (not shown).
TABLE 1.
S. cerevisiae strains
| Straina | Genotype | Source or reference |
|---|---|---|
| W303 | MATaade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | |
| yPO4 | MATagcn5::KanMX4 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | This work |
| SVY084 | MATaspt20::KanMX4 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | This work |
| ySP1090 | MATα cse4-1 ade2-101 his3D-100 leu2-3 LYS2 trp1D ura3-52 | 49 |
| SVY102 | MATα gcn5::KanMX4 cse4-1 ade2-101 his3D-100 leu2-3 LYS2 trp1D ura3-52 | This work |
| ySP1107 | MATα ctf13-30 tet-GFP::LEU2 tetOs::URA3 | P. Hieter |
| SVY081 | MATα ctf13-30 tet-GFP::LEU2 tetOs::URA3 | This work |
| ySP22 | MATα ndc10-1 leu2 trp1 ura3 | J. Kilmartin |
| SVY083 | MATα gcn5::KanMX4 ndc10-1 leu2 trp1 ura3 | This work |
| PDW490 | MATα cep3-1 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | P. De Wulf |
| SVY069 | MATα gcn5::KanMX4 cep3-1 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | This work |
| ySP422 | MATaskp1::TRP1 skp1-3-LEU2 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | 8 |
| SVY082 | MATagcn5::KanMX4 skp1::TRP1 LEU2::skp1-3 ade2-1 trp1-1,112 his3-11,15 ura3 can1-100 ssd1 | This work |
| PDW370 | MATα mif2-3 ade2-1 trp1-1 leu2-3,112 his3-fs11,15 ura3 can1-100 ssd1 | P. De Wulf |
| SVY060 | MATα gcn5::KanMX4 mif2-3 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | This work |
| ySP1717 | MATaCEN 15-GFP ade2 his3 trp1 ura3 leu2 can1 lacI-NLS-GFP::HIS3 LacO::URA3(1.8 kb from centromere) | 16 |
| ySP4427 | MATaCEN 15-GFP gcn5::KanMX4 ade2 his3 trp1 ura3 leu2 can1 lacI-NLS-GFP::HIS3 LacO::URA3 (1.8 kb from centromere) | This work |
| yPO7 | MATα ade2-101 ura3-52 leu2-3,112 his3-D200 lys2 (pTS408-URA3, GAL-GFP-TUB3) | This work |
| yPO8 | MATα gcn5 ade2-101 ura3-52 leu2-3,112 his3 D200 lys2 (pTS408-URA3, GAL-GFP-TUB3) | This work |
| SVY088 | MATaade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 (pDK243, CEN-1xARS) | This work |
| SVY090 | MATagcn5::KanMX4 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 (pDK243, CEN-1xARS) | This work |
| SVY049 | MATaGCN5-9myc-klTRP1 ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3 can1-100 ssd1 | This work |
All strains used are in the W303 background.
Flow cytometric DNA quantification.
Cells were fixed in 70% ethanol for 2 h, washed with 50 mM Tris-HCl (pH 7.8), resuspended in 0.5 ml of 50 mM Tris-HCl (pH 7.8) containing 10 μg/ml RNase A, and incubated at 37°C overnight. After washing with 1 ml fluorescence-activated cell sorter (FACS) buffer (200 mM Tris-HCl [pH 7.5], 211 mM NaCl, 78 mM MgCl2) the cells were stained in 0.5 ml FACS buffer containing 40 μg/ml propidium iodide (P-4170; Sigma-Aldrich). Acquisition of samples was carried out using a FACStar plus flow cytometer (Becton-Dickinson). Results were analyzed with WIN-MDI software.
Cell cycle arrest.
Early-logarithmic-phase cultures grown at 25°C were incubated for 3 h in 4 μg/ml α-factor (T6901; Sigma-Aldrich) for G1 arrest. After blocking, the cells were washed twice and then released in YEPD at 28°C. Samples were collected at different time points and examined by FACS, microscopic analysis, chromatin immunoprecipitation (ChIP), and immunoblotting.
Staining and fluorescence microscopy.
Ethanol-fixed nuclei were visualized by staining DNA with DAPI (4′,6′-diamidino-2-phenylindole) or propidium iodide (P-4170; Sigma-Aldrich) using the FACS preparation protocol. Three independent experiments were performed; values were collected from 400 cells and then plotted. Chromatid separation was analyzed by observing fluorescent spots of the LacI-green fluorescent protein (GFP) signals at 1.8 kb from CEN15 (16). Spindle formation was analyzed in cells containing GAL1 promoter GFP-tubulin plasmid, pTS337 (3), by collecting phase-contrast and fluorescence images (every 3 min) with a confocal microscope. In situ immunofluorescence was performed as described by Fraschini et al. (12). Tubulin was detected by immunostaining with the YOL34 monoclonal antibody (Serotec) and then by indirect immunofluorescence using rhodamine-conjugated anti-rat antibody (1:100; Pierce Chemical Co.). Microscopic observations were performed using a fluorescence microscope (Zeiss Axioskop) and a confocal microscope (Leica TCS SP2 laser [Ar/Kr, Gre/Ne, He/Ne traditional phase contrast plus Nomarski contrast]).
Chromatin analysis.
Chromatin preparation was performed by the nystatin method (54). Cells were harvested from an exponentially growing culture (optical density [OD] at 600 nm, 0.3 to 0.6) to obtain in total 50 OD (OD at 600 × ml). Cells were subjected to in vivo chromatin digestion with DraI at 0, 50, 100, and 150 U/ml at 37°C for 30 min in nystatin buffer (50 mM NaCl, 1.5 mM CaCl2, 20 mM MgCl2, 20 mM Tris-HCl [pH 8], 1 M sorbitol). Digestion was stopped with stop solution (50 mM EDTA, 1% sodium dodecyl sulfate); after deproteinization and RNase treatment, the DNA was fully digested with EcoRI at 37°C overnight. The chromatin preparation from EcoRI/DraI digestion was hybridized simultaneously with a 536-bp CEN3 fragment (map position, 113757 to 114293) and 587-bp GLT1 fragment (154748 to 154161). The degree of DraI digestion was calculated with a Packard Instant-Imager as percent cpm of the EcoRI/DraI band of either CEN3 or GLT1 in relation to the total cpm signal of CEN3 (E/E [5.1 kb] + E/D [2.2 kb]) or of GLT1 (E/E [4.2 kb] + E/D [3.5 kb]) (not shown).
Minichromosome stability assay.
Chromosome loss rates were determined from five independent experiments with cells with pDK243 (18). After growth in YPD for 12 generations at 28°C and 37°C, the same number of cells was plated on complete medium and replicated on selective SD plates. The plasmid-containing growing colonies were quantified and the average plasmid loss per generation calculated.
ChIP analysis.
ChIP analysis was performed as described elsewhere (7) with the following modifications. Samples carrying the GCN5-9Myc-tagged version were fixed for 60 min and washed twice in 20 ml of Tris-buffered saline (20 mM Tris-HCl [pH 7.5], 150 mM NaCl). Cells were lysed in 500 μl of ice cold lysis buffer (1 mM EDTA, 50 mM HEPES, 140 mM NaCl, 1% Triton X-100, 1 mg/ml Na deoxycholate) with glass beads by vortexing them 10 times for 30 s in a cold room. Chromatin was sheared by sonicating it four times for 30 s using a Branson Digital Sonifier (Branson Ultrasonic Corporation, Danbury, CT) on 10% impulse (average fragment size, 300 to 500 bp) and clarified for 5 min in a microcentrifuge. Mouse monoclonal anti-Myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-Cep3 (a kind gift of P. De Wulf) were used for immunoprecipitations with protein A-conjugated Dyna beads (Dynal Biotech, Lake Success, NY). Primers used to amplify the CEN3 locus (244 bp) were CEN3F (5′-GATCAGCGCCAAACAATATGG-3′) and CEN3R (5′-AACTTCCACCAGTAAACGTTTC-3′), those for the CEN16 locus (312 bp) were CEN16F (5′-GGTTGAAGCCGTTATGTTGTCG-3′) and CEN16R(5′-ACCATGGTGTCACTTCCCC-3′), and those for the TEL locus (249 bp) were TEL6F (5′-CCACTCAAAGAGAAATTTACTGG-3′) and TEL6R (5′-TGACATATCCTTCACGAATATTGTTAGA-3′) (1). Taq polymerase (New England Biolabs, Beverly, MA) was used for all PCR amplifications. For PCR analyses, input and immunoprecipitated template concentrations were titrated into the linear range. Fivefold serial dilutions of the crude lysates (input) and immunoprecipitated DNA are shown in all figures.
RESULTS
Deletion of Gcn5p results in slow G2/M progression and temperature sensitivity.
Temperature sensitivity, a lengthened doubling time, and a prolonged G2 phase are distinct features of a gcn5Δ strain (34, 66). Accordingly, FACS analysis of asynchronous gcn5Δ cells grown at permissive temperature showed an increase in the G2/M cell population (Fig. 1A). In order to better understand specific cell growth defects, gcn5Δ cells were tested at different temperatures (Fig. 1B). Deletion of GCN5 induced defective growth with respect to the wild-type strain, especially at 25°C and 37°C. In addition, we tested whether the growth phenotype depends on ADA or SAGA complexes by deleting Spt20 to disrupt the integrity of the SAGA complex (35). Normal growth of the spt20Δ strain demonstrated that the effect of temperature on gcn5Δ cells is linked to the activity of the ADA complex, since no phenotype was found when SAGA integrity was destroyed. To determine whether a specific cell cycle stage was affected, we analyzed cell morphology and bud shape in the asynchronous cell population at 28°C and 37°C. We found a marked reduction in single G1 cells and an increase in G2/M cell doublets (Fig. 1C). In addition, abnormally shaped cells with elongated buds that were more abundant at 37°C were found exclusively in the gcn5Δ strain. These findings confirmed previous observations reporting that the lack of Gcn5p induces a specific delay in G2 progression but also demonstrated that the lack of a defective phenotype in the spt20Δ strain indicates that this phenotype depends on the ADA complex.
FIG. 1.

Deletion of Gcn5p induces G2 delay and temperature sensitivity. (A) gcn5Δ strain accumulates in G2/M. Flow cytometry analysis of wild-type (WT) (W303) and gcn5Δ (yPO4) strains is shown. (B) Fivefold serial dilutions of WT (W303), gcn5Δ (yPO4), and SAGA-deficient spt20Δ (SVY084) strains spotted on YEPD and grown at 25°C, 28°C, or 37°C for 48 h. (C) Analysis of cell morphology in WT (W303) and gcn5Δ (yPO4) strains grown at 28°C or 37°C. Percentages of G1 cells (white bars), G2/M doublets (gray bars), and G2/M doublets with elongated buds (black bars) were determined (n = 450). Error bars indicate standard deviations.
Cells lacking Gcn5p show altered nuclear migration and increased frequency of minichromosome loss.
To assess whether gcn5Δ defects are correlated with improper nuclear segregation, we analyzed the nuclear distribution in G2 cell doublets in logarithmically growing cells at permissive temperature. The migration of duplicated nuclei was monitored at the microscope by calculating the percentages of large-budded cells carrying two divided nuclei, cells with one duplicated nucleus passing through the bud neck, and aberrant G2 cells with a single, unsegregated nucleus in one cell body (Fig. 2A). The histogram illustrates a remarkable decrease in cells with segregated nuclei and a sharp increase in G2 cells bearing unsegregated nuclei after deletion of Gcn5p (Fig. 2B). We then investigated whether subsequent mitotic loss similarly occurred in a gcn5Δ strain by measuring the propagation efficiency of a CEN-based minichromosome after prolonged growth of yeast cultures in nonselective medium (18). Figure 2C shows the percentage of minichromosome loss in the wild-type and gcn5Δ strains calculated after 12 generations of growth in rich medium at 28°C and 37°C. These results demonstrated that loss of the minichromosome is strongly enhanced in gcn5Δ cells, with a further increase at 37°C.
FIG. 2.
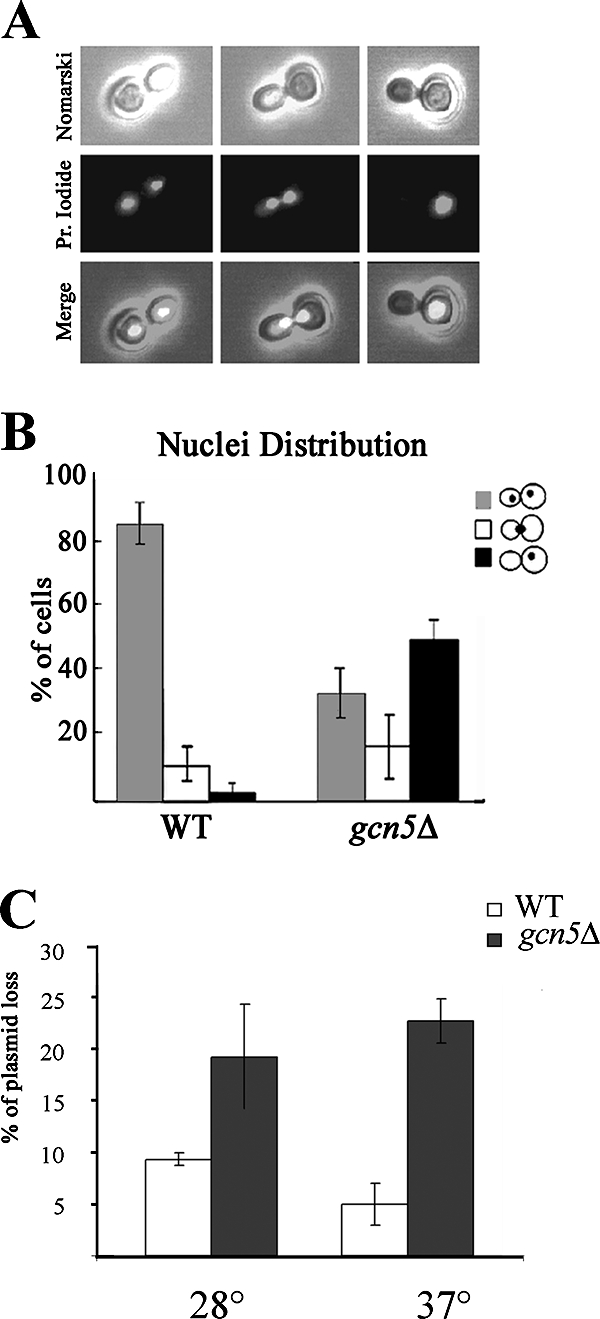
Defective nuclear migration and minichromosome loss in a gcn5Δ strain. Cells were grown at 28°C in YEPD, and nuclei were stained with propidium iodide. (A) Microscopic samples of large-budded doublets carrying two segregated nuclei, duplicated nuclei passing through the bud neck, and unsegregated nuclei in dividing cells. (B) Nuclear distribution in wild-type (WT) (W303) and gcn5Δ (yPO4) strains. (C) Percentage of centromeric plasmid loss (pDK243) measured in WT (W303) and gcn5Δ (yPO4) strains at either 28°C or 37°C after growth for 12 generations in nonselective medium. Error bars indicate standard deviations.
gcn5Δ cells are hypersensitive to microtubule-depolymerizing drugs.
Mutations affecting chromosome stability and efficient chromosome segregation produce hypersensitivity to the microtubule-destabilizing drug benomyl. This phenotype was also reported to occur in mutants of other chromatin effectors (e.g., components of the NuA4 complex and deacetylase Hda3) (23, 29). These findings and the observed defective nucleus migration in the gcn5Δ strain prompted us to test whether Gcn5p and ADA might affect mitotic spindle function. To do this, we tested cell sensitivity to the spindle poison benomyl in a gcn5Δ strain (Fig. 3A). Growth spot assay showed that the gcn5Δ strain was hypersensitive to the presence of benomyl at increasing concentrations. This phenotype was still controlled by ADA, since no effect was observed in the spt20Δ strain, clearly indicating that Gcn5p is required for growth upon contact with the antimicrotubule drug and in spindle shock conditions. We then wanted to follow spindle elongation in a gcn5Δ strain. Wild-type and gcn5Δ spindles tagged with GFP-tubulin (3) and elongating into the daughter cell were monitored by fluorescence microscopy during cell division (Fig. 3B). A time-lapse experiment (over 240 min) was conducted in order to encompass the slow duplication time of gcn5Δ. Under our experimental conditions, the spindle behavior of the dividing wild-type cells was normal, whereas the gcn5Δ strain revealed relevant defects in spindle dynamics: the spindle was short, failed to elongate, and did not move into the neck, even after a prolonged duplication time (240 min). This demonstrates that deletion of Gcn5p induces hypersensitivity to spindle poison and delays mitotic spindle elongation into the daughter cell.
FIG. 3.

Absence of Gcn5p causes sensitivity to microtubule-depolymerizing agents and spindle elongation defects. (A) Fivefold serial dilutions of wild-type (WT) (W303), gcn5Δ (yPO4), and spt20Δ (SVY084) strains were grown on YEPD supplemented with increasing benomyl concentrations (5, 10, and 15 μg/ml) and grown at 28°C for 60 h. (B) In vivo microscopy of spindle elongation visualized with GFP-tubulin (upper fluorescent panels). WT (W303) and gcn5Δ (yPO4) cells growing in SD medium were observed on microscope slides for 240 min. A selection of photographs collected every 3 min is shown. Nomarski pictures are given (bottom row) to show cell septation in a gcn5Δ strain.
Effects of gcn5 mutation on spindle dynamics.
To further investigate whether Gcn5p is required in spindle dynamics, we measured budding spindle elongation and sister chromatid separation at the GFP-tagged CEN15 (16) region after release from G1 block in wild-type and gcn5Δ cells. FACS profiles showed a pronounced delay in recovery from G1 arrest in the absence of Gcn5p (Fig. 4A). In addition, while budding was only slightly affected, centromeric sister chromatid separation and spindle elongation were drastically delayed in the gcn5Δ strain (Fig. 4B); metaphase spindles remained short and failed to elongate until 110 min after block release. Samples collected at maximal spindle elongation and centromeric dot separation (wild type, 90 min; gcn5Δ, 130 min) displayed an accumulation of large-budded cells with short metaphasic spindles and unsegregated nuclei in the gcn5Δ strain (Fig. 4C). Misorientation of short spindles (7% of large-budded gcn5Δ cells) (Fig. 4D) is very likely a consequence of extremely prolonged and unsuccessful metaphases. Collectively, our results suggest that Gcn5p affects the metaphase-to-anaphase transition, with implications for faithful chromosome segregation.
FIG. 4.

Deletion of Gcn5p induces strong delay of G2, spindle elongation, and chromatid separation. (A) FACS analysis of cells released in YEPD medium after α-factor blockage, collected every 10 min, and examined. WT, wild type. (B) After release, we calculated the percentages of budding cells (n = 250) (open circles), elongated anaphasic spindles (triangles), and separated centromeric GFP-tagged dots (closed circles) taken at 10-minute intervals. (C) Spindle (tubulin immunostaining) and nuclear (DAPI) images of WT (W303, left panel) and gcn5Δ (yPO4, right panel) G2/M large-budded cells collected at 90 min and 130 min, respectively, showing normal (WT) or unaligned short (gcn5Δ) spindles. (D) Distribution of short unaligned spindles in WT and gcn5Δ strains.
gcn5 mutation interacts genetically with mutations affecting kinetochore components.
The correct assembly of the kinetochore on the centromeric region constitutes the machinery for linking chromosomes to spindle microtubules and ensures chromosome segregation fidelity (26, 36). Following observation of the mitotic defects, we performed a genetic interaction assay between gcn5Δ and mutants of DNA-bound kinetochore components to test whether we could obtain epistatic links (45). In order to evaluate growth efficiency, GCN5 was replaced with a KanMX4 cassette in a collection of kinetochore mutants for growth analysis of single and double mutants (Fig. 5) at permissive and semipermissive temperatures by deleting GCN5 in temperature-sensitive kinetochore mutants. In this way, a strong interaction between Gcn5p and the centromeric histone H3 variant Cse4p was obtained. As Cse4p recruits numerous kinetochore proteins at the centromere, we assayed another subgroup of kinetochore proteins belonging to the CBF3 complex localized at the inner layer and assembled in proximity of the variant nucleosome (13). Positive interactions were found also with CBF3 complex mutations skp1-3, cep3-1, and ctf13-30, while ndc10-1 and mif2-3 showed no strong interaction. These data show that Gcn5p deletion is functionally linked not only to the histone H3 variant Cse4p but also to the inner kinetochore components functionally involved in chromosome segregation and cell division (36). Remarkably, significant phenotypic convergence of the cse4-1 mutant phenotype with the gcn5Δ strain (e.g., accumulation of large-budded cells with unsegregated nuclei, metaphasic spindles, and elevated chromosome loss) (49) provides further evidence for their close functional interaction.
FIG. 5.

A gcn5Δ strain displays genetic interaction with DNA-bound kinetochore components. Yeast strains of genotypes W303, gcn5Δ (yPO4), cse4-1 (ySP1090), cse4-1, gcn5Δ (SVY102), mif2-3 (PDW370), mif2-3, gcn5Δ (SVY060), skp1-3(ySP422), skp1-3 gcn5Δ (SVY082), cep3-1 (PDW490), cep3-1 gcn5Δ (SVY069), ctf13-30 (ySP1107), ctf13-30, gcn5Δ (SVY081), ndc10-1 (ySP22), and ndc10-1 gcn5Δ (SVY083) (see Table 1) were grown to log phase at 28°C. Fivefold serial dilutions were plated on YEPD and incubated at the indicated temperature for 48 h.
Centromeric chromatin is altered in gcn5Δ mutants.
Acetylation of histone N termini induces chromatin changes that enhance nuclease accessibility (41). In addition, depletion of histones and mutations of DNA-bound kinetochore components alter the accessibility of underlying chromatin by modifying the centromeric nucleosome conformation (20, 43, 44). To gain evidence for a direct effect of Gcn5p on the centromeric nucleosome, we tested the accessibility of chromatin to in vivo digestion with DraI endonuclease followed by EcoRI digestion after DNA purification in wild-type and gcn5Δ strains (Fig. 6A). Compared to the wild type, accessibility of the CDEII internal DraI sites (Fig. 6B) was enriched in the gcn5Δ strain, as demonstrated by a 2.2-kb band at the DraI sites versus the 5.1-kb upper band of the uncut EcoRI-EcoRI sites (Fig. 6C). As a comparison, accessibility of the GLT1 gene on chromosome IV, which was used as a noncentromeric genomic probe for normalization of unspecific endonuclease cleavage (data not shown), was tested under the same conditions. The plot in Fig. 6D clearly indicates that a specific modification of the core centromeric DNA region produced hypersensitivity to DraI cuts inside the core centromeric sequences. This finding demonstrates that Gcn5p induces a localized modification of the variant nucleosome and adds further evidence for both genetic and functional interactions between Gcn5p and the histone H3 variant Cse4p, a major component of the centromeric nucleosome.
FIG. 6.

Gcn5p physically interacts with the centromere and is required for the structure of variant centromeric nucleosome. (A) Schematic restriction map of the CEN3 region. The locations of DraI (black triangles) in the CDEII-CEN3 (II) region and EcoRI flanking sites (E) are indicated. The probe used for panel C is shown as gray bar. (B) Ethidium bromide staining of in vivo restriction of chromatin (wild type [WT] [W303] and gcn5Δ [yPO4] strains) at increasing DraI concentrations (0, 50, 100, and 150 U/ml), followed by EcoRI digestion after DNA extraction. (C) CEN3 accessibility to DraI cuts, showing the full-sized E/E 5.1-kb band and the E/D band of 2.2 kb corresponding to in vivo CDEII-CEN3 accessibility in WT and gcn5Δ strains. (D) Percentages of DraI cuts on the CDEII-CEN3 region and on a noncentromeric gene sequence (GLT1), used as an internal standard of digestion. Error bars indicate standard deviations. (E) ChIP of the inner kinetochore component Cep3p and 9Myc-Gcn5 in an epitope-tagged Gcn5-9Myc strain (SVY049). PCR amplification carried out with primers external to the core centromeric locus produced a 300-bp band spanning CEN3 and CEN16. As control, a telomeric sequence (TEL) was used in all ChIP samples.
HAT Gcn5p directly interacts with centromeric DNA.
Having determined a structural modification of the CEN3 nucleosome, we next wanted to ascertain whether Gcn5p had a direct effect on the centromere and kinetochore by testing its physical interaction with centromeric DNA sequences. To do this, we used a ChIP technique (Fig. 6E); chromatin extracted from a strain carrying C-terminal 9Myc-tagged Gcn5p was cross-linked, processed (see Materials and Methods), and immunoprecipitated using anti-Myc antibody and anti-Cep3p as a kinetochore-bound control protein (11). PCR of total, anti-Myc, anti-Cep3, and no-antibody fractions was carried out with primers spanning the two core centromeric sequences CEN3 and CEN16 and a telomeric region chosen as an additional control. No amplification was obtained without antibody, whereas an increasing linear band was obtained in anti-Myc-Gcn5 and anti-Cep3 immunoprecipitations. No PCR products were consistently obtained with a telomeric sequence (1), unambiguously demonstrating a physical association between Gcn5p and the centromere. This showed for the first time a direct association between HAT Gcn5p and the core centromere, thus highlighting its involvement as a direct determinant of centromere/kinetochore function. Having observed defective cell progression in the Gcn5p-deleted strain, we also wanted to follow Gcn5p localization at the centromere in the cells released from G1 synchronization during the cell cycle. The results demonstrated an almost constant presence of Gcn5p at the centromere (data not shown). Nonetheless, a discrete increase at S phase suggests a possible targeted recruitment at the centromere. This result will need to be taken into account in a further analysis to clarify recruitment time and additional implications.
DISCUSSION
Gcn5p is known for acetylating histone H3 and H4; however, recent data have highlighted a much broader role than originally thought for HATs and the acetylation process (65). So far, the effects of Gcn5p on growth and cell cycle progression have never been investigated in much detail. Therefore, the present investigation was undertaken to better characterize the slow-growth phenotype of the GCN5-deleted strain. By employing different experimental approaches, we describe here, for the first time, a novel role for Gcn5p in determining centromere/kinetochore organization, with a direct implication for the control of faithful chromosome segregation. We demonstrated that the lack of Gcn5p induces a pronounced delay in G2 progression and poor growth at high temperature. Interestingly, GCN5-deleted cells also showed sensitivity to microtubule-depolymerizing drugs; to our knowledge, this phenotype had never been reported so far.
We have shown that after release from G1 block, the gcn5Δ strain progresses slowly through G2, with a pronounced delay in separation of centromeric regions and spindle elongation. Moreover, a defective nuclear distribution and high rate of chromosome loss account for improper chromosomal segregation in a gcn5Δ strain. Remarkably, all these mitotic phenotypes resemble the defects induced by mutation of the centromere-specific histone H3 variant Cse4p (49, 67). In budding yeast, centromeres are required for directing the assembly and association of kinetochore DNA-bound subcomplexes such as CBF3 and Mif2 in close proximity to the centromeric nucleosome (38). To determine whether deletion of Gcn5p also affected kinetochore function, we tested the genetic interaction between Gcn5p and inner kinetochore components. The positive genetic interactions obtained with Cse4p and CBF3 components indicated a relevant function of Gcn5p at the kinetochore, not only demonstrating that Gcn5p is required at the centromere but also suggesting a role of Gcn5p in determining specific kinetochore function.
Chromatin structure and epigenetic and kinetochore function.
Chromatin proteins and nucleosome remodeling correlate with kinetochore function (6, 46, 52). Accordingly, mitotic arrest and spindle phenotypes very similar to those we described for gcn5Δ cells have been reported for mutants of the SWI/SNF complex (61), chromatin deposition factor I (47) and RSC (20, 53). Importantly, previous studies demonstrated that rsc4 mutation is lethal in combination with gcn5 (24), suggesting that RSC function depends on chromatin status and may act in concert with Gcn5p. In the present study we demonstrate that Gcn5p is physically linked to the centromere. This was demonstrated at the centromere of chromosome III (CEN3) and confirmed on a second chromosome (CEN16).
ChIP was further used to measure the recruitment time for Gcn5p at the centromere during the cell cycle. Our data suggest that Gcn5p is constantly recruited at the centromere, although there is a discrete increment at S phase, coincidental with the loading and positioning of newly synthesized nucleosome by the chromatin deposition factor I complex (47). This suggests that Gcn5p may contribute at a functional level with Cse4p to the creation of a specialized nucleosome (4, 6, 15). It cannot be ruled out, however, that an overall hypoacetylation of flanking canonical nucleosomes may affect the centromere and cause segregation defects. Since Cse4p is also involved in de novo kinetochore assembly and function (7), centromeric chromatin may also be an important epigenetic constraint directly regulated by Gcn5p. As Gcn5p and the kinetochore are highly conserved from yeast to humans, the role of Gcn5p in kinetochore maturation and function is likely to be conserved as well. In this regard, our findings may have important implications in the study of tumors with high aneuploidy, where HATs are very often mutated or translocated (42, 62). The identity of the primary target for Gcn5p activity remains a challenging question for the study of epigenetics. Taken together, our results demonstrate the role of Gcn5p in mitosis and its importance for accurate chromosome segregation. The localization of Gcn5p at the centromere further supports a direct role of Gcn5p at the kinetochore, where positive interactions with DNA-bound kinetochore components such as the CBF3 complex occur. Together our data reveal an additional, uninvestigated regulatory pathway of the kinetochore. We believe that this knowledge will prove essential for directing the focus of studies on a novel, more specific structural and cell cycle-dependent role of Gcn5p that goes far beyond its function in the regulation of gene expression.
Acknowledgments
This work was funded by RTL-CNR 2005 to P.F. and by Fondazione Pasteur Istituto Cenci-Bolognetti to P.B. S.V. was supported by Ministero dell'Interno, Dip. P.S.
We thank S. Piatti for helpful advice, for providing the strains, and for help with the experiment shown in Fig. 4. We thank L. Guarente, M. H. Kuo, T. Stearn, S. Berger, B. Stilmann, and P. DeWulf for providing mutant strains and plasmids. We are particularly indebted to E. Marchetti for his skillful technical assistance in confocal microscopy and video time-lapse experiments.
Footnotes
Published ahead of print on 26 November 2007.
REFERENCES
- 1.Agricola, E., L. Verdone, E. Di Mauro, and M. Caserta. 2006. H4 acetylation does not replace H3 acetylation in chromatin remodelling and transcription activation of Adr1-dependent genes. Mol. Microbiol. 621433-1446. [DOI] [PubMed] [Google Scholar]
- 2.Brownell, J. E., J. Zhou, T. Ranalli, R. Kobayashi, D. G. Edmondson, S. Y. Roth, and C. D. Allis. 1996. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84843-851. [DOI] [PubMed] [Google Scholar]
- 3.Carminati, J. L., and T. Stearns. 1997. Microtubules orient the mitotic spindle in yeast through dynein-dependent interactions with the cell cortex. J. Cell Biol. 138629-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, Y., R. E. Baker, K. C. Keith, K. Harris, S. Stoler, and M. Fitzgerald-Hayes. 2000. The N terminus of the centromere H3-like protein Cse4p performs an essential function distinct from that of the histone fold domain. Mol. Cell. Biol. 187037-7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung, P., C. D. Allis, and P. Sassone-Corsi. 2000. Signaling to chromatin through histone modifications. Cell 103263-271. [DOI] [PubMed] [Google Scholar]
- 6.Cleveland, D. W., Y. Mao, and K. F. Sullivan. 2003. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112407-421. [DOI] [PubMed] [Google Scholar]
- 7.Collins, K., A. R. Castillo, S. Y. Tatsutani, and S. Y. Biggins. 2005. De novo kinetochore assembly requires the centromeric histone H3 variant. Mol. Biol. Cell 165649-5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connelly, C., and P. Hieter. 1996. Budding yeast SKP1 encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell 26275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davie, J. R. 1998. Covalent modifications of histones: expression from chromatin templates. Curr. Opin. Genet. Dev. 8173-178. [DOI] [PubMed] [Google Scholar]
- 10.Ekwall, K., T. Olsson, B. M. Turner, G. Cranston, and R. C. Allshire. 1997. Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell 911021-1032. [DOI] [PubMed] [Google Scholar]
- 11.Espelin, C. W., K. B. Kaplan, and P. K. Sorger. 1997. Probing the architecture of a simple kinetochore using DNA-protein crosslinking. J. Cell Biol. 1391383-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraschini, R., E. Formenti, G. Lucchini, and S. Piatti. 1999. Budding yeast Bub2 is localized at spindle pole bodies and activates the mitotic checkpoint via a different pathway from Mad2. J. Cell Biol. 145979-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardner, R. D., A. Poddar, C. Yellman, P. A. Tavormina, M. C. Monteagudo, and D. J. Burke. 2001. The spindle checkpoint of the yeast Saccharomyces cerevisiae requires kinetochore function and maps to the CBF3 domain. Genetics 1571493-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georgakopoulos, T., and G. Thireos. 1992. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. EMBO J. 114145-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glowczewski, L., P. Yang, T. Kalashnikova, M. S. Santisteban, and M. M. Smith. 2000. Histone-histone interactions and centromere function. Mol. Cell. Biol. 205700-5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goshima, G., and M. Yanagida. 2000. Establishing biorientation occurs with precocious separation of the sister kinetochores, but not the arms, in the early spindle of budding yeast. Cell 100619-633. [DOI] [PubMed] [Google Scholar]
- 17.Govind, C. K., F. Zhang, H. Qiu, K. Hofmeyer, and A. G. Hinnebusch. 2007. Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol. Cell 2531-42. [DOI] [PubMed] [Google Scholar]
- 18.Hogan, E., and D. Koshland. 1992. Addition of extra origins of replication to a minichromosome supresses its mitotic loss in cdc6 and cdc14 mutants of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 893098-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howe, L. A., D. Auston, P. Grant, S. John, R. G. Cook, J. L. Workman, and L. Pillus. 2001. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 153144-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu, J. M., J. Huang, P. B. Meluh, and B. C. Laurent. 2003. The yeast RSC chromatin-remodeling complex is required for kinetochore function in chromosome. Mol. Cell. Biol. 93202-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janke, C., M. M., Magiera, N. Rathfelder, C. Taxis, S. Reber, H. Maekawa, A. Moreno-Borchart, G. Doenges, E. Schwob, E. Schiebel, and M. Knop. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 11947-962. [DOI] [PubMed] [Google Scholar]
- 22.John, S., L. Howe, S. T. Tafrov, P. A. Grant, R. Sternglanz, and J. L. Workman. 2000. The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev. 141196-1208. [PMC free article] [PubMed] [Google Scholar]
- 23.Kanta, H., L. Laprade, A. Almutairi, and I. Pinto. 2006. Suppressor analysis of a histone defect identifies a new function for the hda1 complex in chromosome segregation. Genetics 173435-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasten, M., H. Szerlog, H. Erdjument-Bromage, P. Tempst, M. Werner, and B. R. Cairns. 2004. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 231348-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kingstone, R. E., and G. J. Narlikar. 1999. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 132339-2352. [DOI] [PubMed] [Google Scholar]
- 26.Kitagawa, K., and P. Hieter. 2001. Evolutionary conservation between budding yeast and human kinetochores. Nat. Rev. Mol. Cell Biol. 9678-687. [DOI] [PubMed] [Google Scholar]
- 27.Kouzarides, T. 1999. Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 940-48. [DOI] [PubMed] [Google Scholar]
- 28.Krebs, J. E., C. J. Fry, M. L. Samuels, and C. L. Peterson. 2000. Global role for chromatin remodeling enzymes in mitotic gene expression. Cell 102587-598. [DOI] [PubMed] [Google Scholar]
- 29.Krogan, N. J., K. Baetz, M. C. Keogh, N. Datta, C. Sawa, T. C. Kwok, N. J. Thompson, M. G. Davey, J. Pootoolal, T. R. Hughes, A. Emili, S. Buratowski, P. Hieter, and J. F. Grrenblatt. 2004. Regulation of chromosome stability by the histone H2A variant Htz1, the Swr1 chromatin remodeling complex, and the histone acetyltransferase NuA4. Proc. Natl. Acad. Sci. USA 10113513-13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo, M. H., J. Zhou, P. Jambeck, M. E. Churchill, and C. D. Allis. 1998. Histone acetyltransferase activity of yeast Gcn5p is required for the activation of target genes in vivo. Genes Dev. 12627-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuo, M. H., E. vom Baur, K. Struhl, and C. D. Allis. 2000. Gcn4 activator targets Gcn5 histone acetyltransferase to specific promoters independently of transcription. Mol. Cell 61309-1320. [DOI] [PubMed] [Google Scholar]
- 32.Lin, W., G. Srajer, Y. A. Evrard, H. M. Phan, Y. Furuta, and S. Y. Dent. 2007. Developmental potential of Gcn5(−/−) embryonic stem cells in vivo and in vitro. Dev. Dyn. 2361547-1557. [DOI] [PubMed] [Google Scholar]
- 33.Longtine, M. S., A. McKenzie, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14953-961. [DOI] [PubMed] [Google Scholar]
- 34.Marcus, G. A., N. Silverman, S. L. Berger, J. Horiuchi, and L. Guarente. 1994. Functional similaritiy and physical association between GCN5 and ADA2: putative transcriptional adaptors. EMBO J. 134807-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcus, G. A., J. Horiuchi, N. Silverman, and L. Guarente. 1996. ADA5/SPT20 links the ADA and SPT genes, which are involved in yeast transcription. Mol. Cell. Biol. 163197-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McAinsh, A. D., J. D. Tytell, and P. K. Sorger. 2003. Structure, function, and regulation of budding yeast kinetochores. Annu. Rev. Cell Dev. Biol. 19519-539. [DOI] [PubMed] [Google Scholar]
- 37.Meluh, P. B., and D. Koshland. 1997. Budding yeast centromere composition and assembly as revealed by in vivo cross-linking. Genes Dev. 113401-3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meluh, P. B., P. Yang, L. Glowczewski, D. Koshland, and M. M. Smith. 1998. Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell 94607-613. [DOI] [PubMed] [Google Scholar]
- 39.Minoda, A., S. Saitoh, K. Takahashi, and T. Toda. 2005. BAF53/Arp4 homolog Alp5 in fission yeast is required for histone H4 acetylation, kinetochore-spindle attachment, and gene silencing at centromere. Mol. Biol. Cell 16316-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu, L., A. Burgess, D. P. Fairlie, H. Leonard, P. G. Parsons, and B. G. Gabrielli. 2000. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 62069-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roth, S. Y., J. M. Denu, and C. D. Allis. 2001. Histone acetyltransferases. Annu. Rev. Biochem. 7081-120. [DOI] [PubMed] [Google Scholar]
- 42.Santillan, D. A., C. M. Theisler, A. S. Ryan, R. Popovic, T. Stuart, M. M. Zhou, S. Alkan, and N. J. Zeleznik-Le. 2006. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia phenotype. Cancer Res. 661003-1009. [DOI] [PubMed] [Google Scholar]
- 43.Saunders, M., M. Fitzgerald-Hayes, and K. Bloom. 1988. Chromatin structure of altered yeast centromeres. Proc. Natl. Acad. Sci. USA 85175-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saunders, M. J., E. Yeh, M. Grunstein, and K. Bloom. 1990. Nucleosome depletion alters the chromatin structure of Saccharomyces cerevisiae centromeres. Mol. Cell. Biol. 105721-5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segrè, D., A. DeLuna, G. M. Church, and R. Kishony. 2005. Modular epistasis in yeast metabolism. Nat. Genet. 3777-83. [DOI] [PubMed] [Google Scholar]
- 46.Sharp, J. A., and P. D. Kaufman. 2003. Chromatin proteins are determinants of centromere function. Curr. Top. Microbiol. Immunol. 27423-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharp, J. A., A. A. Franco, M. A. Osley, and P. D. Kaufman. 2002. Chromatin assembly factor I and Hir proteins contribute to building functional kinetochores in S. cerevisiae. Genes Dev. 1685-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sterner, D. E., P. A. Grant, S. M. Roberts, L. J. Duggan, R. Belotserkovskaya, L. A. Pacella, F. Winston, J. L. Workman, and S. L. Berger. 1999. Functional organization of the yeast SAGA complex: distinct components involved in structural integrity, nucleosome acetylation, and TATA-binding protein interaction. Mol. Cell. Biol. 1986-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stoler, S., K. C. Keith, K. E. Curnick, and M. Fitzgerald-Hayes. 1995. A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev. 9573-586. [DOI] [PubMed] [Google Scholar]
- 50.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 40341-45. [DOI] [PubMed] [Google Scholar]
- 51.Struhl, K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12599-606. [DOI] [PubMed] [Google Scholar]
- 52.Sullivan, K. F. 2001. A solid foundation: functional specialization of centromeric chromatin. Curr. Opin. Genet. Dev. 11182-188. [DOI] [PubMed] [Google Scholar]
- 53.Tsuchiya, E., Hosotani, T., and T. Miyakawa. 1998. A mutation in NPS1/STH1, an essential gene encoding a component of a novel chromatin-remodeling complex RSC, alters the chromatin structure of Saccharomyces cerevisiae centromeres. Nucleic Acids Res. 263286-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venditti, S., and G. Camilloni. 1994. In vivo analysis of chromatin following nystatin mediated import of active enzymes into Saccharomyces cerevisiae. Mol. Gen. Genet. 24293-97. [DOI] [PubMed] [Google Scholar]
- 55.Vogelauer, M., J. Wu, N. Suka, and M. Grunstein. 2000. Global histone acetylation and deacetylation in yeast. Nature 408495-498. [DOI] [PubMed] [Google Scholar]
- 56.Wigge, P. A., O. N. Jensen, S. Holmes, S. Soues, M. Mann, and J. V. Kilmartin. 1998. Analysis of the Saccharomyces spindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J. Cell Biol. 141967-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu, J., and M. Grunstein. 2000. 25 years after the nucleosome model: chromatin modifications. Trends Biochem. Sci. 12619-623. [DOI] [PubMed] [Google Scholar]
- 58.Wu, P. Y., and F. Winston. 2002. Analysis of Spt7 function in the Saccharomyces cerevisiae SAGA coactivator complex. Mol. Cell. Biol. 155367-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu, W. S., G. Perez, L. Ngo, C. Y. Gui, and P. A. Marks. 2005. Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effects. Cancer Res. 657832-7839. [DOI] [PubMed] [Google Scholar]
- 60.Xu, W., D. G. Edmondson, Y. A. Evrard, M. Wakamiya, R. R. Behringer, and S. Y. Roth. 2000. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 26229-232. [DOI] [PubMed] [Google Scholar]
- 61.Xue, Y., J. C. Canman, C. S. Lee, Z. Nie, D. Yang, G. T. Moreno, M. K. Young, E. D. Salmon, and W. Wang. 2000. The human SWI/SNF-B chromatin-remodeling complex is related to yeast rsc and localizes at kinetochores of mitotic chromosomes. Proc. Natl. Acad. Sci. USA 9713015-13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang, X. J. 2004. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 32959-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuen, W. Y., B. Montpetit, and P. Hieter. 2005. The kinetochore and cancer: what's the connection? Curr. Opin. Cell Biol. 17576-582. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, K., W. Lin, J. A. Latham, G. M. Riefler, J. M. Schumacher, C. Chan, k. Tatchell, D. H. Hawke, R. Kobayashi, and S. Y. Dent. 2005. The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell 122723-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang, K., and S. Y. Dent. 2005. Histone modifying enzymes and cancer: going beyond histones. J. Cell Biochem. 961137-1148. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, W., J. R. Bone, D. G. Edmonson, B. A. Turner, and S. Y. Roth. 1998. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 173155-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang, W., B. G. Mellone, and G. H. Karpen. 2007. A specialized nucleosome has a “point” to make. Cell 1291047-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]