

Abstract
Mitochondrial complex II is a tumor suppressor comprised of four subunits (SdhA, SdhB, SdhC, and SdhD). Mutations in any of these should disrupt complex II enzymatic activity, yet defects in SdhA produce bioenergetic deficiency while defects in SdhB, SdhC, or SdhD induce tumor formation. The mechanisms underlying these differences are not known. We show that the inhibition of distal subunits of complex II, either pharmacologically or via RNA interference of SdhB, increases normoxic reactive oxygen species (ROS) production, increases hypoxia-inducible factor alpha (HIF-α) stabilization in an ROS-dependent manner, and increases growth rates in vitro and in vivo without affecting hypoxia-mediated activation of HIF-α. Proximal pharmacologic inhibition or RNA interference of complex II at SdhA, however, does not increase normoxic ROS production or HIF-α stabilization and results in decreased growth rates in vitro and in vivo. Furthermore, the enhanced growth rates resulting from SdhB suppression are inhibited by the suppression of HIF-1α and/or HIF-2α, indicating that the mechanism of SdhB-induced tumor formation relies upon ROS production and subsequent HIF-α activation. Therefore, differences in ROS production, HIF proliferation, and cell proliferation contribute to the differences in tumor phenotype in cells lacking SdhB as opposed to those lacking SdhA.
Mitochondrial complex II (succinate-ubiquinone oxidoreductase [Sdh]) is comprised of four subunits that are encoded in the nuclear genome (SdhA, SdhB, SdhC, and SdhD). Genetic defects in complex II are associated with a diverse collection of disorders. Mutations in SdhA are rare and have been linked to severe metabolic disorders resulting from decreased activity of the Krebs cycle, impaired oxidative phosphorylation, and bioenergetic deficiency (38). These autosome-recessive conditions manifest as childhood encephalopathy, myopathy, adult optic atrophy, and Leigh syndrome. However, mutations in subunit SdhB, SdhC, or SdhD are linked to tumorigenesis in the form of autosome-dominant familial paragangliomas and pheochromocytomas (1, 3, 16, 19, 33). Paragangliomas are benign, highly vascular tumors located within sympathetic paraganglia in the head and neck, and they are derived from neural crest cells (38). These include tumors of the carotid body (36), which normally is responsible for sensing oxygen levels in arterial blood. Furthermore, the numbers of carotid body paragangliomas are increased in populations living at higher altitudes (2), which suggests that the pathways activated by Sdh mutations overlap with those activated by hypoxia. These observations suggest that defects in SdhB, SdhC, or SdhD, but not SdhA, lead to an abnormal activation of hypoxia-inducible factor 1α (HIF-1α), which regulates the expression of vascular mitogens, including vascular endothelial growth factor (VEGF) (19, 46).
We previously reported that mitochondrial reactive oxygen species (ROS) signals arising from complex III trigger HIF-1α stabilization during hypoxia (10, 21, 31). It therefore is possible that increases in ROS production at complex II caused by defects in SdhB, SdhC, or SdhD could activate HIF by mimicking the hypoxia signaling pathway, thereby promoting cell proliferation, angiogenesis, and the tumor phenotype observed clinically.
Complex II links succinate dehydrogenase activity in the Krebs cycle to the mitochondrial electron transport chain. Complex II oxidizes succinate to fumarate and transfers the electrons to ubiquinone through a sequence of steps involving a flavin moiety in SdhA, a set of iron-sulfur clusters in SdhB, a heme group in SdhC, and the ubiquinone binding site in SdhC and SdhD (Fig. 1) (1, 9). When complex II is disrupted at SdhB, SdhC, or SdhD, the succinate dehydrogenase activity within SdhA can remain intact even though overall complex II function is disabled (51). In that situation, the oxidation of succinate and electron transfer to the flavin group in SdhA may be normal, while subsequent transfer to the iron-sulfur clusters and to ubiquinone is impaired (38). An impairment in the normal removal of electrons from the flavin group in SdhA could promote superoxide generation through the autoxidation of the reduced flavin group by O2 in the matrix (34, 38, 46, 51). Consistent with this model, Ishii et al. detected increases in superoxide production, along with an increase in tumorigenesis, in cells with a mutated SdhC gene (26). In contrast, we predict that defects that impair the ability of SdhA to oxidize succinate could inhibit overall complex II function without increasing ROS production, because the flavin group would remain oxidized. Therefore, the differences in tumor phenotype observed clinically with defects in SdhA, as opposed to those in SdhB, SdhC, or SdhD, could be explained by the differential effects of these defects on ROS production from SdhA and the consequent effects on HIF-α stabilization. However, no analysis comparing ROS production, HIF-α stabilization, and tumor growth in SdhA, SdhB, SdhC, or SdhD defects has been reported.
FIG. 1.

Mitochondrial complex II. The relationships among the four subunits are shown, along with the sites of activation or inhibition.
HIF-1α stability is regulated by its hydroxylation by a 2-oxoglutarate-dependent dioxygenase, the HIF prolyl hydroxylase (PHD). Selak et al. reported that complex II deficiency achieved by the suppression of SdhD induces normoxic HIF-1α stabilization via the accumulation of succinate without inducing a detectable increase in ROS production (43, 44). According to that model, the inhibition of complex II should promote HIF-1α stability through the succinate-mediated inhibition of PHD regardless of which subunit is interrupted. A similar model of substrate-mediated PHD inhibition has been described (23, 37, 42) in which the loss of fumarate hydratase activity leads to the accumulation of fumarate, which also can inhibit PHD competitively. A study by Lee et al. reported that prolyl inhibition by succinate, in the setting of Sdh mutations, contributes to pheochromocytoma pathogenesis through an inhibition of apoptosis (29). While intriguing, those reports still do not address the observed differences in tumor phenotype associated with defects in SdhB, SdhC, and SdhD and those associated with defects in SdhA.
All functional mutations in complex II should cause cellular succinate levels to increase, regardless of which subunit is responsible for the decrease in activity. However, mutations in SdhB, SdhC, and SdhD in complex II have been more strongly associated with increased HIF-1α stabilization and tumorigenesis than have defects in SdhA. We therefore set out to address this question by using in vitro and in vivo models employing genetic and pharmacological tools to modify SdhA and SdhB activity in multiple tumor cell lines. We show that the inhibition of distal subunits of complex II, either pharmacologically or via RNA interference of SdhB, increases normoxic ROS production, increases HIF-α stabilization in an ROS-dependent manner, and increases growth rates in vitro and in vivo without affecting the hypoxia-mediated activation of HIF-α. Proximal pharmacologic inhibition or RNA interference of complex II at SdhA, however, does not increase normoxic ROS production or HIF-α stabilization but rather results in decreased growth rates in vitro and in vivo. Furthermore, the enhanced growth rates resulting from SdhB suppression are inhibited by the suppression of HIF-1α and/or HIF-2α, indicating that the mechanism of SdhB-induced tumor formation relies upon ROS production and subsequent HIF-α activation.
MATERIALS AND METHODS
Cell culture and reagents.
Hep3B human hepatoma cells, A549 human alveolar epithelium-derived tumor cells, and 143B human osteosarcoma cells were obtained from the ATCC and maintained in a humidified incubator at 37°C and 5% CO2. Medium was formulated according to manufacturer's recommendations. Cells with stable suppression of SdhB or SdhA were supplemented with uridine and pyruvate. Thenoyl-trifluoroacetone (TTFA), 3-nitropropionic acid (3-NPA), malonate (MA), methyl-MA (MMA), ebselen, 4,4′-diisothiocyano-2,2′-disulfonic acid (DIDS), and dimethyl succinate (DMS) were purchased from Sigma Chemicals. The selection of cells with stable transfection was achieved using G418 at 0.6 mg/ml (Hep3B cells), 0.7 mg/ml (143B cells), or 1.0 mg/ml (A549 cells), while puromycin was used at 1 μg/ml for simultaneous selection in the HIF-1α and HIF-2α suppression studies.
RNA interference.
RNA interference constructs for complex II subunit B (SdhB) and subunit A (SdhA) in human 143B, A549, and Hep3B cells were generated by using six primers generated from mRNA sequences identified using the RNAi Oligoretriever application at http://katahdin.cshl.org:9331/RNAi/html/rnai.html. We employed a previously described PCR-based strategy (35) to design six different small hairpin RNA (shRNA) PCR primers. This primer then was used with an SP6 primer to PCR amplify the shRNA hairpin directly downstream of the U6 promoter, using pGEM-U6 as a template. After TOPO cloning and sequencing the PCR product, we cloned the U6-shRNA cassette into pPNT using EcoRI (pPNT-shRNA). pPNT confers resistance to G418.
Cells were electroporated with a pPNT or pPNT-shRNA vector containing a single shRNA sequence, and after 24 h cells were selected with G418 (0.7 mg/ml for 143B cells, 1.0 mg/ml for A549 cells, and 0.6 mg/ml for Hep3B cells). Stable G418-resistant clones were maintained in medium containing G418, uridine, and pyruvate until clonal colonies emerged, at which time clones were collected. Clones generated from each of the six shRNA vectors or the empty (wild-type) pPNT vector were screened via immunoblotting for the expression of SdhB or SdhA, and clones demonstrating a significant reduction of SdhB or SdhA expression were further analyzed. The wild-type control cells shown in the figures were stably transfected with pPNT and are resistant to G418.
The stable suppression of HIF-1α and/or HIF-2α was carried out by infecting SdhB shRNA 143B cells with retroviruses encoding HIF-1α shRNA and/or HIF-2α shRNA sequences driven by a U6 promoter. Selection was carried out using puromycin, and resistant clones were selected, screened, and used for experiments.
Western blotting.
Whole-cell lysates were generated by washing cells once or twice with phosphate-buffered saline (PBS) and then lysing attached cells with a whole-cell extract buffer (50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 5 mM EDTA, 4 mg/ml β-glycerophosphate) containing freshly added protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, 10 mM sodium fluoride, and 250 μM sodium orthovanadate. Cell extracts were vigorously vortexed and incubated on ice for 20 min. Extracts then were centrifuged at 14,000 rpm (16,000 × g) for 20 min, and the supernatant was stored at −70°C. Protein extracts were run at 100 V on a sodium dodecyl sulfate-polyacrylamide gel and were transferred to a nitrocellulose membrane at 100 V for 1 h. Transfers were verified with Ponceau S staining, and membranes were blocked with a solution of Tris-buffered saline (TBS), 0.1% Tween-20 (TBS-T) containing 5% nonfat milk. Primary antibodies against human HIF-1α (BD Biosciences), SdhB, SdhA (Molecular Probes), and β-actin (Abcam) were added to a solution of TBS-T, 5% milk, 0.01% azide and were incubated with the membrane overnight at 4°C. Membranes were washed with TBS-T, and secondary antibodies conjugated with horseradish peroxidase were added to the membrane with TBS-T and 5% milk for 2 h at room temperature. Membranes were washed with TBS-T, stained with ECL reagent (Amersham), and exposed to film. Exposed blots were scanned, and densitometric analysis was carried out using Image J (NIH).
Complex II assay.
For the complex II assay, cells (2 × 106) were collected by trypsinization and washed in PBS. The cells then were resuspended in a KH2PO4 solution (25 mM) with 0.1% Triton X-100. Succinate (20 mM) was added to the cell suspension. After 15 min at room temperature, azide (10 mM), antimycin A (AA; 1 μg/ml), rotenone (1 μg/ml), and decylubiquinone (50 μM) were added. Sodium 2,6-dichloroindophenolate DCIP (50 μM) then was added, and the absorbance at 600 nm was monitored for 2 to 3 min. TTFA (50 μM) then was added to determine the nonspecific reduction rates of DCIP. The TTFA-sensitive rate of complex II-mediated DCIP reduction then was calculated.
HRE-luciferase reporter gene assay.
For the hypoxia response element (HRE)-luciferase reporter gene assay, cells were transfected for 3.5 h with a plasmid encoding the HRE-luciferase reporter gene as well as a plasmid carrying a cytomegalovirus (CMV)-β-galactosidase gene using Lipofectamine 2000. After the transfection reagent was removed, cells were allowed to recover overnight before starting the indicated treatments. Cells were lysed with a reporter lysis buffer (Promega) and cleared of debris by centrifugation at 14,000 rpm for 15 min at 4°C, and luciferase and β-galactosidase activities were measured (Promega).
ROS measurements.
For measurements of ROS using the ratiometric redox-sensitive probe roGFP, cells were plated on glass coverslips and infected with an adenovirus expressing roGFP for 24 h before the start of the experiment. Cells were imaged using an epifluorescence microscope while continuously being superfused with balanced salt solutions at 37°C, and they were excited at a wavelength of 400 nm followed by one at 485 nm, while emission was measured at 530 nm. The roGFP ratio was determined by dividing the 485-nm excitation/530-nm emission intensity by the 400-nm excitation/530-nm emission intensity. As the roGFP thiols are chemically reduced, the roGFP ratio increases, whereas the roGFP ratio decreases upon thiol oxidation (15). The percentage of oxidation/reduction in the cells was determined by first measuring the roGFP ratio, after which the probe was calibrated by perfusion with dithiothreitol (1 mM) to maximally reduce the protein sensor, followed by the addition of t-butyl hydroperoxide (TBP; 1 mM) to maximally oxidize the protein sensor. Values are expressed as percentages of reduction or oxidation of the probe, with 100% being fully reduced and 1% being fully oxidized.
For dihydroethidium (DHE) and Mito-SOX experiments, cells were treated with the appropriate conditions for 5 to 6 h before the addition of DHE (10 μM) or Mito-SOX (1 μM) to the culture medium. Cells were incubated for an additional 1.5 h before being trypsinized and resuspended. Fluorescence was measured via flow cytometry, and a minimum of 2,000 cells were counted for each sample.
In vitro growth assay.
For the in vitro growth assay, cells (2 × 104) were plated in individual wells of a 6-well plate. Cells were trypsinized, collected, and counted in duplicate every 24 h from a single well. Trypan blue was used to stain nonviable cells, and only unstained cells were counted.
Nude mouse xenograft tumor experiments.
For the nude mouse xenograft tumor experiments, cells (1 × 106) were resuspended in 100 μl PBS and injected subcutaneously into the flank of 6-week-old male athymic nude mice (Foxn; Harlan) anesthetized with isoflurane. Mice were monitored for 25 to 35 days, during which time tumors were measured in two dimensions. Tumors then were excised, measured in three dimensions, weighed, and homogenized for Western blot analysis. All mouse procedures were carried out after institutional review and approval by the Animal Care and Use Committee at Northwestern University.
RESULTS
Inhibitors of SdhD, but not SdhA, induce ROS production and normoxic HIF-1α stabilization.
Pharmacological agents that inhibit SdhB, SdhC, or SdhD should mimic genetic defects in these subunits in terms of increasing ROS and HIF-1α stabilization, whereas agents that interfere with SdhA should fail to produce that effect. TTFA inhibits complex II at the ubiquinol binding site in SdhD (Fig. 1). When applied to Hep3B cells, TTFA elicits a dose-dependent increase in HIF-1α stabilization during normoxia without interfering with the response to hypoxia (Fig. 2A). TTFA also elicits an increase in normoxic HIF-dependent transcription of the HRE-luciferase reporter gene (Fig. 2D). In contrast, 3-NPA, MA, and MMA all inhibit complex II at the succinate binding site on SdhA (41). Both 3-NPA and MMA have no significant effect on HIF-1α stabilization during normoxia or hypoxia in Hep3B cells (Fig. 2B and C) or on normoxic HRE-luciferase expression (Fig. 2D). Unlike TTFA, both 3-NPA and MMA tend to inhibit the baseline normoxic stabilization of HIF-1α (Fig. 2B and C) at the highest doses tested. Furthermore, 3-NPA and MMA inhibit the normoxic HIF-1α stabilization induced by TTFA (Fig. 2E), suggesting that the effect of TTFA is specific to its action on complex II and that electrons must enter the SdhA subunit for TTFA to have an effect on HIF-1α stabilization.
FIG. 2.

HIF-1α stabilization and ROS production during pharmacological inhibition of complex II. Wild-type Hep3B cells were treated with TTFA (200 μM) (A), 3-NPA (100 μM) (B), MMA (1 mM) (C), or combinations of these drugs (E) for 4 h under normoxic or hypoxic conditions. HIF-1α and β-actin were detected via immunoblotting. (D) Wild-type 143B cells were transfected with plasmids containing HRE-luciferase (HRE-Luc) and CMV-β-galactosidase and then treated with TTFA (200 μM), 3-NPA (100 μM), or MMA (1 mM) for 4 h. Lysates were analyzed for luciferase activity and corrected for transfection efficiency using β-galactosidase. *, P < 0.05. (F) 143B cells were infected with an adenovirus encoding roGFP, allowed to recover for 24 h, and imaged every 60 s during perfusion with buffer containing the indicated compounds for 30 min. The ratio of baseline oxidation of roGFP is indicated as 100%; values greater than 100% represent oxidation, whereas values less than 100% represent the reduction of the level of the sensor relative to baseline redox levels. (G) Superoxide production as assessed by DHE fluorescence in wild-type 143B cells treated with AA (1 μM), TTFA (200 μM), 3-NPA (100 μM), or MMA (1 mM). After treatment for 5 h, DHE (1 μM) was added for an additional hour. Cells were trypsinized, collected, and analyzed by flow cytometry. Values are shown as percentages relative to values for controls. *, P < 0.05. (H) Cells were treated with the indicated compounds for 5 h, after which Mito-SOX (1 μM) was added for an additional hour. Cells were trypsinized, collected, and analyzed by flow cytometry. Cont, control. Values are shown as percentages relative to values for controls. *, P < 0.05.
Mitochondrial ROS production has been implicated in the stabilization of HIF-α during hypoxia (21, 31), and the flavin group in SdhA is capable of mediating the production of superoxide (51). We therefore tested whether TTFA stabilizes HIF-1α by triggering ROS generation. Using a novel, engineered, ratiometric fluorescent protein redox sensor (roGFP) (15, 22) expressed in cells by using a recombinant adenovirus, we find that the acute administration of TTFA significantly increases the rate of protein thiol oxidation compared to that induced by 3-NPA, MMA (Fig. 2F), or MA (data not shown). TTFA-induced ROS production also was detected by using the cytosolic oxidant-sensitive probe DHE (Fig. 2G) and the mitochondrial ROS sensor Mito-SOX red, a derivative of DHE targeted to the mitochondrial matrix (Fig. 2H). Oxidation of Mito-SOX under all conditions is inhibited by the superoxide dismutase (SOD)-mimetic Mn(III)tetrakis(4-benzoic acid) porphyrin chloride (TBAP), confirming that the signal detected by this probe is superoxide. Interestingly, 3-NPA and MMA increase Mito-SOX oxidation but not DHE oxidation, suggesting that these compounds generate a small ROS signal within the mitochondrial matrix that does not escape into the cytosol, whereas the larger oxidant signal elicited by TTFA is sufficient to escape to the cytosol. Furthermore, the stabilization of HIF-1α by TTFA is inhibited by the administration of the mitochondrial antioxidant Mito-Q (40; also data not shown), suggesting that mitochondrial ROS production is required for TTFA-induced HIF-1α stabilization. No increase in cell death was detected in 143B or Hep3B cells after 6 h of incubation with TTFA, 3-NPA, or MMA, indicating that these drugs were not toxic at the concentrations used in this study (data not shown).
Genetic suppression of SdhB expression leads to normoxic HIF-1α stabilization through ROS signaling.
To inhibit complex II activity, SdhB expression was stably suppressed using RNA interference in 143B osteosarcoma, A549, and Hep3B cell lines (SdhB shRNA). Clones generated from shRNA constructs demonstrate significant suppression of SdhB activity; the same clones exhibit increases in normoxic HIF-1α stabilization (Fig. 3A and B) and HIF-2α stabilization (data not shown). Stable suppression of SdhB also causes increased normoxic expression of VEGF mRNA, as detected by quantitative real-time PCR (data not shown). In contrast, cells stably transfected with a mock shRNA vector show no decrease in SdhB and no increase in normoxic HIF-1α protein (data not shown) or HRE-luciferase reporter gene expression (Fig. 3E). Hypoxia-induced stabilization of HIF-1α is normal in all of these cells (data not shown).
FIG. 3.

Complex II activity, oxidant stress, normoxic HIF-1α stabilization, and HRE-luciferase reporter gene activity in A549, Hep3B, and 143B cells during the suppression of SdhB expression. (A) Complex II activity of 143B clone 4F or 5E, A549 clone 4-2, and Hep3B polyclonal line 2, compared to that of the respective wild-type (WT) cells, given as percentages. *, P < 0.05. (B) Normoxic levels of HIF-1α, SdhB, β-actin, Rieske iron-sulfur protein (RISP), and cytochrome oxidase subunit IV (Cox IV) were measured via immunoblotting in 143B WT cells and SdhB shRNA clones 4F and 5E. (C) Normoxic levels of HIF-1α measured by immunoblotting in A549 WT cells and SdhB shRNA clone 4-2 in the presence of the antioxidant ebselen (25 μM), 3-NPA (100 μM), or MMA (10 mM). Also shown are normoxic levels of HIF-1α measured in WT Hep3B cells and SdhB shRNA polyclonal cell line 2, in the presence of DMS (20 mM) or ebselen (25 μM), via immunoblotting. (D) Normoxic levels of HIF-1α measured by immunoblotting in WT 143B and A549 cells and SdhB shRNA clones in the presence (+) or absence of (−) 3-NPA (100 μM) or Mito-Q (1 μM). Immunoblots were quantified via densitometry and are presented as the increase (n-fold) above levels for WT normoxia controls. *, P < 0.05. (E) Normoxic HRE-luciferase expression analyzed in 143B WT, 143B mock shRNA, 143B SdhB shRNA clone 5E, A549 WT, and A549 SdhB clone 4-2 cells. Data are presented as the increase (n-fold) above the level for the cell-type-specific WT control. *, P < 0.05.
To determine whether the response to SdhB suppression is unique to 143B cells, we generated additional clones with stable RNA interference of SdhB in A549 and Hep3B cell lines. We also tested whether normoxic HIF-1α stabilization is dependent on ROS generated by the SdhA subunit. Using the antioxidant ebselen and SdhA inhibitors 3-NPA and MMA, we find that the increased normoxic HIF-1α stabilization in A549 SdhB shRNA clones, especially in clone 4-2, is dependent upon ROS and on electron transfer from succinate into SdhA (Fig. 3C). Significant decreases in complex II activity are evident in A549 shRNA clones and in mass culture lines of Hep3B cells stably transfected with SdhB shRNA construct 2 (Fig. 3A). The Hep3B SdhB shRNA polyclonal line also exhibits increased normoxic HIF-1α stabilization that is inhibited by ebselen (Fig. 3C).
Densitometric analysis of multiple experiments using 143B SdhB shRNA cells and A549 SdhB shRNA cells shows that the addition of the mitochondrion-specific antioxidant Mito-Q results in a significant attenuation of normoxic HIF-1α stabilization (Fig. 3D). Similar to the case of HIF stabilization induced by TTFA, the elevated levels of normoxic HIF-1α also are significantly attenuated in these same SdhB shRNA lines by 3-NPA, again suggesting that electrons need to enter complex II in order to induce HIF-1α stabilization during SdhB suppression (Fig. 3D). Suppression of SdhB expression in 143B and A549 cells also results in increased normoxic HRE-luciferase reporter gene expression (Fig. 3E), which is similarly inhibited with antioxidants and 3-NPA (data not shown). These data are consistent with the conclusion that the increased HIF-1α stabilization during SdhB suppression is the result of increased ROS generation from SdhA.
If ROS production from SdhA increases when SdhB, SdhC, or SdhD is disrupted, then increasing the amount of succinate available for electron transfer into SdhA might increase ROS production and HIF-1α stabilization in cells with decreased SdhB levels. In contrast, normal cells should cope with increased succinate levels by augmenting the overall rate of electron flux through complex II to ubiquinone. The addition of the cell-permeable succinate analog DMS produces a slight increase in normoxic HIF-1α stabilization in wild-type cells and a larger increase in HIF-1α stabilization in Hep3B SdhB construct 2 cells (Fig. 3C). This response is abolished by the antioxidant Mito-Q (see Fig. 5E), indicating that increased electron flux through complex II, under conditions in which SdhB activity is limited, causes HIF activation in a ROS-dependent manner. Therefore, in cells with SdhB inhibition, normoxic HIF-1α stabilization may be a consequence of increased intracellular succinate and ROS, both of which act in combination to inhibit HIF PHD.
FIG. 5.

Effects of shRNA suppression of SdhA on ROS production and HIF-α stabilization. (A) 143B cells stably transfected with SdhA shRNA demonstrated a significant decrease in SdhA expression compared to that of wild-type (WT) or SdhB shRNA cells. Levels of SdhA and HIF-1α under normoxic conditions were detected by immunoblotting and were quantified using densitometric analysis (Image J). Cells were either left untreated or cotransfected with HRE-luciferase and CMV-β-galactosidase reporter plasmids in order to assess HRE-luciferase expression, as described in Materials and Methods. *, P < 0.05 compared to levels for WT 143B cells; #, P < 0.05 compared to levels for SdhB shRNA clone 5E. (B) Complex II activity of a polyclonal SdhA shRNA line (SdhA polyclonal line 3) and a monoclonal SdhA shRNA line (SdhA polyclonal line 3C). Complex II activity was measured as described in Materials and Methods, and activity is expressed as the percentage of the level for WT 143B cells. *, P < 0.05. (C) Mito-SOX fluorescence in normoxic cells, determined as previously described. *, P < 0.05. (D) Percent reduction of the roGFP cytosolic redox sensor in mock shRNA, SdhA polyclonal line 3, and SdhA clone 3C cells under normoxia. No significant differences were detected. (E) 143B WT and SdhA shRNA cells were exposed to DMS (20 mM) or DMS plus Mito-Q (1 μM) under normoxic conditions. Normoxic HIF-1α levels were measured via immunoblotting and were quantified using densitometric analysis (Image J). Data are presented as percentages of normoxic HIF-1α expression for each cell type. *, P < 0.05.
Effect of SdhB shRNA on ROS production.
We tested whether the suppression of SdhB expression elicits an increase in ROS production by assessing cytosolic protein thiol oxidation using adenovirus-mediated expression of the roGFP sensor (15). In both polyclonal and monoclonal Hep3B shRNA cell lines exhibiting a significant decrease in complex II activity, an increase in basal oxidation of the roGFP probe is evident under normoxic conditions (Fig. 4A). Under baseline conditions, the amount of roGFP probe primarily is reduced, and therefore the oxidation of the probe can be expressed as a decrease in the amount of reduced probe relative to the amount of probe in wild-type cells. The calibration of the sensor was achieved in each study by comparing baseline fluorescence ratios to those obtained after the maximal reduction of the probe (dithiothreitol; 1 mM) followed by maximal oxidation (TBP; 1 mM). This permits the calculation of the percent oxidation/reduction of the protein under baseline conditions. The administration of exogenous TBP (20 μM) or TTFA produces the partial oxidation (decreased reduction) of roGFP (Fig. 4A). In 143B cells, mock shRNA does not change basal levels of roGFP oxidation, whereas SdhB shRNA significantly increased basal oxidation of the roGFP sensor (Fig. 4B). These results demonstrate that SdhB suppression specifically increases basal cytosolic oxidant stress. The calibration of the roGFP response to oxidants shows that the oxidation seen in SdhB shRNA cells is equivalent to low micromolar concentrations of cytosolic oxidant signals (data not shown).
FIG. 4.

Increased ROS production in cells with shRNA suppression of SdhB. (A) Cells were infected with an adenovirus expressing the cytosolic oxidant-sensitive roGFP sensor, and the baseline oxidation of roGFP was measured and expressed as percent reduction. Oxidation of roGFP therefore causes a decrease in the reduction of the probe, as indicated by arrows to the right. The percent reduction of the roGFP probe relative to the level of probe of wild-type (WT) controls is presented in conjunction with complex II activity levels in Hep3B SdhB shRNA clones. All cell lines are monoclonal, except for Hep3B SdhB lines 2 and 4, which are polyclonal cell lines. TBP (20 μM) and TTFA (200 μM) are shown as controls. *, P < 0.05. (B) Oxidant stress assessed by the cytosolic oxidant-sensitive roGFP sensor in 143B wild-type cells, 143B cells with stable expression of mock shRNA, or 143B SdhB shRNA clone 5E cells, presented in conjunction with complex II activity levels. *, P < 0.05. (C) Normoxic ROS production in the mitochondrial matrix detected using the probe Mito-SOX red in WT 143B cells, 143B cells with stable expression of mock shRNA, or 143B SdhB shRNA clone 5E cells. Fluorescence intensity was measured by flow cytometry. Incubation with the SOD-mimetic TBAP was performed to assess the specificity of Mito-SOX for superoxide. Data are presented as percentages of WT control levels. *, P < 0.05. (D) Normoxic ROS production in the mitochondrial matrix detected using Mito-SOX red in the presence or absence of DMS (20 mM) and measured by flow cytometry in 143B cell lines. *, P < 0.05 compared to WT 143B cells and P < 0.05 for SdhB mutant cells with DMS compared to 143B WT cells with DMS.
Mito-SOX also was used to detect changes in matrix oxidant stress under normoxic conditions. In 143B cells, SdhB shRNA results in an increase in baseline Mito-SOX oxidation, which is abolished and restored to wild-type baseline levels by the SOD-mimetic TBAP (Fig. 4C). Cells stably transfected with a mock shRNA vector show no increase in Mito-SOX oxidation. As stated earlier, the addition of DMS to SdhB shRNA cells increases the level of normoxic HIF-1α stabilization compared to that of wild-type controls (Fig. 3C). We therefore tested whether exogenous succinate also increases ROS production. The addition of DMS results in a significant increase in Mito-SOX oxidation in all cells tested, but SdhB shRNA cells demonstrate a significantly greater oxidation of Mito-SOX than do wild-type controls when given DMS (Fig. 4D). These results strengthen the conclusion that the generation of superoxide increases when the electron flux through complex II is increased and that even greater increases in ROS occur when SdhB also is suppressed. These increases in basal ROS production, likely in combination with increases in succinate, contribute to the normoxic stabilization of HIF.
Suppression of SdhA expression does not increase ROS generation, HIF-1α stabilization, or activity during normoxia.
In a previous study, the suppression of SdhD expression led to an increase in HIF-1α stabilization, a response that was attributed to the inhibition of PHD by succinate that accumulated as a consequence of the impairment in complex II activity (43). SdhA suppression therefore should yield the same HIF response as SdhB suppression if succinate is solely responsible for the phenotype (8). On the other hand, if increases in ROS signals from SdhA contribute to the inhibition of PHD when SdhB is suppressed, then the ROS and HIF-α stabilization responses should be different when SdhA is suppressed compared to that when SdhB is suppressed. We therefore tested whether genetic suppression of SdhA produces the same cellular phenotype as SdhB suppression. In 143B cells, we generated stable SdhA shRNA cell lines with significant suppression of SdhA that were either polyclonal (SdhA clone 3) or monoclonal (SdhA clones 3C and 3L). These clones demonstrate a significant decrease in SdhA as detected by Western blotting (Fig. 5A) or by complex II activity (Fig. 5B). We find no significant increase in HIF-1α stabilization via Western blotting during normoxia in any of these lines (Fig. 5A). Although an increase in normoxic reporter gene expression using HRE-luciferase is evident in the monoclonal lines but not in the polyclonal line, this increase is small compared to that seen in SdhB shRNA cells (Fig. 5A). No significant increase in normoxic Mito-SOX oxidation was detected (Fig. 5C), with the exception of SdhA clone 3C, which again shows a small increase relative to the level of oxidation of SdhB shRNA cells. Using the roGFP sensor to assess the protein-thiol redox state in the cytosol, no significant change in oxidant stress was detected in 143B SdhA shRNA clones compared to levels for mock shRNA cells (Fig. 5D). Furthermore, SdhA shRNA cells do not demonstrate an increase in HIF-1α stabilization in response to DMS, indicating that an intact SdhA is necessary for DMS-mediated HIF-1α stabilization and that both ROS and succinate are required for PHD inhibition in response to exogenous succinate (Fig. 5E). Both SdhB and SdhA shRNA cell lines respond normally to hypoxia (data not shown), indicating that the disruption of complex II does not affect the oxygen-dependent regulation of HIF-1α and HIF-2α during hypoxia, which is mediated by ROS production from complex III (21, 31). Collectively, these data reveal that the suppression of SdhA produces a different cellular phenotype than does SdhB suppression, indicating that factors other than a loss of overall complex II function contribute to the phenotypes observed during the stable suppression of the individual subunits.
Suppression of SdhB causes increased cell growth in vitro.
HIF-dependent genes stimulate cell proliferation (45), and the suppression of HIF-1α has been shown to decrease cell growth rates in vitro and in vivo (14). To determine whether the HIF activation observed in normoxic SdhB shRNA cells stimulates proliferation, we compared the growth rates of cells in vitro after the stable suppression of SdhB to those after the stable suppression of SdhA. Stable transfection with a mock shRNA vector does not significantly affect growth in vitro (Fig. 6A). However, the stable suppression of SdhB significantly increases growth rates in 143B cells (Fig. 6A) and tends to increase growth in A549 cells (data not shown). In contrast, the suppression of SdhA leads to a decrease in cell growth rates in culture (Fig. 6B). These data suggest that the increases in ROS and normoxic HIF-1α stabilization in the SdhB shRNA cells contribute to an increased rate of proliferation in vitro, while the lack of ROS and HIF-1α stabilization in SdhA shRNA cells, in combination with decreased complex II activity, leads to decreased rates of growth in vitro.
FIG. 6.
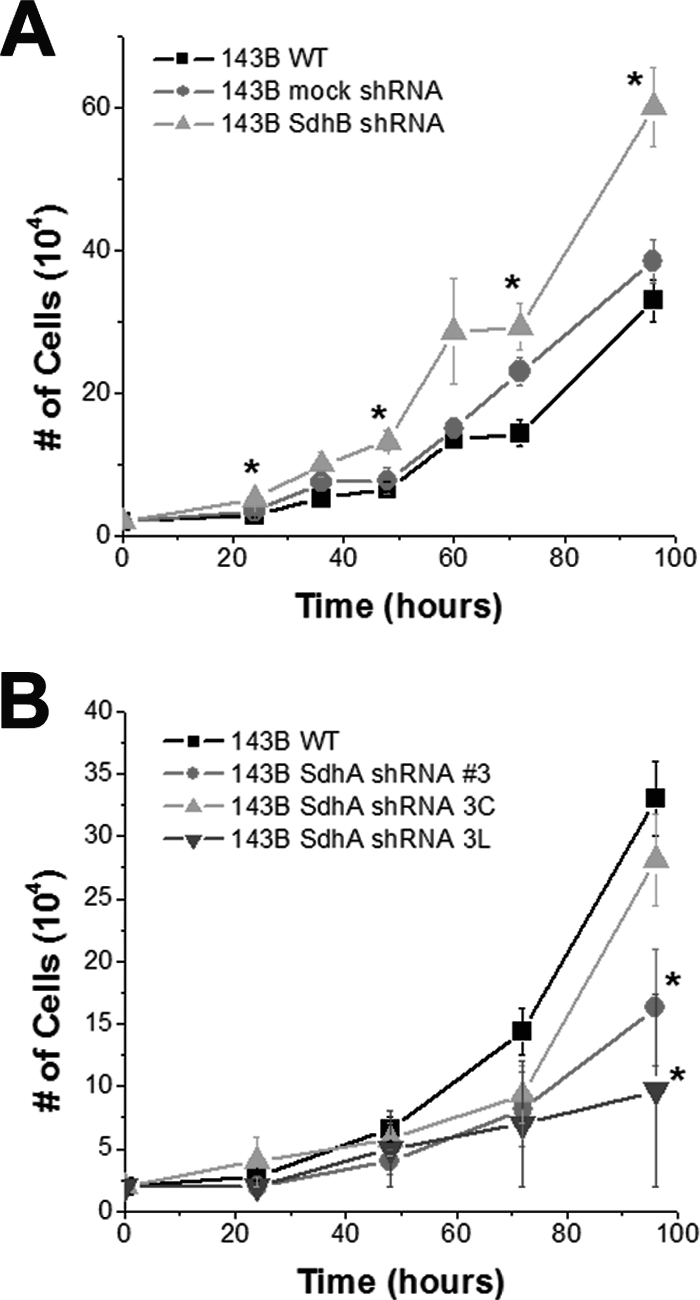
Increased rate of cell growth in vitro in SdhB shRNA, but not SdhA shRNA, cells. (A) 143B wild-type (WT), mock shRNA, and SdhB shRNA cells plated at 20,000 cells/well in 6-well plates were trypsinized, collected, and counted every 12 to 24 h. (B) 143B WT, SdhA shRNA polyclonal line 3, SdhA shRNA clone 3C, and SdhA shRNA clone 3L cells plated at 20,000 cells/well in 6-well plates were trypsinized, collected, and counted every 24 h. *, P < 0.05.
Suppression of SdhB causes increased tumor growth in a mouse xenograft model.
To determine whether the differences in growth rates of SdhB- and SdhA-suppressed cells in vitro is recapitulated in vivo, athymic nude mice were injected subcutaneously in the flank with identical numbers of cells, and tumor growth was monitored over time. Stable suppression of SdhB results in a marked increase in tumor dimensional growth rates and tumor mass (Fig. 7A to C), whereas mock shRNA cells do not exhibit a detectable change in growth rate or tissue mass compared to those of wild-type controls. By comparison, cells with stable suppression of SdhA generate tumors that grow more slowly (Fig. 7B and C) than wild-type controls or SdhB shRNA cells. Protein lysates from SdhB shRNA-derived tumors, but not SdhA shRNA-derived tumors, exhibit increased levels of HIF-1α, HIF-2α, and VEGF protein expression, as determined by immunoblotting (Fig. 7D). We also find that SdhB shRNA cells are not different from wild-type cells in their susceptibility to death induced by staurosporine (24 h) or anoxia (48 h) (data not shown), indicating that the increased cell and tumor growth rates are not likely to be the result of an enhanced resistance to apoptosis. These data indicate that the suppression of SdhB, but not SdhA, results in increased tumor growth, in association with an increase in HIF-α stabilization.
FIG. 7.

Increased rate of tumor growth and increased HIF-α stabilization in SdhB shRNA cell-derived tumors. (A and B) Athymic nude mice were injected with 1 × 106 cells (143B wild type [WT], 143B mock shRNA, 143B SdhB shRNA clone 5E, or 143B SdhA shRNA polyclonal line 3 and clone 3C). Tumor growth was monitored for 35 days. Growth rates for SdhA shRNA polyclonal line 3 and 3C cell lines were virtually identical and have been combined in this graph. *, P < 0.05. (C) Upon excision, tumors were washed in cold PBS, and their mass was measured. *, P < 0.05. (D) Portions of the tumors were homogenized and lysed. Protein levels were measured by immunoblotting. Densitometric analysis of blots was carried out (Image J), corrected for β-actin levels, and normalized to WT levels. *, P < 0.05. Avg., average.
Tumor growth of SdhB shRNA cells is HIF dependent.
To determine the roles of HIF-1α and HIF-2α in promoting the rate of cell growth in vitro and tumor growth in vivo, we generated stable suppression of HIF-1α, HIF-2α, or HIF-1α and HIF-2α (referred to as HIF-1α+HIF-2α hereafter) in the 143B SdhB shRNA cell line using retrovirally delivered HIF-1α and/or HIF-2α shRNA constructs. A control line of SdhB shRNA cells was stably transfected with a Drosophila melanogaster HIF shRNA construct (dHIF), which differs in sequence from that of the human HIF shRNA and therefore is ineffective in suppressing HIF-1α expression. Compared to hypoxic HIF-α stabilization responses of SdhB dHIF controls, cells with suppression of HIF-1α and/or HIF-2α show attenuation of hypoxic HIF-α stabilization responses in various clones (Fig. 8A). Suppression of HIF-1α, HIF-2α, and HIF-1α+HIF-2α results in a significant decrease of cell growth in vitro (Fig. 8B), significantly slower tumor growth in vivo (Fig. 8C), and significantly smaller tumor mass at harvest (Fig. 8D) compared to those of the SdhB dHIF control cells. These results demonstrate that both HIF-1α and HIF-2α are necessary for the increased rates of growth in vitro and in vivo in SdhB shRNA cells.
FIG. 8.

Effect of HIF-α suppression on cell growth and tumor formation. (A) 143B SdhB shRNA cells were stably infected with retroviruses expressing shRNA constructs targeting dHIF as controls or human HIF-1α and/or HIF-2α. HRE-luciferase expression and HIF-1α and HIF-2α protein levels were measured during hypoxia (1.5% O2). Values are normalized to those of the SdhB dHIF shRNA controls. *, P < 0.05. Ln 2, line 2. (B) Comparison of cell growth rates in cultures of SdhB shRNA cells with simultaneous dHIF, HIF-1α, and/or HIF-2α shRNA suppression. Cells plated at 20,000 cells/well in 6-well plates were trypsinized, collected, and counted every 24 h. *, P < 0.05. (C) Athymic nude mice were injected with 1 × 106 cells (143B SdhB shRNA plus dHIF, HIF-1α, and/or HIF-2α shRNA suppression). Tumor growth was monitored for 25 days. *, P < 0.05. Avg., average. (D) Tumor weight in 143B SdhB shRNA cells with simultaneous dHIF, HIF-1α, and/or HIF-2α shRNA suppression after 25 days of growth in vivo. *, P < 0.05.
DISCUSSION
Overview.
Five mechanisms have been proposed to explain the linkage between succinate dehydrogenase (Sdh) mutations and tumorigenesis. The first model suggests that Sdh inhibition arising from mutations in the B, C, or D subunit leads to an increase in ROS production, resulting in oxidative damage to DNA, genomic instability, and tumorigenesis. The second model proposes that Sdh mutations lead to an increase in mitochondrial ROS production, but these oxidants act as signal transduction messengers to trigger HIF-1α stabilization by inhibiting PHD, which regulates the stability of the protein through the hydroxylation of two proline residues. Activation of HIF then would drive tumorigenesis through the enhanced expression of genes involved in cell proliferation, tumor angiogenesis, cell migration, and cell survival (32). A third model suggests that HIF-1α is stabilized by an increase in succinate, which accumulates when Sdh activity declines. Succinate inhibits PHD competitively, so this mechanism would mimic the hypoxic response without the need for a decrease in [O2] or an increase in ROS. A fourth model proposes that Sdh inhibition leads to an increase in ROS generation, which mediates the nonenzymatic decarboxylation of α-ketoglutarate and oxaloacetate. Decarboxylation of oxaloacetate yields MA, which further inhibits Sdh, while succinate produced by the decarboxylation of α-ketoglutarate transits to the cytosol and inhibits PHD. A fifth model suggests that neuronal precursor cells with mutations in Sdh fail to undergo normal apoptosis in response to growth factor withdrawal during embryogenesis. This apoptosis requires PHD; succinate accumulation resulting from Sdh inhibition impairs its activity while contributing to later pheochromocytoma progression by amplifying HIF-1α stabilization. Investigators have examined these mechanisms in diverse model systems, and controversy has arisen with respect to (i) whether or not an ROS increase is involved in the response and (ii) why tumorigenesis is associated with mutations in the B, C, and D subunits but not the A subunit. In the present study, we used stable shRNA inhibition of SdhA or SdhB in multiple oncogenic cell lines to address these issues in cell culture and tumor xenograft models.
Generation of ROS in response to Sdh inhibition.
We found that the inhibition of SdhB caused an increase in oxidant stress during normoxia, which was detected in the cytosol by the redox-sensitive protein roGFP and in the mitochondrial matrix with the fluorescent probe Mito-Sox red. The ratiometric behavior of the roGFP sensor permits its calibration in live cells, which facilitates a comparison of the percent oxidation under basal conditions across various cell lines with stable genetic suppression of different Sdh subunits. The increase in oxidant stress in SdhB knockdown clones was small but statistically significant, while no evidence of increased cytosolic oxidant stress was detected in the SdhA knockdown cells. Similar increases in cytosolic oxidant stress also were detected in response to TTFA, which inhibits electron flux at SdhD (47). In both cases, these changes in oxidant stress were sufficient to trigger HIF-1α stabilization, indicating their biological significance. Other studies with diverse model systems also indicate that mutations in complex II can cause increases in ROS production (24, 25, 46, 51, 54).
Superoxide generated at complex II should be released into the mitochondrial matrix, based on the assembly of Sdh subunits in that compartment. Accordingly, SdhB knockdown cells demonstrated significant increases in Mito-SOX oxidation by superoxide. Oxidant stress in the matrix was amplified in all cell lines when DMS, a cell-permeable succinate analog, was administered to enhance enzymatic activity and electron flux in Sdh. However, the greatest increases in ROS production, compared to the ROS production of wild-type or mock knockdown cells, were seen when DMS was administered to SdhB knockdown cells. These findings demonstrate that SdhB suppression is associated with an increase in basal superoxide generation in the matrix compartment. Hydrogen peroxide (H2O2) arising from the dismutation of superoxide by Mn-SOD appears to escape to the cytosol in SdhB shRNA cells, where it can be detected by the roGFP sensor and where it inhibits PHD and triggers HIF-α stabilization.
Using DHE, Selak and coworkers were unable to detect oxidant stress in HEK293 cells that had been subjected to SdhD suppression by shRNA transfection (43, 44). Based on those results, they concluded that redox stress is not essential for the phenotype observed in cells with Sdh inhibition. However, although DHE is rapidly oxidized by superoxide, it is insensitive to H2O2. Because DHE remains in the cytosol and nuclear compartments, it would not detect superoxide within the mitochondrial matrix. Like Selak et al., we were unable to detect increased oxidant stress using DHE in our SdhB knockdown cells (data not shown). We therefore suspect that their inability to detect increased DHE oxidation during SdhD suppression was due to the lack of mitochondrial matrix superoxide escape to the cytosol. As positive controls they used AA (44) or the overexpression of p53 (43) to validate their ability to detect ROS with DHE. Those interventions would be expected to increase cytosolic superoxide levels, because AA generates superoxide at the outer surface of the inner mitochondrial membrane (50) while p53 induces the expression of a variety of nonmitochondrial oxidase systems (53). Indeed, by using these approaches, their DHE measurements successfully detected an increase in ROS production. We therefore suggest that the differences in ROS production between our study and theirs relates to the methods used to detect oxidant production during Sdh inhibition. However, we cannot rule out the possibility that HIF accumulation associated with SdhD deficiency proceeds by a different mechanism than that associated with SdhB suppression.
Sites of ROS production in complex II.
Controversy exists regarding the sites in complex II at which ROS generation occurs when electron flux is disrupted as a consequence of alterations in the B, C, or D subunit. Some evidence suggests that the flavin adenine dinucleotide site in SdhA is the principal source of superoxide generation, due to the exposure of that moiety to the aqueous environment and the high electron density that develops when the electron flux downstream from that site is obstructed (34, 51). However, other studies suggest that mutations in SdhB, SdhC, or SdhD influence superoxide generation at the ubiquinone binding site in SdhD through intermolecular interactions (20, 48). Our data are consistent with the former model, as increases in ROS were detected in response to the suppression of SdhB expression, implying that the backup of electrons at SdhA was responsible. However, our results certainly do not refute the possibility that mutations in SdhB, SdhC, or SdhD could influence ROS production at the D subunit through the latter mechanism. In either case, the increase in ROS production likely would occur within the matrix compartment.
SdhB inhibition triggers normoxic HIF-1α and HIF-2α stabilization through an ROS-dependent mechanism.
The stable suppression of SdhB, but not SdhA, caused HIF-1α stabilization under normoxia. Likewise, TTFA, which inhibits SdhD, produced an increase in roGFP oxidation and dose-dependent HIF-1α stabilization. In contrast, proximal inhibitors 3-NPA and MMA, which bind at SdhA, failed to augment cytosolic oxidant stress or to stabilize HIF-α. In fact, proximal inhibitors of Sdh suppressed the normoxic stabilization of HIF-1α by TTFA, as did the antioxidants ebselen and Mito-Q. The SdhA inhibitors probably abolish the TTFA response by limiting electron entry at the SdhA subunit and thereby decreasing ROS production, whereas Mito-Q acts by scavenging mitochondrial ROS. The effects of Sdh inhibition on HIF-1α protein stabilization were further confirmed by HRE-luciferase reporter assays. However, pharmacologic or genetic manipulation of SdhA failed to produce any increase in cytosolic ROS and failed to augment HIF-1α stabilization under normoxia. Collectively, these results support the conclusion that oxidant stress generated by the inhibition of SdhD or genetic alterations in SdhB is sufficient to mimic the response to hypoxia in terms of HIF-α stabilization and HIF-dependent transcription.
Role of increases in succinate.
HIF-1α stabilization in the setting of complex II dysfunction has been attributed to an inhibition of PHD by succinate (8, 13, 37, 43). Decreases in Sdh activity should cause an accumulation of succinate, which can transit to the cytosol through the dicarboxylate carrier. There, it potentially can inhibit PHD and cause HIF-α stabilization through a pseudohypoxia pathway that does not require ROS (43, 44). However, for a given level of Sdh inhibition, one would expect to find a similar increase in succinate regardless of which subunit is responsible for the inhibition in overall enzyme activity. However, SdhB, SdhC, and SdhD mutations are associated with a tumor phenotype, whereas SdhA mutations are not. Some authors have explained this paradox by suggesting that two isoforms of SdhA exist (8, 49) and that distinct genetic loci encode these two SdhA variants (49). However, genetic analysis reveals that the two variants arise from a single, highly polymorphic SdhA gene, resulting from the persistence of two distinct haplogroups in the human genome (4). Therefore, the different phenotypic responses associated with SdhA mutations and SdhB, SdhC, or SdhD mutations cannot be explained by the rescue of SdhA activity by a second isoform. In light of this information, the factors responsible for the phenotypic difference are not established.
Our results suggest that the additional activation induced by ROS signaling in cells with SdhB, SdhC, or SdhD mutations will amplify the pseudohypoxic response beyond that seen in SdhA mutants. We agree that increases in succinate represent an important contribution to the stabilization of HIF-α in cells with Sdh inhibition (43). However, mutations in SdhB, SdhC, or SdhD would be expected to cause both an ROS-mediated and a succinate-mediated effect, whereas SdhA mutations would act by augmenting succinate alone. Mutations in SdhA may be sufficient to induce nuclear translocation of HIF-1α (8), although the present study did not detect any increase in HIF-1α stabilization or tumor progression in the SdhA shRNA cells. We suggest that the additional activation mediated by ROS can explain the difference in phenotype that has been noted. In this regard, our findings are consistent with those of the model proposed by Szeto et al., who suggested, based on experiments with Saccharomyces cerevisiae, that mutations in Sdh promote tumor progression by contributing to both ROS production and succinate accumulation (48).
Studies by Lee et al. related pheochromocytomas to germ line mutations in Sdh by observing that the apoptosis of neuronal precursor cells, induced by the withdrawal of neural growth factor, requires c-Jun-dependent apoptosis mediated by the PHD EglN3 (29). Mutations in Sdh that inhibit enzyme function impair this apoptosis by causing succinate accumulation, which inhibits EglN3. While interesting, these findings do not address why phenotypic differences occur with SdhA mutations and SdhB, SdhC, or SdhD mutations, as a mutation in any subunit that affects succinate levels (and, thus, EglN3 activity) also would affect Krebs cycle function and bioenergetics, yet SdhA defects are not tumorigenic. We assessed the ability of SdhA and SdhB knockdown cells to undergo apoptosis in response to serum withdrawal or staurosporine, but we were unable to detect any differences. We therefore conclude that the augmented tumor growth in our SdhB shRNA cells was not due to a decrease in apoptotic potential.
ROS-induced increases in succinate accumulation.
Another possibility is that increases in ROS production resulting from genetic mutations in SdhB, SdhC, or SdhD could contribute to the accumulation of succinate through a mechanism involving H2O2-mediated nonenzymatic decarboxylation of α-ketoglutarate and oxaloacetate. As described by Fedotcheva et al., mitochondrial treatment with H2O2 led to an increase in α-ketoglutarate oxidation to succinate and oxaloacetate decarboxylation to MA (17). The former reaction would promote the formation of succinate, while MA released by the latter reaction would inhibit succinate oxidation by Sdh. The net effect would be to augment succinate accumulation, which then could exit to the cytosol and inhibit PHD. While our data are not inconsistent with this model, our results suggest that MA inhibition at SdhA tends to inhibit ROS production at complex II by preventing succinate dehydrogenation and electron entry into the system. Also, the concentration of exogenous H2O2 needed to produce this effect was relatively high (100 to 500 μM), and it is not clear whether these levels of ROS are achieved in the mitochondria.
Tumor progression and the role of HIF-1 and HIF-2.
We observed a stark contrast between the effects of SdhA suppression and those of SdhB suppression on cell growth in vitro and tumor growth in vivo. The stable suppression of SdhB enhanced xenograft tumor formation, which mirrors the clinical tumor syndromes linked to mutations in SdhB, SdhC, or SdhD (5). In contrast, cell proliferation and tumor growth rates in SdhA shRNA cells were inhibited, consistent with results of clinical studies linking SdhA mutations to a bioenergetic deficiency rather than a tumor phenotype (7). Transgenic cell lines with a mutated SdhC gene also exhibit increased ROS production and increased tumor formation rates in a nude mouse model (26). The enhanced tumor progression in our SdhB cells can be attributed to the effects of HIF-1 and HIF-2, because simultaneous stable shRNA suppression of HIF-1α or HIF-2α led to a reversal of that response. The data reveal that HIF-1α and HIF-2α are required for supporting the growth of tumors derived from the SdhB shRNA cells, such that the loss of either factor slows growth measurably. These results underscore the importance of HIF activation for cell proliferation and tumor progression in these cell lines. This is consistent with previous observations that the activation of HIF activity through its overexpression (28) or chronic hypoxia (52) leads to increased tumor growth. Conversely, decreasing HIF transcriptional activity through antisense, dominant-negative, or short interfering RNA constructs (12, 14, 27, 30, 39) slows tumor progression. ROS signals are sufficient to trigger the stabilization of HIF-α (21, 31), and a recent study indicates that antioxidant therapy significantly inhibits tumor progression in vivo by inhibiting HIF activation (18). Collectively, these observations are consistent with a model in which SdhB, SdhC, or SdhD mutations lead to increased tumorigenicity through an ROS-mediated enhancement of HIF activation.
Relationship to oxygen sensing.
Previous studies have implicated complex III in the cellular O2-sensing pathway involved in the regulation of HIF-α stability during hypoxia (10, 21). Genetic and pharmacologic interventions that limit ROS production at complex III inhibit the ability of cells to respond to acute hypoxia without limiting their ability to respond to interventions that mimic hypoxia (6, 10, 11, 21, 31). None of our complex II inhibitors prevented the stabilization of HIF-1α during hypoxia, and SdhB shRNA cells responded normally to hypoxia in terms of HIF-1α stabilization. Consistent with that finding, O2 sensing was not abolished by the genetic deletion of SdhD in a previous study (36). The observation that O2 sensing is maintained despite the loss of complex II is expected, because the oxygen-sensing mechanism functions by releasing ROS from complex III. However, our results indicate that mutations in SdhB, SdhC, or SdhD leading to increases in ROS production by complex II can mimic the ROS signal that arises from complex III during hypoxia, thereby activating the hypoxic response under normoxic conditions. Complex II has been classified as a tumor suppressor, but our results suggest that it functions as a protooncogene capable of activating HIF when defects in SdhB, SdhC, or SdhD cause an increase in ROS production, leading to the stimulation of HIF-dependent gene expression and cell proliferation.
Acknowledgments
This work was supported by NHLBI grants HL35440, HL079650, and HL32646 to P.T.S. and by the Katten Muchin Rosenman Travel Scholarship Award from the Robert H. Lurie Comprehensive Cancer Center of Northwestern University to R.D.G.
Footnotes
Published ahead of print on 29 October 2007.
REFERENCES
- 1.Ackrell, B. A. C. 2000. Progress in understanding structure-function relationships in respiratory chain complex II. FEBS Lett. 4661-5. [DOI] [PubMed] [Google Scholar]
- 2.Astrom, K., J. E. Cohen, J. E. Willett-Brozick, C. E. Aston, and B. E. Baysal. 2003. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum. Genet. 113228-237. [DOI] [PubMed] [Google Scholar]
- 3.Baysal, B. E., R. E. Ferrell, J. E. Willett-Brozick, E. C. Lawrence, D. Myssiorek, A. Bosch, A. van der Mey, P. E. Taschner, W. S. Rubinstein, E. N. Myers, C. W. Richard III, C. J. Cornelisse, P. Devilee, and B. Devlin. 2000. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287848-851. [DOI] [PubMed] [Google Scholar]
- 4.Baysal, B. E., E. C. Lawrence, and R. E. Ferrell. 2007. Sequence variation in human succinate dehydrogenase genes: evidence for long-term balancing selection on SDHA. BMC Biol. 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baysal, B. E., and E. N. Myers. 2002. Etiopathogenesis and clinical presentation of carotid body tumors. Microsc. Res. Tech. 59256-261. [DOI] [PubMed] [Google Scholar]
- 6.Bell, E. L., T. A. Klimova, J. Eisenbart, C. T. Moraes, M. P. Murphy, G. R. Budinger, and N. S. Chandel. 2007. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 1771029-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourgeron, T., P. Rustin, D. Chretien, M. Birch-Machin, M. Bourgeois, E. Viegas-Pequignot, A. Munnich, and A. Rotig. 1995. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 11144-149. [DOI] [PubMed] [Google Scholar]
- 8.Brière, J. J., J. Favier, P. Benit, V. El Ghouzzi, A. Lorenzato, D. Rabier, M. F. Di Renzo, A. P. Gimenez-Roqueplo, and P. Rustin. 2005. Mitochondrial succinate is instrumental for HIF1α nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum. Mol. Genet. 143263-3269. [DOI] [PubMed] [Google Scholar]
- 9.Cecchini, G. 2003. Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 7277-109. [DOI] [PubMed] [Google Scholar]
- 10.Chandel, N. S., E. Maltepe, E. Goldwasser, C. E. Mathieu, M. C. Simon, and P. T. Schumacker. 1998. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 9511715-11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandel, N. S., D. S. McClintock, C. E. Feliciano, T. M. Wood, J. A. Melendez, A. M. Rodriguez, and P. T. Schumacker. 2000. Reactive oxygen species generated at mitochondrial complex III stabilize HIF-1-alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 27525130-25138. [DOI] [PubMed] [Google Scholar]
- 12.Chang, Q., R. Qin, T. Huang, J. Gao, and Y. Feng. 2006. Effect of antisense hypoxia-inducible factor 1α on progression, metastasis, and chemosensitivity of pancreatic cancer. Pancreas 32297-305. [DOI] [PubMed] [Google Scholar]
- 13.Dahia, P. L., K. N. Ross, M. E. Wright, C. Y. Hayashida, S. Santagata, M. Barontini, A. L. Kung, G. Sanso, J. F. Powers, A. S. Tischler, R. Hodin, S. Heitritter, F. Moore, R. Dluhy, J. A. Sosa, I. T. Ocal, D. E. Benn, D. J. Marsh, B. G. Robinson, K. Schneider, J. Garber, S. M. Arum, M. Korbonits, A. Grossman, P. Pigny, S. P. Toledo, V. Nose, C. Li, and C. D. Stiles. 2005. A HIF1α regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 172-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dang, D. T., F. Chen, L. B. Gardner, J. M. Cummins, C. Rago, F. Bunz, S. V. Kantsevoy, and L. H. Dang. 2006. Hypoxia-inducible factor-1α promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res. 661684-1936. [DOI] [PubMed] [Google Scholar]
- 15.Dooley, C. T., T. M. Dore, G. T. Hanson, W. C. Jackson, S. J. Remington, and R. Y. Tsien. 2004. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 27922284-22293. [DOI] [PubMed] [Google Scholar]
- 16.Douwes Dekker, P. B. D., P. C. W. Hogendoorn, N. Kuipers-Dijkshoorn, F. A. Prins, S. G. Van Duinen, P. E. M. Taschner, A. G. L. Van der Mey, and C. J. Cornelisse. 2003. SDHD mutations in head and neck paragangliomas result in destabilization of complex II in the mitochondrial respiratory chain with loss of enzymatic activity and abnormal mitochondrial morphology. J. Pathol. 201480-486. [DOI] [PubMed] [Google Scholar]
- 17.Fedotcheva, N. I., A. P. Sokolov, and M. N. Kondrashova. 2006. Nonenzymatic formation of succinate in mitochondria under oxidative stress. Free Radic. Biol. Med. 4156-64. [DOI] [PubMed] [Google Scholar]
- 18.Gao, P., H. Zhang, R. Dinavahi, F. Li, Y. Xiang, V. Raman, Z. M. Bhujwalla, D. W. Felsher, L. Cheng, J. Pevsner, L. A. Lee, G. L. Semenza, and C. V. Dang. 2007. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12230-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gimenez-Roqueplo, A. P., J. Favier, P. Rustin, J. J. Mourad, P. F. Plouin, P. Corvol, A. Roetig, and X. Jeunemaitre. 2001. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am. J. Hum. Genet. 691186-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo, J., and B. D. Lemire. 2003. The ubiquinone-binding site of the Saccharomyces cerevisiae succinate-ubiquinone oxidoreductase is a source of superoxide. J. Biol. Chem. 27847629-47635. [DOI] [PubMed] [Google Scholar]
- 21.Guzy, R. D., B. Hoyos, E. Robin, H. Chen, L. Liu, K. D. Mansfield, M. C. Simon, U. Hammerling, and P. T. Schumacker. 2005. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1401-408. [DOI] [PubMed] [Google Scholar]
- 22.Hanson, G. T., R. Aggeler, D. Oglesbee, M. Cannon, R. A. Capaldi, R. Y. Tsien, and S. J. Remington. 2004. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 27913044-13053. [DOI] [PubMed] [Google Scholar]
- 23.Isaacs, J. S., Y. J. Jung, D. R. Mole, S. Lee, C. Torres-Cabala, Y. L. Chung, M. Merino, J. Trepel, B. Zbar, J. Toro, P. J. Ratcliffe, W. M. Linehan, and L. Neckers. 2005. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8143-153. [DOI] [PubMed] [Google Scholar]
- 24.Ishiguro, H., K. Yasuda, N. Ishii, K. Ihara, T. Ohkubo, M. Hiyoshi, K. Ono, N. Senoo-Matsuda, O. Shinohara, F. Yosshii, M. Murakami, P. S. Hartman, and M. Tsuda. 2001. Enhancement of oxidative damage to cultured cells and Caenorhabditis elegans by mitochondrial electron transport inhibitors. IUBMB Life 51263-268. [DOI] [PubMed] [Google Scholar]
- 25.Ishii, N., M. Fujii, P. S. Hartman, M. Tsuda, K. Yasuda, N. Senoo-Matsuda, S. Yanase, D. Ayusawa, and K. Suzuki. 1998. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394694-697. [DOI] [PubMed] [Google Scholar]
- 26.Ishii, T., K. Yasuda, A. Akatsuka, O. Hino, P. S. Hartman, and N. Ishii. 2005. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 65203-209. [PubMed] [Google Scholar]
- 27.Jensen, R. L., B. T. Ragel, K. Whang, and D. Gillespie. 2006. Inhibition of hypoxia inducible factor-1α (HIF-1α) decreases vascular endothelial growth factor (VEGF) secretion and tumor growth in malignant gliomas. J. Neurooncol. 78233-247. [DOI] [PubMed] [Google Scholar]
- 28.Kondo, Y., J. Hamada, C. Kobayashi, R. Nakamura, Y. Suzuki, R. Kimata, T. Nishimura, T. Kitagawa, M. Kunimoto, N. Imura, and S. Hara. 2005. Over expression of hypoxia-inducible factor-1α in renal and bladder cancer cells increases tumorigenic potency. J. Urol. 1731762-1766. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S., E. Nakamura, H. Yang, W. Wei, M. S. Linggi, M. P. Sajan, R. V. Farese, R. S. Freeman, B. D. Carter, W. G. Kaelin, Jr., and S. Schlisio. 2005. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8155-167. [DOI] [PubMed] [Google Scholar]
- 30.Li, J., M. Shi, Y. Cao, W. Yuan, T. Pang, B. Li, Z. Sun, L. Chen, and R. C. Zhao. 2006. Knockdown of hypoxia-inducible factor-1α in breast carcinoma MCF-7 cells results in reduced tumor growth and increased sensitivity to methotrexate. Biochem. Biophys. Res. Commun. 3421341-1351. [DOI] [PubMed] [Google Scholar]
- 31.Mansfield, K. D., R. D. Guzy, Y. Pan, R. M. Young, T. P. Cash, P. T. Schumacker, and M. C. Simon. 2005. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 1393-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maxwell, P. H., C. W. Pugh, and P. J. Ratcliffe. 2001. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 11293-299. [DOI] [PubMed] [Google Scholar]
- 33.McWhinney, S. R., R. T. Pilarski, S. R. Forrester, M. C. Schneider, M. M. Sarquis, E. P. Dias, and C. Eng. 2004. Large germline deletions of mitochondrial complex II subunits SDHB and SDHD in hereditary paraganglioma. J. Clin. Endocrinol. Metab. 895694-5699. [DOI] [PubMed] [Google Scholar]
- 34.Messner, K. R., and J. A. Imlay. 2002. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 27742563-42571. [DOI] [PubMed] [Google Scholar]
- 35.Paddison, P. J., A. A. Caudy, and G. J. Hannon. 2002. Stable suppression of gene expression by RNAi in mammalian cells. Proc. Natl. Acad. Sci. USA 991443-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piruat, J. I., C. O. Pintado, P. Ortega-Saenz, M. Roche, and J. Lopez-Barneo. 2004. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell. Biol. 2410933-10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollard, P. J., J. J. Briere, N. A. Alam, J. Barwell, E. Barclay, N. C. Wortham, T. Hunt, M. Mitchell, S. Olpin, S. J. Moat, I. P. Hargreaves, S. J. Heales, Y. L. Chung, J. R. Griffiths, A. Dalgleish, J. A. McGrath, M. J. Gleeson, S. V. Hodgson, R. Poulsom, P. Rustin, and I. P. Tomlinson. 2005. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 142231-2239. [DOI] [PubMed] [Google Scholar]
- 38.Rustin, P., and A. Roetig. 2002. Inborn errors of complex II—unusual human mitochondrial diseases. Biochim. Biophys. Acta Bio-Energetics 1553117-122. [DOI] [PubMed] [Google Scholar]
- 39.Ryan, H. E., M. Poloni, W. McNulty, D. Elson, M. Gassmann, J. M. Arbeit, and R. S. Johnson. 2000. Hypoxia-inducible factor-1α is a positive factor in solid tumor growth. Cancer Res. 604010-4015. [PubMed] [Google Scholar]
- 40.Sanjuán-Pla, A., A. M. Cervera, N. Apostolova, R. Garcia-Bou, V. M. Victor, M. P. Murphy, and K. J. McCreath. 2005. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1α. FEBS Lett. 5792669-2674. [DOI] [PubMed] [Google Scholar]
- 41.Scallet, A. C., R. L. Haley, D. M. Scallet, H. M. Duhart, and Z. K. Binienda. 2003. 3-Nitropropionic acid inhibition of succinate dehydrogenase (complex II) activity in cultured Chinese hamster ovary cells: antagonism by l-carnitine. Ann. N. Y. Acad. Sci. 993305-312. [DOI] [PubMed] [Google Scholar]
- 42.Schofield, C. J., and P. J. Ratcliffe. 2004. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5343-354. [DOI] [PubMed] [Google Scholar]
- 43.Selak, M. A., S. M. Armour, E. D. MacKenzie, H. Boulahbel, D. G. Watson, K. D. Mansfield, Y. Pan, M. C. Simon, C. B. Thompson, and E. Gottlieb. 2005. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 777-85. [DOI] [PubMed] [Google Scholar]
- 44.Selak, M. A., R. V. Duran, and E. Gottlieb. 2006. Redox stress is not essential for the pseudo-hypoxic phenotype of succinate dehydrogenase deficient cells. Biochim. Biophys. Acta Bio-Energetics 1757567-572. [DOI] [PubMed] [Google Scholar]
- 45.Semenza, G. L. 1999. Perspectives on oxygen sensing. Cell 98281-284. [DOI] [PubMed] [Google Scholar]
- 46.Senoo-Matsuda, N., K. Yasuda, M. Tsuda, T. Ohkubo, S. Yoshimura, H. Nakazawa, P. S. Hartman, and N. Ishii. 2001. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J. Biol. Chem. 27641553-41558. [DOI] [PubMed] [Google Scholar]
- 47.Sun, F., X. Huo, Y. J. Zhai, A. J. Wang, J. X. Xu, D. Su, M. Bartlam, and Z. H. Rao. 2005. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 1211043-1057. [DOI] [PubMed] [Google Scholar]
- 48.Szeto, S. S., S. N. Reinke, B. D. Sykes, and B. D. Lemire. 2007. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J. Biol. Chem. 28227518-27526. [DOI] [PubMed] [Google Scholar]
- 49.Tomitsuka, E., Y. Goto, M. Taniwaki, and K. Kita. 2003. Direct evidence for expression of type II flavoprotein subunit in human complex II (succinate-ubiquinone reductase). Biochem. Biophys. Res. Commun. 311774-779. [DOI] [PubMed] [Google Scholar]
- 50.Turrens, J. F., A. Alexandre, and A. L. Lehninger. 1985. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 237408-414. [DOI] [PubMed] [Google Scholar]
- 51.Yankovskaya, V., R. Horsefield, S. Toernroth, C. Luna-Chavez, H. Miyoshi, C. Leger, B. Byrne, G. Cecchini, and S. Iwata. 2003. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299700-704. [DOI] [PubMed] [Google Scholar]
- 52.Yao, K., J. A. Gietema, S. Shida, M. Selvakumaran, X. Fonrose, N. B. Haas, J. Testa, and P. J. O'Dwyer. 2005. In vitro hypoxia-conditioned colon cancer cell lines derived from HCT116 and HT29 exhibit altered apoptosis susceptibility and a more angiogenic profile in vivo. Br. J. Cancer 931356-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu, J., L. Zhang, P. M. Hwang, C. Rago, K. W. Kinzler, and B. Vogelstein. 1999. Identification and classification of p53-regulated genes. Proc. Natl. Acad. Sci. USA 9614517-14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, L., L. D. Yu, and C. A. Yu. 1998. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem. 27333972-33976. [DOI] [PubMed] [Google Scholar]