

Abstract
Chlamydia trachomatis infection induces inflammatory pathologies in the upper genital tract, potentially leading to ectopic pregnancy and infertility in the affected women. Caspase-1 is required for processing and release of the inflammatory cytokines interleukin-1β (IL-1β), IL-18, and possibly IL-33. In the present study, we evaluated the role of caspase-1 in chlamydial infection and pathogenesis. Although chlamydial infection induced caspase-1 activation and processing of IL-1β, mice competent and mice deficient in caspase-1 experienced similar courses of chlamydial infection in their urogenital tracts, suggesting that Chlamydia-activated caspase-1 did not play a significant role in resolution of chlamydial infection. However, when genital tract tissue pathologies were examined, the caspase-1-deficient mice displayed much reduced inflammatory damage. The reduction in inflammation was most obvious in the fallopian tube tissue. These observations demonstrated that although caspase-1 is not required for controlling chlamydial infection, caspase-1-mediated responses can exacerbate the Chlamydia-induced inflammatory pathologies in the upper genital tract, suggesting that the host caspase-1 may be targeted for selectively attenuating chlamydial pathogenicity without affecting the host defense against chlamydial infection.
Chlamydia trachomatis is an obligate intracellular bacterial pathogen consisting of four biovars and more than 15 different serovars. The trachoma biovar, including serovars A to C, mainly infects human eyes, potentially leading to preventable blindness (45). The trachomatis biovar, including serovars D to K, infects the human urogenital tract, which can potentially lead to severe complications, such as ectopic pregnancy and infertility (43). The lymphogranuloma venereum biovar, including serovars L1 to L3, has recently caused various outbreaks in humans (2, 39). The mouse biovar of C. trachomatis, formerly known as mouse pneumonitis agent (designated MoPn), is now classified as a new species, Chlamydia muridarum. C. muridarum has been and still is extensively used to study C. trachomatis pathogenesis and immunology in mouse models (1, 8, 23, 28, 30, 35, 51).
Using various mouse models, much has been learned about C. trachomatis pathogenic mechanisms and host and chlamydial determinants important in C. trachomatis infection. Both C. trachomatis human serovars and murine C. muridarum strains have been used to infect mice via multiple routes, including the intravenous (20, 44), intranasal (3, 13, 23), intrabursa (30), and intravaginal (19, 25-27, 35, 36) routes. It has consistently been shown that gamma interferon (IFN-γ)-mediated immunity is a major protective mechanism for mice to control chlamydial infection regardless of the infection type and route. However, various versions of mouse models have also led to some conflicting observations. For example, interleukin-6 (IL-6) was found to play a significant role in host defense against mouse lung infection (48), but not against genital tract infection (34), by C. muridarum. Since C. trachomatis-caused diseases in humans are in the urogenital tract and the C. muridarum-induced genital tract pathologies closely resemble those in the human genital tract induced by C. trachomatis infection (32, 40, 46, 50), the C. muridarum urogenital infection mouse model has been widely used to study C. trachomatis pathogenesis and immune responses. Using this model, several groups have successfully mapped host adaptive immune components during resolution of chlamydial infection (25, 26, 33) and determined the role of Toll-like receptors in the innate immunity against chlamydial infection and the development of Chlamydia-induced pathologies (10). Furthermore, it was found that although the inducible nitric oxide synthase did not prevent chlamydial infection in mice (17, 37), it protected mice from the chlamydial infection-induced genital tract pathologies (38). In contrast, host matrix metalloproteinases (MMPs) seem to exacerbate chronic inflammatory pathologies resulting from urogenital C. muridarum infection, although they also do not affect the course of infection (18). It appears that MMP-7 is not required for the MMP-mediated exacerbation of inflammation since mice deficient in MMP-7 did not show any significant reduction in Chlamydia-induced urogenital tract pathologies (31).
The role of inflammatory caspases, including caspase-1, in microbial infection and pathogenesis has recently received more attention as a better understanding of the activation and regulation of the inflammatory caspases has been obtained. It is now known that caspase-1 is activated in the context of the inflammasome, a multiprotein complex consisting of the intracellular adaptor protein ASC (apoptosis-associated specklike protein containing a caspase activation recruitment domain) and the intracellular sensor protein NALP3 (NACHT-, leucine-rich repeat- and pyrin-domain-containing protein 3) in addition to procaspase-1 (24). Binding of microbial or cellular components to the leucine-rich repeat in NALP3 can cause a conformational change in the protein complex, leading to activation of caspase-1. Many pathogens, including the intracellular organisms C. trachomatis and Francisella tularensis, have been shown to activate caspase-1 (21, 29, 47, 49), although different types of pathogens may rely on different signaling pathways for activating caspase-1-containing inflammasomes (15). The activated caspase-1 can then process IL-1β, IL-18, and possibly IL-33 and promote maturation and secretion of these cytokines. Since IL-1β, IL-18, and IL-33 are frequently expressed and activated during microbial infection, we evaluated the role of caspase-1 in chlamydial infection and pathogenesis using the mouse model of C. muridarum infection of the urogenital tract in the current study. We found that although chlamydial infection induced caspase-1 activation and processing of IL-1β, a deficiency in caspase-1 did not alter the course of infection in mouse urogenital tracts after either a primary or secondary chlamydial infection. However, when the genital tract tissue pathologies were examined, caspase-1-deficient mice displayed much less inflammatory damage, and the reduction in inflammation in the fallopian tube tissues was statistically significant, especially following the primary infection. These observations demonstrated that although caspase-1 is not required for controlling chlamydial infection, the caspase-1-mediated responses can significantly contribute to the inflammatory pathologies in the upper genital tract during chlamydial infection.
MATERIALS AND METHODS
Chlamydial infection.
C. trachomatis serovar L2 was used to infect HeLa cells (human cervical carcinoma epithelial cells; ATCC catalog number CCL2), and C. muridarum was used to infect mice and mouse macrophages (Mφs). Both organisms were propagated, purified, aliquoted, and stored as described previously (6). To infect HeLa cells, HeLa cells grown in tissue flasks containing Dulbecco's modified Eagle's medium (GIBCO BRL, Rockville, MD) with 10% fetal calf serum (GIBCO BRL) at 37°C in an incubator supplied with 5% CO2 were inoculated with serovar L2 cells as described previously (6). The infected cultures were harvested at different time points after infection for Western blot analyses as described below. To infect mice, female NOD mice with [NOD.129S2(B6)-Casp1tm/Sesh/LtJ; stock number 004947; 13 mice] or without (NOD/Ltj; stock number 001976; 14 mice) caspase-1 gene knockout (KO) that were 5 to 6 weeks old were purchased from Jackson Laboratories (Bar Harbor, ME). Each mouse was inoculated intravaginally with 1 × 104 inclusion-forming units (IFUs) of live C. muridarum in 20 μl of sucrose-phosphate-glutamate buffer (218 mM sucrose, 3.76 mM KH2PO4, 7.1 mM K2HPO4, 4.9 mM glutamate; pH 7.2). Five days prior to infection, each mouse was inoculated with 2.5 mg Depo-provera (Pharmacia Upjohn, Kalamazoo, MI) subcutaneously to synchronize the menstrual cycles and to increase mouse susceptibility to chlamydial infection. For some mice, a secondary infection was performed similarly on day 51 after the first infection. Depo-provera was also given to the mice 5 days prior to the secondary infection. For in vitro infection of Mφs, mouse Mφs were collected from the peritoneal cavity as described previously (56). Briefly, 4 to 5 ml of cold Hanks buffer (2.5 mM HEPES [pH 7.4], 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM Na2HPO4, 25 mM glucose, 0.05% bovine serum albumin) was inoculated into the mouse peritoneal cavity using a 27-gauge needle. After gentle massaging, the solution was slowly withdrawn from the mouse peritoneal cavity using a 20-gauge needle. After the total number of viable cells was determined, the peritoneal cavity-derived cells were resuspended in RPMI 1640 with 10% fetal calf serum and 10 μg/ml gentamicin, and 2 × 105 cells were added to each well of 48-well plates. The plates were incubated at 37°C for 2 h in a CO2 incubator to allow Mφs to adhere. After nonadherent cells were washed away, fresh medium was added to each well and incubation was continued. The adherent Mφs were cultured overnight prior to chlamydial inoculation. Chlamydiae diluted in cell growth medium were directly inoculated onto the cell monolayers at a multiplicity of infection of 5. The infected cultures were incubated for 24 h at 37°C in a CO2 incubator before they were harvested for measurement of cytokines by an enzyme-linked immunosorbent assay (ELISA).
Western blot assay.
The Western blot assay was carried out as described elsewhere (11, 12, 14, 42, 52). Briefly, the cell samples were solubilized in 2% sodium dodecyl sulfate sample buffer and loaded onto sodium dodecyl sulfate-polyacrylamide gel wells. After electrophoresis, the proteins were transferred to nitrocellulose membranes, and the blots were detected with primary antibodies, including rabbit anti-caspase-1 (catalog number 06-503; Upstate, Chicago, IL) and anti-IL-1β (catalog number SC-2022; Santa Cruz Biotech, Santa Cruz, CA) and mouse anti-chlamydial major outer membrane protein (clone MC22) (53) and anti-mammalian heat shock protein 70 (catalog number SC-24; clone w27; Santa Cruz Biotech). The primary antibody binding was probed with a horseradish peroxidase-conjugated secondary antibody (either goat anti-rabbit or anti-mouse immunoglobulin G; Jackson Immunologicals, Westgrove, PA) and visualized with an enhanced chemiluminescence kit (Santa Cruz Biotech).
ELISA.
After the infected Mφs were cultured, the supernatants were collected in order to measure secreted cytokines, while after two washes with warm medium the remaining cell monolayers were collected and lysates were prepared by sonication in an equal amount of medium in order to measure cell-associated or intracellular cytokines using commercially available ELISA kits. The kits used for mouse IL-1β (catalog number DY400), IL-6 (catalog number DY406), tumor necrosis factor alpha (catalog number DY410), and macrophage inflammatory protein 2 (mouse homolog of human IL-8) (catalog number DY452) were all obtained from R&D Systems, Inc. (Minneapolis, MN). The ELISA was carried out by following the instructions provided by the manufacturer or instructions described elsewhere (41, 54, 55). Briefly, 96-well ELISA microplates (Nunc, Rochester, NY) were coated with a capture antibody, and after blocking, the cytokine samples or standards were added to the coated plates, followed by a biotin-conjugated detection antibody. The antibody binding was measured with horseradish peroxidase-conjugated Avidin plus a soluble colorimetric substrate [2,2′-azino-di-(3-ethylbenzthiazolinesulfonic acid) (ABTS)]. The absorbance at 405 nm was determined using a microplate reader (Molecular Devices Corporation, Sunnyvale, CA). The cytokine concentrations were calculated using absorbance values, cytokine standards, and sample dilution factors and were expressed in ng or pg per ml.
Monitoring mouse shedding of live chlamydiae.
To monitor the shedding of live organisms, vaginal swabs were taken once every 3 to 4 days after inoculation for the first 30 days and once per week thereafter. Each swab was dissolved and sonicated in 500 μl of sucrose-phosphate-glutamate buffer and then titrated using HeLa cell monolayers in duplicate as described previously (27). Briefly, serially diluted swab samples were inoculated onto HeLa cell monolayers grown on coverslips in 24-well plates. After incubation for 24 h in the presence of 2 μg/ml cycloheximide, the cultures were fixed with 2% paraformaldehyde dissolved in phosphate-buffered saline for 30 min at room temperature, followed by permeabilization with 2% saponin for an additional 1 h. After washing and blocking, the cells were stained with Hoechst stain (Sigma, St. Louis, MO) (blue) to visualize nuclear DNA and a mouse anti-chlamydial lipopolysaccharide antibody (clone MB5H9) (unpublished observations) plus goat anti-mouse immunoglobulin G conjugated with Cy3 (red) (Jackson ImmunoResearch) to visualize chlamydial inclusions. The inclusions were counted using an Olympus AX-70 fluorescence microscope equipped with multiple filter sets (Olympus, Melville, NY). Five random fields were counted per coverslip. For coverslips containing less than 1 IFU per field, the entire coverslip was counted. Coverslips showing obvious cytotoxicity of HeLa cells were not taken into account. The total number of IFUs per swab was calculated based on the number of IFUs per field, the number of fields per coverslip, the dilution factor, and the inoculation and total sample volumes. An average was calculated using the serially diluted and duplicate samples for a given swab. The calculated total number of IFUs/swab was converted into a log10 value, and the log10 IFU values were used to calculate the mean and standard deviation for each group at each time point.
Evaluating mouse genital tract tissue pathologies and histological scoring.
Eighty days after the primary infection, all mice were sacrificed, and the mouse urogenital tract tissues were isolated. Before the tissues were removed from a mouse body, an in situ gross examination was performed to obtain evidence of hydrosalpinx formation and any other related abnormalities. The excised tissues were then fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned longitudinally (5-μm sections). A effort was made to include the cervix and both uterine horns and oviducts, as well as luminal structures of each tissue, in each section. The sections were stained with hematoxylin and eosin (H&E) as described elsewhere (40). The H&E-stained sections were assessed by a certified pathologist (I-T.Y) blinded to mouse treatment, and the severity of inflammation and pathologies was scored based on the modified schemes established previously (18, 27, 40). The uterine horns and fallopian tubes were scored separately. The scores for dilatation of the uterine horn or fallopian tube were as follows: 0, no significant dilatation; 1, mild dilatation of a single cross section; 2, one to three dilated cross sections; 3, more than three dilated cross sections; and 4, confluent pronounced dilation. The scores for inflammatory cell infiltrates (at the chronic stage of infection, the infiltrates mainly contained mononuclear cells) were as follows: 0, no significant infiltration; 1, infiltration at a single focus; 2, infiltration at two to four foci; 3, infiltration at more than four foci; and 4, confluent infiltration. Scores assigned to individual mice were used to calculate the mean ± standard error for each group of animals (n = 5 to 9).
Statistical analysis.
The chi-square test (Microsoft Excel) was used to analyze qualitative (categorical incidence) data. An analysis of variance (http://www.physics.csbsju.edu/stats/anova.html) was performed to analyze quantitative data from multiple groups, and a two-tailed Student t test (Microsoft Excel) was used to compare two groups.
RESULTS
Caspase-1 is activated during C. trachomatis infection.
We monitored the processing of both caspase-1 and IL-1β during infection of human cervical carcinoma epithelial cells with C. trachomatis (Fig. 1A) or C. muridarum (Fig. 1B). The 45-kDa procaspase-1 was both up-regulated and cleaved into the active p20 fragment at 12 h, and the processing continued to increase up to 48 h for C. trachomatis and up to 36 h for C. muridarum after infection (Fig. 1, panels a). The caspase-1 cleavage was accompanied by processing of pro-IL-1β (panels b), indicating that the cleaved caspase-1 was active in processing its substrates. The chlamydial major outer membrane protein became detectable 24 h after infection (panels c), and the total amount of protein loaded into each lane was monitored by simultaneous detection of host heat shock protein 70 (panels d). The results described above confirm the previous observation that infection with either C. trachomatis or C. muridarum can lead to activation of caspase-1 and processing of IL-1β and IL-18 (21, 29, 49).
FIG. 1.

Activation of caspase-1 by chlamydial infection. HeLa cells infected with C. trachomatis (A) or C. muridarum (MoPn) (B) were harvested at various time points after infection as indicated at the top in order to monitor caspase-1 (panels a) and IL-1β (panels b) processing in a Western blot. The anti-caspase-1 and IL-1β antibodies indicated on the left detected both the pro and mature forms of caspase-1 and IL-1β, as indicated on the right. A mouse anti-chlamydial major outer membrane protein (MOMP) monoclonal antibody was used to monitor chlamydial infection (panels c), and an anti-mammalian heat shock protein 70 (HSP70) antibody was used to monitor total protein loading (panels d). The sample harvested at zero time was normal HeLa cells not infected with chlamydiae (lane 1). Note that both caspase-1 processing and IL-1β processing were induced by chlamydial infection. MW, molecular mass; ns, not significant.
Caspase-1 is not required for host defense against chlamydial infection.
After confirming that caspase-1 is activated during C. trachomatis infection, we next determined whether the caspase-1 activation could contribute to host defense against chlamydial challenge and infection. Groups of mice with or without a caspase-1 gene deficiency were intravaginally infected with C. muridarum, and the vaginal shedding of live organisms was monitored over the course of infection (Fig. 2). All mice, regardless of the genotype, were infected with C. muridarum and shed live organisms up to 2 weeks after infection. Although 3 and 7 of the 14 wild-type mice no longer shed live organisms at days 17 and 21 postinfection, respectively, the average numbers of live organisms shed from both caspase-1 KO and wild-type mice were not significantly different at these two time points. By day 24, no live organisms could be detected for any mouse. To test whether caspase-1 can contribute to the adaptive immunity against Chlamydia infection, six and five mice from the caspase-1 KO and wild-type mouse groups, respectively, were reinfected with C. muridarum on day 51. As expected, the time course following reinfection was greatly shortened and lasted only about 1 week. This was true for both groups of mice, and no significant difference in the shedding of live organisms between the caspase-1 KO and wild-type mice was observed. The observations described above suggest that caspase-1 is not required for either innate or adaptive immunity against chlamydial infection. When the expression and secretion of cytokines by mouse Mφs were measured, we found that a deficiency in caspase-1 did not alter the ability of mouse Mφs to produce and secrete other inflammatory cytokines in response to chlamydial infection, although caspase-1-deficient Mφs did not process IL-1β (Fig. 3).
FIG. 2.

Effect of caspase-1 deficiency on live chlamydia shedding following chlamydial infection. Mice deficient (open bars) or competent (filled bars) for caspase-1 were infected intravaginally with C. muridarum, and vaginal swabs were taken during the course of infection as indicated on the x axis to determine the number of live organisms (expressed in IFUs) shed from the urogenital tract. The number of IFU obtained from each swab was converted into a log10 value, and the log10 IFU values were used to calculate the mean and standard deviation for each mouse group at each time point, as shown on the y axis. At the start of the experiment the caspase-1 KO group contained 13 mice, while the wild-type (wt) group contained 14 mice. The number of mice with detectable IFUs at each time point is indicated above the horizontal line at the top. On day 51 after the primary infection, six and five mice in the caspase-1 KO and wild-type groups, respectively, were reinfected with C. muridarum. All mice were sacrificed on day 80 after the primary infection. The log10 IFU values for the KO and wild-type groups at each time point were analyzed using a two-tailed Student t test, and no statistically significant differences were found. Note that the course of infection was dramatically shortened following the secondary infection.
FIG. 3.
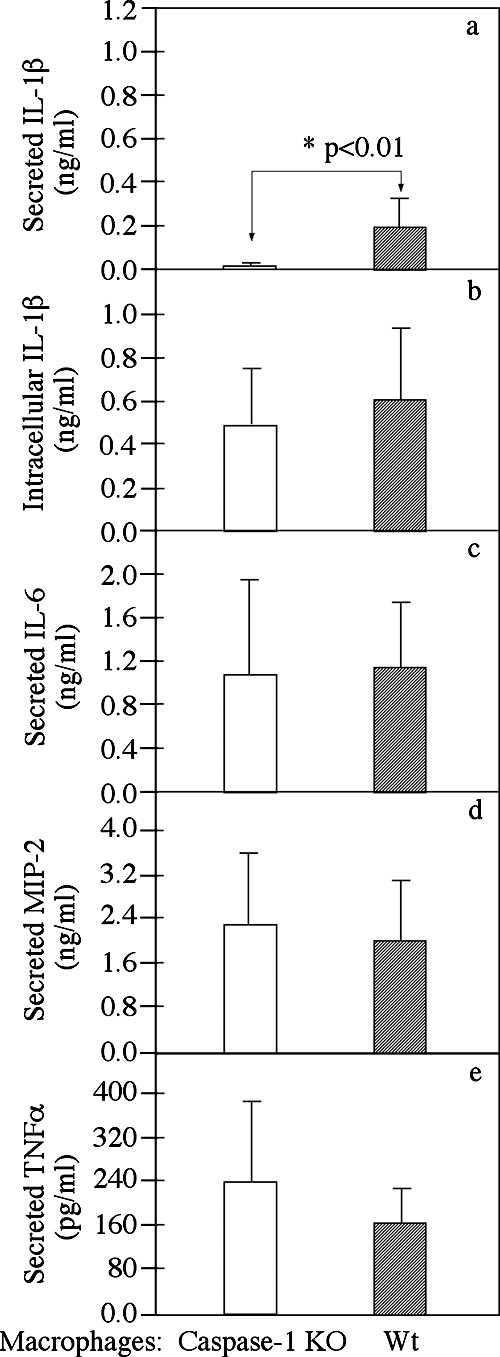
Effect of caspase-1 deficiency on cytokine production and secretion by mouse Mφs after Chlamydia infection. Peritoneal Mφs harvested from either caspase-1 KO (open bars) or wild-type (filled bars) mice were infected with chlamydiae for 24 h in 48-well plates. The intracellular IL-1β (b), secreted IL-1β (a), IL-6 (c), macrophage inflammatory protein 2 (MIP-2) (mouse IL-8 homolog) (d), and tumor necrosis factor alpha (TNFα) (panel e) levels were measured using a commercially available ELISA kit. The data were obtained using seven to nine mice in each group and were expressed in ng or pg per ml of culture supernatant; the bars indicate means, and the error bars indicate standard deviations. The asterisk indicates a statistically significant difference (P < 0.01) in the level of secreted IL-1β between the caspase-1 KO and wild-type Mφs (both infected with chlamydiae).
Deficiency in caspase-1 protected mice from upper genital tract pathologies following C. muridarum infection.
At 80 days after infection, all mice were sacrificed in order to evaluate the pathologies of mouse genital tract tissues. Inflammatory infiltrates and luminal dilation as determined with a microscope, together with hydrosalpinx formation as determined by gross appearance, are hallmarks of the urogenital tract pathologies caused by chlamydial vaginal infection in mice (40). We found that the wild-type NOD mice developed these typical pathological changes in their genital tracts upon chlamydial intravaginal infection (Fig. 4A). When the gross appearance of the isolated genital tracts was examined, we found that the incidence of hydrosalpinx formation was significantly lower in caspase-1 KO mice after reinfection (Table 1) (P < 0.05, as determined by a chi-square test). The H&E-stained histological sections were viewed with a microscope, and inflammation and luminal dilation were semiquantitatively scored. Reduced levels of inflammatory infiltrates and luminal dilation were observed in both the uterine horn and fallopian tube tissue sections from the caspase-1-deficient mice (compared to the sections from wild-type mice) after either primary or secondary infection (Table 2). It is worth noting that none of the NOD mice showed uterine horn luminal dilation despite reinfection. Due to the low number of samples in each score category, we reclassified the samples with a positive inflammation or dilation score (≥1) as inflammation or dilation positive and compared the rates of positivity for the wild-type and caspase-1 KO mouse groups. We found that the rates of positive inflammation in both uterine horn and fallopian tube tissue samples were significantly lower in caspase-1-deficient mice (P < 0.05 and P < 0.01, respectively, as determined by a chi-square test). Furthermore, the caspase-1-deficient mice also displayed a significantly lower rate of fallopian tube dilation after the secondary infection (P < 0.05). This observation is consistent with the significant difference in the incidence of hydrosalpinx formation shown in Table 1. The same mouse samples with hydrosalpinx formation as determined by gross appearance were also scored positive for fallopian tube luminal dilation using the microscope. When the means and standard errors of the inflammation scores for the wild-type and caspase-1-deficient mice were compared using the Student t test, we found that the capspase-1-deficient mice had significantly less inflammation in the fallopian tube tissues (Fig. 4C) (P < 0.01). However, the reduction in inflammation disappeared after reinfection. This was mainly due to the dramatic increase in inflammation induced by the secondary infection in the caspase-1-deficient mice (P < 0.05).
FIG. 4.

Effect of caspase-1 deficiency on the development of inflammatory pathologies in the mouse urogenital tract following chlamydial infection. (A) When urogenital tract tissues from wild-type NOD mice with (panels b and d) or without (panels a and c) a chlamydial infection were examined at the level of gross appearance (panels a and b) and with a microscope (panels c and d), obvious inflammatory pathologies were noted in the samples from the infected mouse but not in the samples from the normal mouse, and these pathologies included hydrosalpinx formation (panel b, white arrow), extensive infiltration of mononuclear cells (panel d, arrows), and fallopian tube luminal dilation (panel d, arrows). Different types of tissues in a NOD mouse urogenital tract with a normal gross appearance are indicated in panel a (arrows). (B) Mouse genital tract tissues were sectioned and analyzed using a microscope after H&E staining. A representative image of a histological section from either the uterine horn (panels a, c, e, g, I, and k) or fallopian tube (panels b, d, f, h, j, and l) tissues is shown for each group of mice with or without a primary or secondary infection. The asterisks indicate inflammatory cell infiltration, the number sign indicates fibrosis, and the ampersand indicates luminal dilation. Scores based on the severity of inflammatory infiltration are indicated in some of the images. Note that chlamydial infection induced more severe inflammatory changes in wild-type mice. (C) Inflammation scores assigned to individual mice were used to calculate the means (bars) and standard errors (error bars) for different groups. The various tissue and mouse groups are indicated on the x axis. The open bars indicate tissue samples from caspase-1 KO mice, while the filled bars indicate tissue samples from wild-type mice. An analysis of variance test was used to analyze differences among different groups, and a two-tailed Student t test was used to analyze differences between the caspase-1 KO and wild-type groups. There was a highly significant difference in inflammation scores (P < 0.01, Student t test) between the caspase-1 KO and wild-type fallopian tissues, and there was a significant difference in the caspase-1 KO fallopian tube tissue inflammation (P < 0.05, Student t test) between the primary and secondary infections.
TABLE 1.
In situ macroscopic examination of genital tissues for incidence of hydrosalpinx formationa
| Infection | Mice | No. | No. with hydrosalpinx formation in:
|
|
|---|---|---|---|---|
| One oviduct | Both oviducts | |||
| Primary | Caspase-1 KO | 7 | 0 | 0 |
| Wild type | 9 | 2 | 0 | |
| Secondary | Caspase-1 KO | 6 | 2 | 1 |
| Wild type | 5 | 4 | 0 | |
The gross appearance of mouse urogenital tract tissues was inspected to determine the presence of hydrosalpinx formation. Mice with hydrosalpinx formation in only one oviduct (unilateral) or in both oviducts (bilateral) were identified separately. However, for statistical analysis, mice with hydrosalpinx formation that was either unilateral or bilateral were classified as hydrosalpinx positive. A chi-square test showed that the incidence of hydrosalpinx formation was significantly higher in wild-type mice (four of five mice) than in caspase-1 KO mice (three of six mice) following the second infection (P < 0.05).
TABLE 2.
Microscopic examination of sections for incidence of inflammation and dilation scoresa
| Infection | Mice | No. | Uterus horn
|
Fallopian tube
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. with inflammation score of:
|
No. dilation positive | No. with inflammation score of:
|
No. with dilation score of:
|
||||||||||||
| 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | 4 | 0 | 3 | 4 | ||||
| Primary | Caspase-1 KO | 7 | 6 | 1 | 0 | 6 | 1 | 7 | |||||||
| Wild type | 9 | 4 | 1 | 3 | 1 | 0 | 1 | 4 | 3 | 1 | 7 | 2 | |||
| Secondary | Caspase-1 KO | 6 | 4 | 2 | 0 | 2 | 1 | 1 | 2 | 3 | 2 | 1 | |||
| Wild type | 5 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 2 | 1 | 2 | 2 | ||
The extent of microscopic inflammation was semiquantitatively evaluated for each tissue sample based on the criteria described in Materials and Methods, and the samples were assigned inflammation and dilation scores. However, for statistical analysis, all samples with an inflammation or dilation score of 1 or more were classified as inflammation or dilation positive. A chi-square test showed that the rate of positive inflammation scores for both uterus horn (P < 0.05) and fallopian tube (P < 0.01) tissues was significantly higher for wild-type mice than for caspase-1 KO mice following the primary infection. The rate of positive fallopian tube luminal dilation scores was higher for wild-type mice after the secondary infection (P < 0.05).
DISCUSSION
C. trachomatis infection induces inflammatory pathologies in the urogenital tract, which may lead to long-term sequelae, such as infertility and ectopic pregnancy (7). Despite the tremendous efforts made to understand chlamydial pathogenic mechanisms, the molecular basis of the chlamydial infection-induced inflammatory damage in the upper urogenital tract remains unknown. Here, we demonstrated that caspase-1 significantly contributes to the Chlamydia-induced inflammation in the upper urogenital tract, especially in the fallopian tube tissues. First, caspase-1 was activated during chlamydial infection of a human cervical epithelial cell line (Fig. 1). This observation is consistent with a previous report that both caspase-1 processing and IL-18 processing were detected in Chlamydia-infected epithelial cells (21). Second, the rate of hydrosalpinx formation in caspase-1-deficient mice was significantly lower than that in caspase-1-competent mice (Table 1). This observation was confirmed by microscopic analysis of corresponding tissue sections stained with H&E, which revealed that the caspase-1-deficient mice displayed a significantly decreased rate of fallopian tube luminal dilation (Table 2). Third, using a qualitative analysis with the chi-square test, we found that the rate of inflammatory infiltration in both uterine horn and fallopian tube samples was significantly lower in caspase-1-deficient mice after primary infection, although the reduction was attenuated after reinfection (Table 2). Finally, a more careful quantitative analysis further confirmed that there was a profound reduction in inflammatory infiltration in the fallopian tube tissues following primary infection of the caspase-1-deficient mice (Fig. 4C).
Although we present convincing evidence that caspase-1 has a strong role in exacerbating the fallopian tube inflammation induced by a primary infection, caspase-1's contribution to inflammation was not significant during a secondary infection. The diminished contribution by caspase-1 was probably due to the greatly amplified inflammatory responses mediated by adaptive immunity during the secondary infection. Clearly, the adaptive immunity-mediated inflammation was caspase-1 independent since reinfection significantly increased the fallopian tube inflammation in the caspase-1-deficient mice (P < 0.05) (Fig. 4C). It is clear that caspase-1 plays a dominant role in Chlamydia-induced oviduct inflammation at the innate adaptive immunity stages but not at the adaptive immunity stages. This conclusion is consistent with the fact that caspase-1-containing inflammasomes are directly activated by microbial pathogens via the innate immunity receptors, such as Toll-like receptors and Nod-like receptors, including NALP3 (24). It is worth noting that following the secondary infection, the courses of infection were dramatically shortened. Unfortunately, this powerful protection against infection also significantly exacerbated pathologies in the oviducts, which is consistent with the well-accepted concept that the whole chlamydia-induced immunity can lead to both short-term protection against infection and exacerbation of tissue damage.
Although inflammation is generally considered a double-edged sword, contributing to both host defense and tissue damage, in the current study we found that caspase-1-dependent inflammation significantly contributed only to Chlamydia-induced inflammatory damage and did not affect the host defense against chlamydial infection. This biased role of caspase-1 in chlamydial pathogenesis may be due to the fact that Chlamydia can activate caspase-1 inflammasomes but the caspase-1-mediated inflammatory responses are unable to affect chlamydial growth. Indeed, the most important host factor identified so far for controlling chlamydial infection is IL-12/IFN-γ (19, 33) and not caspase-1-mediated responses. IFN-γ has been shown to restrict chlamydial growth both in vitro and in vivo, and a lack of IL-12 or IFN-γ dramatically increases host susceptibility to chlamydial infection (9, 33). The caspase-1-mediated responses do not appear to positively affect either IL-12- or IFN-γ-mediated antichlamydial activity. Of the cytokines activated by caspse-1 (including IL-1β, IL-18, and IL-33), IL-18 is the only one that can induce T, B, and NK cells to produce IFN-γ. However, it has been shown that IL-18 is not required for clearance of chlamydial infection (22). IL-1β-triggered responses are mediated by the type I receptor of IL-1, which can induce a wide spectrum of inflammatory cytokines and chemokines, such as IL-6 and IL-8, but not IFN-γ (4). IL-33 activates ST2-mediated signaling pathways (5), which mainly promotes Th2 cytokine production (16). Since Th2 cytokines can suppress Th1 responses and inhibit IFN-γ production, it is not likely that IL-33 plays a significant role in blocking chlamydial infection. Although caspase-1-mediated pathways can lead to the production of many other effector molecules, these effector molecules may not contribute to the host defense against chlamydial infection. For example, IL-6, nitric oxide, and MMPs, which are inducible by the caspase-1-activated IL-1β, have been shown to play little role in clearing chlamydial infection in the urogenital tract (18, 34). Together, the analyses described above suggest that caspase-1-mediated responses are unable to affect chlamydial infection. This conclusion not only is consistent with the results obtained in the current study but also is supported by the observation that caspase-1-deficient mice are as susceptible to C. muridarum pulmonary infection as caspase-1-competent mice (22).
Because caspase-1-mediated responses can induce multiple effectors, it is difficult to predict the precise effector molecules responsible for the reduced pathologies in caspase-1-deficient mice without further experiments. It has been shown that inhibition of the IL-1β-inducible MMPs with chemical inhibitors, although failing to alter the course of chlamydial infection, can significantly suppress inflammatory pathologies in mouse oviducts during chlamydial infection (18), which very much mimics the phenotype of caspase-1 deficiency. However, because MMPs can also be induced by pathways independent of caspase-1, it is unlikely that the redundantly regulated MMPs are responsible for the caspase-1-dependent exacerbation of oviduct inflammation. Nevertheless, we are in the process of evaluating the levels of MMPs in the urogenital tracts of caspase-1-deficient mice. At the same time, mice deficient in IL-1β, IL-18, or IL-33 will be used to further define the pathogenic effectors that are dependent on caspase-1. Regardless of the effector molecules responsible for the caspase-1-dependent pathogenic phenotype, the finding that caspase-1 contributes significantly to inflammatory pathologies during chlamydial primary infection has provided important information for developing anti-inflammatory drugs for minimizing Chlamydia-induced oviduct pathologies in humans. We are planning to evaluate various cell-permeable caspase-1-specific small-molecule inhibitors to determine their abilities to attenuate Chlamydia-induced inflammation in the mouse model of urogenital infection.
Acknowledgments
This work was supported in part by grants to G.Z. from the National Institutes of Health.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 19 November 2007.
REFERENCES
- 1.Barteneva, N., I. Theodor, E. M. Peterson, and L. M. de la Maza. 1996. Role of neutrophils in controlling early stages of a Chlamydia trachomatis infection. Infect. Immun. 644830-4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauwens, J. E., H. Orlander, M. P. Gomez, M. Lampe, S. Morse, W. E. Stamm, R. Cone, R. Ashley, P. Swenson, and K. K. Holmes. 2002. Epidemic Lymphogranuloma venereum during epidemics of crack cocaine use and HIV infection in the Bahamas. Sex. Transm. Dis. 29253-259. [DOI] [PubMed] [Google Scholar]
- 3.Bilenki, L., S. Wang, J. Yang, Y. Fan, A. G. Joyee, and X. Yang. 2005. NK T cell activation promotes Chlamydia trachomatis infection in vivo. J. Immunol. 1753197-3206. [DOI] [PubMed] [Google Scholar]
- 4.Boraschi, D., and A. Tagliabue. 2006. The interleukin-1 receptor family. Vitam. Horm. 74229-254. [DOI] [PubMed] [Google Scholar]
- 5.Carriere, V., L. Roussel, N. Ortega, D. A. Lacorre, L. Americh, L. Aguilar, G. Bouche, and J. P. Girard. 2007. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 104282-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, C., D. Chen, J. Sharma, W. Cheng, Y. Zhong, K. Liu, J. Jensen, R. Shain, B. Arulanandam, and G. Zhong. 2006. The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infect. Immun. 744826-4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, C. R., and R. C. Brunham. 1999. Pathogenesis of Chlamydia induced pelvic inflammatory disease. Sex. Transm. Infect. 7521-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotter, T. W., Q. Meng, Z. L. Shen, Y. X. Zhang, H. Su, and H. D. Caldwell. 1995. Protective efficacy of major outer membrane protein-specific immunoglobulin A (IgA) and IgG monoclonal antibodies in a murine model of Chlamydia trachomatis genital tract infection. Infect. Immun. 634704-4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotter, T. W., K. H. Ramsey, G. S. Miranpuri, C. E. Poulsen, and G. I. Byrne. 1997. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect. Immun. 652145-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darville, T., J. M. O'Neill, C. W. Andrews, Jr., U. M. Nagarajan, L. Stahl, and D. M. Ojcius. 2003. Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J. Immunol. 1716187-6197. [DOI] [PubMed] [Google Scholar]
- 11.Dong, F., M. Pirbhai, Y. Xiao, Y. Zhong, Y. Wu, and G. Zhong. 2005. Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect. Immun. 731861-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong, F., Y. Zhong, B. Arulanandam, and G. Zhong. 2005. Production of a proteolytically active protein, chlamydial protease/proteasome-like activity factor, by five different Chlamydia species. Infect. Immun. 731868-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong-Ji, Z., X. Yang, C. Shen, H. Lu, A. Murdin, and R. C. Brunham. 2000. Priming with Chlamydia trachomatis major outer membrane protein (MOMP) DNA followed by MOMP ISCOM boosting enhances protection and is associated with increased immunoglobulin A and Th1 cellular immune responses. Infect. Immun. 683074-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan, T., H. Lu, H. Hu, L. Shi, G. A. McClarty, D. M. Nance, A. H. Greenberg, and G. Zhong. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187487-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchi, L., T. D. Kanneganti, G. R. Dubyak, and G. Nunez. 2007. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 28218810-18818. [DOI] [PubMed] [Google Scholar]
- 16.Gadina, M., and C. A. Jefferies. 2007. IL-33: a sheep in wolf's clothing? Sci. STKE 2007pe31. [DOI] [PubMed] [Google Scholar]
- 17.Igietseme, J. U., L. L. Perry, G. A. Ananaba, I. M. Uriri, O. O. Ojior, S. N. Kumar, and H. D. Caldwell. 1998. Chlamydial infection in inducible nitric oxide synthase knockout mice. Infect. Immun. 661282-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imtiaz, M. T., J. H. Schripsema, I. M. Sigar, J. N. Kasimos, and K. H. Ramsey. 2006. Inhibition of matrix metalloproteinases protects mice from ascending infection and chronic disease manifestations resulting from urogenital Chlamydia muridarum infection. Infect. Immun. 745513-5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito, J. I., and J. M. Lyons. 1999. Role of gamma interferon in controlling murine chlamydial genital tract infection. Infect. Immun. 675518-5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lampe, M. F., C. B. Wilson, M. J. Bevan, and M. N. Starnbach. 1998. Gamma interferon production by cytotoxic T lymphocytes is required for resolution of Chlamydia trachomatis infection. Infect. Immun. 665457-5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu, H., C. Shen, and R. C. Brunham. 2000. Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J. Immunol. 1651463-1469. [DOI] [PubMed] [Google Scholar]
- 22.Lu, H., X. Yang, K. Takeda, D. Zhang, Y. Fan, M. Luo, C. Shen, S. Wang, S. Akira, and R. C. Brunham. 2000. Chlamydia trachomatis mouse pneumonitis lung infection in IL-18 and IL-12 knockout mice: IL-12 is dominant over IL-18 for protective immunity. Mol. Med. 6604-612. [PMC free article] [PubMed] [Google Scholar]
- 23.Lu, H., and G. Zhong. 1999. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect. Immun. 671763-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mariathasan, S., and D. M. Monack. 2007. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 731-40. [DOI] [PubMed] [Google Scholar]
- 25.Morrison, R. P., K. Feilzer, and D. B. Tumas. 1995. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect. Immun. 634661-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison, S. G., and R. P. Morrison. 2005. A predominant role for antibody in acquired immunity to chlamydial genital tract reinfection. J. Immunol. 1757536-7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murthy, A. K., J. P. Chambers, P. A. Meier, G. Zhong, and B. P. Arulanandam. 2007. Intranasal vaccination with a secreted chlamydial protein enhances resolution of genital Chlamydia muridarum infection, protects against oviduct pathology, and is highly dependent upon endogenous gamma interferon production. Infect. Immun. 75666-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murthy, A. K., J. Sharma, J. J. Coalson, G. Zhong, and B. P. Arulanandam. 2004. Chlamydia trachomatis pulmonary infection induces greater inflammatory pathology in immunoglobulin A deficient mice. Cell. Immunol. 23056-64. [DOI] [PubMed] [Google Scholar]
- 29.Ojcius, D. M., P. Souque, J. L. Perfettini, and A. Dautry-Varsat. 1998. Apoptosis of epithelial cells and macrophages due to infection with the obligate intracellular pathogen Chlamydia psittaci. J. Immunol. 1614220-4226. [PubMed] [Google Scholar]
- 30.Pal, S., E. M. Peterson, and L. M. de la Maza. 2005. Vaccination with the Chlamydia trachomatis major outer membrane protein can elicit an immune response as protective as that resulting from inoculation with live bacteria. Infect. Immun. 738153-8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pal, S., A. P. Schmidt, E. M. Peterson, C. L. Wilson, and L. M. de la Maza. 2006. Role of matrix metalloproteinase-7 in the modulation of a Chlamydia trachomatis infection. Immunology 117213-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patton, D. L., D. V. Landers, and J. Schachter. 1989. Experimental Chlamydia trachomatis salpingitis in mice: initial studies on the characterization of the leukocyte response to chlamydial infection. J. Infect. Dis. 1591105-1110. [DOI] [PubMed] [Google Scholar]
- 33.Perry, L. L., K. Feilzer, and H. D. Caldwell. 1997. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J. Immunol. 1583344-3352. [PubMed] [Google Scholar]
- 34.Perry, L. L., K. Feilzer, and H. D. Caldwell. 1998. Neither interleukin-6 nor inducible nitric oxide synthase is required for clearance of Chlamydia trachomatis from the murine genital tract epithelium. Infect. Immun. 661265-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perry, L. L., K. Feilzer, S. Hughes, and H. D. Caldwell. 1999. Clearance of Chlamydia trachomatis from the murine genital mucosa does not require perforin-mediated cytolysis or Fas-mediated apoptosis. Infect. Immun. 671379-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry, L. L., H. Su, K. Feilzer, R. Messer, S. Hughes, W. Whitmire, and H. D. Caldwell. 1999. Differential sensitivity of distinct Chlamydia trachomatis isolates to IFN-gamma-mediated inhibition. J. Immunol. 1623541-3548. [PubMed] [Google Scholar]
- 37.Ramsey, K. H., G. S. Miranpuri, C. E. Poulsen, N. B. Marthakis, L. M. Braune, and G. I. Byrne. 1998. Inducible nitric oxide synthase does not affect resolution of murine chlamydial genital tract infections or eradication of chlamydiae in primary murine cell culture. Infect. Immun. 66835-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsey, K. H., I. M. Sigar, S. V. Rana, J. Gupta, S. M. Holland, and G. I. Byrne. 2001. Role for inducible nitric oxide synthase in protection from chronic Chlamydia trachomatis urogenital disease in mice and its regulation by oxygen free radicals. Infect. Immun. 697374-7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schachter, J., and J. Moncada. 2005. Lymphogranuloma venereum: how to turn an endemic disease into an outbreak of a new disease? Start looking. Sex. Transm. Dis. 32331-332. [DOI] [PubMed] [Google Scholar]
- 40.Shah, A. A., J. H. Schripsema, M. T. Imtiaz, I. M. Sigar, J. Kasimos, P. G. Matos, S. Inouye, and K. H. Ramsey. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex. Transm. Dis. 3249-56. [DOI] [PubMed] [Google Scholar]
- 41.Sharma, J., A. M. Bosnic, J. M. Piper, and G. Zhong. 2004. Human antibody responses to a Chlamydia-secreted protease factor. Infect. Immun. 727164-7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma, J., F. Dong, M. Pirbhai, and G. Zhong. 2005. Inhibition of proteolytic activity of a chlamydial proteasome/protease-like activity factor by antibodies from humans infected with Chlamydia trachomatis. Infect. Immun. 734414-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherman, K. J., J. R. Daling, A. Stergachis, N. S. Weiss, H. M. Foy, S. P. Wang, and J. T. Grayston. 1990. Sexually transmitted diseases and tubal pregnancy. Sex. Transm. Dis. 17115-121. [DOI] [PubMed] [Google Scholar]
- 44.Starnbach, M. N., W. P. Loomis, P. Ovendale, D. Regan, B. Hess, M. R. Alderson, and S. P. Fling. 2003. An inclusion membrane protein from Chlamydia trachomatis enters the MHC class I pathway and stimulates a CD8+ T cell response. J. Immunol. 1714742-4749. [DOI] [PubMed] [Google Scholar]
- 45.Taylor, H. R., S. L. Johnson, J. Schachter, H. D. Caldwell, and R. A. Prendergast. 1987. Pathogenesis of trachoma: the stimulus for inflammation. J. Immunol. 1383023-3027. [PubMed] [Google Scholar]
- 46.Tuffrey, M., F. Alexander, C. Inman, and M. E. Ward. 1990. Correlation of infertility with altered tubal morphology and function in mice with salpingitis induced by a human genital-tract isolate of Chlamydia trachomatis. J. Reprod. Fertil. 88295-305. [DOI] [PubMed] [Google Scholar]
- 47.Weiss, D. S., T. Henry, and D. M. Monack. 2007. Francisella tularensis activation of the inflammasome. Ann. N. Y. Acad. Sci. 1105219-237. [DOI] [PubMed] [Google Scholar]
- 48.Williams, D. M., B. G. Grubbs, T. Darville, K. Kelly, and R. G. Rank. 1998. A role for interleukin-6 in host defense against murine Chlamydia trachomatis infection. Infect. Immun. 664564-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolf, K., H. J. Betts, B. Chellas-Gery, S. Hower, C. N. Linton, and K. A. Fields. 2006. Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle. Mol. Microbiol. 611543-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zana, J., D. Thomas, M. Muffat-Joly, J. de Brux, J. J. Pocidalo, J. Orfila, C. Carbon, and J. Salat-Baroux. 1990. An experimental model for salpingitis due to Chlamydia trachomatis and residual tubal infertility in the mouse. Hum. Reprod. 5274-278. [DOI] [PubMed] [Google Scholar]
- 51.Zhang, D., X. Yang, H. Lu, G. Zhong, and R. C. Brunham. 1999. Immunity to Chlamydia trachomatis mouse pneumonitis induced by vaccination with live organisms correlates with early granulocyte-macrophage colony-stimulating factor and interleukin-12 production and with dendritic cell-like maturation. Infect. Immun. 671606-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong, G., F. Castellino, P. Romagnoli, and R. N. Germain. 1996. Evidence that binding site occupancy is necessary and sufficient for effective major histocompatibility complex (MHC) class II transport through the secretory pathway redefines the primary function of class II-associated invariant chain peptides (CLIP). J. Exp. Med. 1842061-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong, G., P. Fan, H. Ji, F. Dong, and Y. Huang. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193935-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong, G., I. Toth, R. Reid, and R. C. Brunham. 1993. Immunogenicity evaluation of a lipidic amino acid-based synthetic peptide vaccine for Chlamydia trachomatis. J. Immunol. 1513728-3736. [PubMed] [Google Scholar]
- 55.Zhong, G. M., and R. C. Brunham. 1990. Immunoaccessible peptide sequences of the major outer membrane protein from Chlamydia trachomatis serovar C. Infect. Immun. 583438-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong, G. M., and L. M. de la Maza. 1988. Activation of mouse peritoneal macrophages in vitro or in vivo by recombinant murine gamma interferon inhibits the growth of Chlamydia trachomatis serovar L1. Infect. Immun. 563322-3325. [DOI] [PMC free article] [PubMed] [Google Scholar]