

Abstract
The attaching and effacing (A/E) bacterial pathogens enteropathogenic Escherichia coli and enterohemorrhagic E. coli and the related mouse pathogen Citrobacter rodentium colonize their hosts' intestines by infecting the apical surfaces of enterocytes, subverting their function, and they ultimately cause diarrhea. Surprisingly, little is known about the interactions of these organisms with goblet cells, which are specialized epithelial cells that secrete the protective molecules Muc2 and trefoil factor 3 (Tff3) into the intestinal lumen. C. rodentium infection leads to dramatic goblet cell depletion within the infected colon, yet it is not clear whether C. rodentium infects goblet cells or if this pathology is pathogen or host mediated. As determined by immunostaining and PCR, both the number of goblet cells and the expression of genes encoding Muc2 and Tff3 were significantly reduced by day 10 postinfection. While electron microscopy and immunostaining revealed that C. rodentium directly infected a fraction of colonic goblet cells, C. rodentium localization did not correlate with goblet cell depletion. To assess the role of the host immune system in these changes, Rag1 knockout (KO) (T- and B-cell-deficient) mice were infected with C. rodentium. Rag1 KO mice did not exhibit the reduction in the number of goblet cells or in mediator (Muc2 and Tff3) expression observed in infected immunocompetent mice. However, reconstitution of Rag1 KO mice with T and B lymphocytes from C57BL/6 mice restored the goblet cell depletion phenotype during C. rodentium infection. In conclusion, these studies demonstrated that while colonic goblet cells can be subject to direct infection and potential subversion by A/E pathogens in vivo, it is the host immune system that primarily modulates the function of these cells during infection.
Enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli (EHEC) are food- and waterborne pathogens that are attaching and effacing (A/E) bacteria, a class of noninvasive enteric bacterial pathogens (49). These bacteria infect the apical surfaces of intestinal epithelial cells, causing a signature histopathology characterized by intimate attachment to the host plasma membrane and localized effacement of the brush border immediately surrounding the attached bacteria (16, 74). As a result of this infection and the resulting host inflammatory response, EPEC and EHEC cause severe diarrhea and other complications, leading to the death of hundreds of thousands of children worldwide each year (7, 28, 49). Although these pathogens are human specific and thus difficult to model in vivo, Citrobacter rodentium is a natural A/E bacterial pathogen of mice that is closely related to EPEC and EHEC (11). C. rodentium infection causes colonic epithelial cell proliferation and crypt elongation, as well as inflammation and diarrhea (20, 38). Because C. rodentium produces A/E lesions that are virtually indistinguishable from those produced by EPEC and EHEC (38), it has been widely used as a model to study A/E bacterial pathogenesis in vivo (11, 12, 21, 25).
During infection by several enteric bacterial pathogens, including A/E pathogens, intestinal epithelial cells can be subject to direct modulation by the pathogens (4, 22, 50, 67). A/E pathogens utilize a type III secretion system (T3SS) to secrete various bacterial effectors encoded in their genomes (e.g., the transmembrane intimin receptor Tir) (18, 31) directly into host cells to cause disease. These virulence factors act in an orchestrated manner to subvert intracellular signaling pathways within host cells, altering various cellular processes, including cytoskeletal (61), organelle (39, 47), and barrier (10, 19, 39) functions. This strategy allows the bacteria not only to intimately attach to and form A/E lesions on epithelial surfaces (6, 32) but also to suppress inflammatory responses and host defenses (44, 65). Through the release of various cytokines, host immune cells can also modulate intestinal epithelial function by altering epithelial cell proliferation (5, 54), migration (8), and permeability (24, 75). Such immunomodulation of epithelial function is thought to represent a critical effector mechanism by which the host is able to mediate clearance of invading enteric pathogens, as demonstrated with diverse classes of pathogens, such as viruses (5) and helminths (8, 35, 48). However, while this mechanism has been characterized best for parasitic infections, the role of immunomodulation of intestinal epithelial cells during enteric bacterial infections, including A/E pathogen infections, remains largely undefined.
Infection by several enteric pathogens, including C. rodentium, leads to a dramatic reduction in the number of phenotypically distinct goblet cells, which is termed “goblet cell depletion” (38). Intestinal goblet cells are highly polarized secretory cells that are present throughout the intestinal tract but are most abundant in the distal colon and rectum (59), where they make up 16% of the total epithelial cell population in mice (29). These specialized epithelial cells are thought to play an important protective role in the intestine by synthesizing and secreting several mediators, including the mucin MUC2 (46) and the small peptide trefoil factor 3 (TFF3) (63). MUC2 (in mice, Muc2) is a high-molecular-weight glycoprotein that is stored within granules in the apical compartment of the cell. Under basal conditions or under the influence of host or bacterial stimuli (14), goblet cells release MUC2-containing granules into the lumen, where they hydrate and form the structural basis for the mucus gel layer overlying the intestinal epithelium (69). This mucus layer plays important physiological roles in the gut; it simultaneously lubricates the intestinal surface, limits passage of luminal molecules into the mucosa, functions as a dynamic defensive barrier against enteric pathogens (14, 59), and acts as a substrate and niche which the commensal flora can colonize and from which this flora can derive nutrients (57). TFF3 (in mice, Tff3) is another goblet cell-derived molecule belonging to a family of small cysteine-rich secretory peptides that are expressed in a region-specific manner throughout the gastrointestinal tract (63). A potent inducer of cell migration and an inhibitor of apoptosis (64), TFF3 plays a critical role in wound healing by promoting epithelial restitution following mucosal injury (45). In addition, TFF3 is thought to synergize with colonic mucins to enhance the protective barrier properties of the mucus layer against bacterial toxins (36). Evidence of the importance of goblet cells in maintaining overall health has come from studies of mice lacking either Muc2 or Tff3. These mice are highly susceptible to experimental colitis (45) or have profound defects in intestinal homeostasis under basal conditions (70).
Considering the critical role that goblet cells appear to play in host defense against enteric pathogens, the observation that these cells are depleted during C. rodentium infection may have important implications regarding the pathogenesis of this infection, as well as infection by clinically important pathogens, including Shigella (53, 60) and Campylobacter (37), where the goblet cell depletion phenotype is also observed. At present, whether the goblet cell depletion seen during C. rodentium infection reflects the death or functional alteration of goblet cells is not clear, nor has the expression of goblet cell mediators been assessed in this model. Similarly, it is not clear if this pathology reflects direct infection and subversion of goblet cell function by C. rodentium, perhaps in an attempt to bypass mucosal defenses, or alternatively, if the goblet cell depletion is mediated by the host as a currently cryptic form of host defense. We hypothesized that goblet cell function during C. rodentium infection is subject to modulation by both the pathogen and components of the host's immune system. With this hypothesis in mind, the current study addressed the mechanisms underlying the intestinal goblet cell depletion that occurs during C. rodentium infection.
MATERIALS AND METHODS
Mice.
Six- to 8-week-old female C57BL/6 mice and Rag1 knockout (KO) mice (with a C57BL/6 background) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were kept in sterilized, filter-topped cages, handled in tissue culture hoods, and fed autoclaved food and water under specific-pathogen-free conditions. Sentinel animals were routinely tested for common pathogens. The protocols employed were approved by the University of British Columbia's Animal Care Committee and were in direct accordance with guidelines drafted the Canadian Council on the Use of Laboratory Animals.
Bacterial strains and infection of mice.
Mice were infected by oral gavage with 0.1 ml of an overnight culture in Luria broth containing approximately 2.5 × 108 CFU of wild-type C. rodentium (formerly Citrobacter freundii biotype 4280 strain DBS100) (33). For transmission electron microscope (TEM) studies, mice were also infected with a mutant ΔescN C. rodentium strain lacking a functional T3SS (12).
Tissue collection.
Uninfected control mice or mice at days 6 and 10 postinfection (p.i.) were anesthetized with Halothane and killed by cervical dislocation, and colons resected for further analysis; the colons were divided in half to separate the proximal and distal portions. Tissues were immediately placed in 10% neutral buffered formalin (Fisher) for histological studies, or they were placed in RNA later (Qiagen) and stored at −86°C for subsequent RNA extraction or in 4% paraformaldehyde for subsequent freezing and cryosectioning. The paraformaldehyde-fixed tissues were washed in phosphate-buffered saline (PBS), incubated in 20% sucrose in PBS overnight at 4°C, and then embedded in Shandon Cryomatrix embedding medium (Thermoelectron Corporation), and the mold was frozen by partial immersion in liquid N2-precooled 2-methylbutane and stored at −20°C until it was used.
Bacterial counting.
Whole mouse colons, including stools, were washed thoroughly in PBS (pH 7.4), placed in 1.5 ml of PBS, and homogenized at 19,000 rpm for 45 s using a Polytron homogenizer (Kinematica). Tissue homogenates were serially diluted in PBS, plated onto Luria broth agar plates, and incubated overnight at 37°C, and bacterial colonies were counted the following day.
RNA extraction and quantitative RT-PCR.
Colon tissues stored in RNAlater (Qiagen) at −86°C were thawed on ice and weighed, and the total RNA was extracted using a Qiagen RNeasy kit by following the manufacturer's instructions. Tissues were homogenized in 1 ml of buffer RLT (supplied with the Qiagen RNeasy kit) using a Polytron homogenizer for 1 min at 26,000 rpm. Total RNA was quantified using a Bio-Rad SmartSpec (Bio-Rad), and 1 to 2 μg of RNA was reverse transcribed using a Qiagen Omniscript reverse transcription (RT) kit (Qiagen) according to the manufacturer's instructions. cDNA was diluted 1:25 in RNase- and DNase-free H2O, and 5 μl was added to a 15-μl PCR mixture. Conventional semiquantitative PCR was carried out with an Eppendorf Mastercycler, using the primers for murine Muc2, Tff3, or β-actin as a housekeeping control. The sequences of all primer sets used, the PCR conditions, and the cycle numbers are shown in Table 1. Agarose gels were stained with SYBR Safe DNA gel stain (Molecular Probes) and visualized with a Chemi Doc XRS system (Bio-Rad). Densitometric analysis was carried out using ImageJ software 1.38x (downloaded from the National Institutes of Health website [http://rsb.info.nih.gov/ij/download.html]). For quantitative PCR, Bio-Rad Supermix was used at a 1:2 dilution, and real-time PCR was carried out using a Bio-Rad MJ Mini-Opticon. Quantitation was carried out using GeneEx Macro OM 3.0 software, which employs the 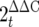 method for real-time quantification of gene expression. Melting point analysis confirmed the specificity for each of the PCRs, and the PCR efficiency for each of primer set was incorporated into the final calculations.
method for real-time quantification of gene expression. Melting point analysis confirmed the specificity for each of the PCRs, and the PCR efficiency for each of primer set was incorporated into the final calculations.
TABLE 1.
Primer sets and PCR conditions used in this study
| Target mRNA | Primera
|
PCR cycle conditions (denaturation/annealing/extension)b | No. of cycles (endpoint PCR only) | |
|---|---|---|---|---|
| Direction | Sequence | |||
| Muc2 | Forward | 5′-CTGACCAAGAGCGAACACAA-3′ | 94°C for 30 s/55°C for 30 s/72°C for 45 s | 23 |
| Reverse | 5′-CATGACTGGAAGCAACTGGA-3′ | |||
| Tff3c | Forward | 5′-CAGATTACGTTGGCCTGTCTCC-3′ | 94°C for 30 s/60°C for 30 s/72°C for 30 s | 30 |
| Reverse | 5′-ATGCTTGCTACCCTTGGACCAC-3′ | |||
| TNF-αd | Forward | 5′-ATGAGCACAGAAAGCATGATC-3′ | 94°C for 30 s/59°C for 30 s/72°C for 45 s | NAe |
| Reverse | 5′-TACAGGCTTGTCACTCGAATT-3′ | |||
| IL-17A (IL-17)f | Forward | 5′-GCTCCAGAAGGCCCTCAGA-3′ | 94°C for 30 s/60°C for 30 s/72°C for 30 s | NA |
| Reverse | 5′-CTTTCCCTCCGCATTGACA-3′ | |||
| β-Actin | Forward | 5′-CAGCTTCTTTGCAGCTCCTT-3′ | 94°C for 30 s/55 to 60°C for 30 s/72°C for 30 s | 23 or 30 |
| Reverse | 5′-CTTCTCCATGTCGTCCCAGT-3′ | |||
The same primer sets were used for end point and quantitative PCR experiments described in text.
In all PCR experiments there was an initial denaturation step of 95°C for 5 min before PCR cycling, and in end point PCR experiments there was an extension step of 72°C for 10 min after the final cycle.
Tff3 primer sequences were obtained from reference 27.
TNF-α primer sequences were obtained from reference 62.
NA, not applicable.
IL-17 primer sequences were obtained from reference 23.
Histological staining.
Briefly, 5-μm paraffin sections were deparaffinized by heating them at 55 to 65°C for 10 min, cleared with xylene, and rehydrated through an ethanol gradient to water. For periodic acid-Schiff (PAS) staining, standard histological techniques were used. For immunostaining, either rabbit polyclonal antisera that recognized the murine colonic mucin Muc2 (1:50; a gift from Jan Dekker), rabbit polyclonal antisera generated against rat Tff3 (1:200; a gift from D. Podolsky), or rat antisera against C. rodentium Tir (1:500; a gift from W. Deng) were used as the primary antibody. The primary antibodies were diluted in PBS containing 1% bovine serum albumin. For immunoperoxidase staining, antigen retrieval was performed by incubating deparaffinized, rehydrated slides in 10 mM citric acid (pH 6.0) at 90 to 100°C for 20 min, followed by cooling to room temperature. Horseradish peroxidase-linked goat anti-rabbit immunoglobulin G (IgG) (1:200; Genetex) was used as the secondary antibody. All antibody incubations were carried out in the presence of 0.2% Triton X-100 (Sigma) to facilitate cell permeabilization. The immunoreaction was developed using SigmaFast DAB substrate (Sigma). Stained sections were washed in water, counterstained with Gill's hematoxylin, dehydrated using ethanol, cleared using xylene, and mounted using Entellan (EM Biosciences). For double-immunofluorescence studies with anti-Muc2 and anti-Tir, no permeabilization or antigen retrieval methods were used to minimize staining for untranslocated Tir still present within the bacteria. To obtain frozen sections, 6-μm sections were cut, placed onto Superfrost/Plus slides (Fisher), and subsequently stained with rat anti-mouse CD3 (clone 17A2; 1:300; BioLegend) overnight at 4°C or with rat anti-mouse CD45R/B220 (clone RA3-6B2; 1:100; Becton Dickinson) for 1 h at room temperature. Immunofluorescent labeling for all stains was carried out with the appropriate secondary antibody using AlexaFluor 488-conjugated goat anti-rat IgG, AlexaFluor 568-conjugated goat anti-rabbit IgG, or AlexaFluor 568-conjugated goat anti-rat IgG (Invitrogen). Tissues were mounted using ProLong Gold Antifade (Invitrogen) that contained 4′,6′-diamidino-2-phenylindole (DAPI) for DNA staining. Sections were viewed at 350, 488, and 594 nm with a Zeiss AxioImager microscope. Images were obtained using a Zeiss AxioImager microscope equipped with an AxioCam HRm camera operating through AxioVision software (version 4.4).
Quantitative histological studies. (i) Goblet cell enumeration.
PAS- and hematoxylin-stained sections prepared at various time points were photographed, and the total numbers of epithelial cells and PAS-positive cells were determined for 20 to 30 longitudinally sectioned crypts per section. The number of goblet cells was expressed as the total number of PAS-positive cells per 100 epithelial cells. Phenotypically mature goblet cells were assessed based on the intensity of staining, the size of the apical region, the location on the crypt base-to-surface axis, and morphology, similar to the method described by Katz and coworkers (30).
(ii) Quantification of infected crypts.
For crypt infection studies involving double immunostaining for Muc2 and Tir, crypts exhibiting goblet cell depletion were defined as crypts with less than three Muc2-positive phenotypically mature goblet cells and with little Muc2 in the crypt lumen. Crypts that did not exhibit goblet cell depletion were defined as crypts with three or more strongly Muc2-positive phenotypically mature goblet cells and an intense secreted Muc2 signal in the crypt lumen. Positively infected crypts were defined as crypts that were positive for Tir staining on the cells of the surface epithelium and on cells in the upper one-third of colonic crypts.
TEM.
Mouse colons were immersed for 3 h in a fixative containing 0.1 M sodium cacodylate, 1.5% paraformaldehyde, and 1.5% glutaraldehyde (pH 7.3). Following fixation, the material was postfixed for 1 h on ice in 1% osmium tetroxide in 0.1 M sodium cacodylate (pH 7.3) and stained with 0.1% uranyl acetate. The material was dehydrated using an ascending alcohol series, followed by incubation in propylene oxide. The blocks were then left in a 1:1 solution of propylene oxide-Polybed overnight. The material was embedded in 100% Polybed, and the resin was polymerized at 60°C for 24 h. Sections were viewed and photographed using a Philips 300 electron microscope operated at 60 kV.
In vivo imaging: bacterial strains and generation of bioluminescent C. rodentium.
Bioluminescent strains of C. rodentium were constructed by introducing plasmid pT7 (E. A. Meighen, Department of Biochemistry, McGill University) carrying the entire lux operon from Photorhabdus luminescens. Bioluminescent colonies were selected on Luria broth agar plates supplemented with 100 μg/ml of ampicillin and were screened using a model 1420 Victor3V multilabel counter (PerkinElmer). For in vivo tissue imaging of bioluminescent C. rodentium at 6 and 10 days p.i., mice were anesthetized and then euthanized. The colons and ceca were removed and opened lengthwise so that the lumen was exposed, and the tissue was washed with sterile PBS. Tissues were then placed in a light-tight specimen chamber that is part of an in vivo imaging system (IVIS; Xenogen, Alameda, CA). The bacterial signals were quantified using the software program LIVING IMAGE (Xenogen) as an overlay on Igor (Wavemetrics, Seattle, WA). To determine the anatomical location, a pseudocolor image showing the light intensity (blue [least intense] to red [most intense]) was generated using LIVING IMAGE software and was superimposed over the grayscale reference image.
Immune cell reconstitution of Rag1 KO mice.
The adaptive immune system was reconstituted in Rag1 KO mice using splenic and mesenteric lymph node (MLN) populations of T and B lymphocytes as previously described (66). In brief, wild-type immunocompetent mice were euthanized, and their spleens and MLNs were aseptically removed. Spleens and MLNs were placed in RPMI medium with 10% fetal bovine serum, mashed to a pulp with the rubber end of the plunger from a 1.0-ml syringe, and then forced through a 70-μm-pore-size filter (BD Biosciences), generating a single-cell suspension. Cells were spun down and resuspended in red blood cell lysis buffer (155 mM NH4Cl, 1 mM KHO3, 0.01 mM Na2EDTA-2H2O; pH 7.4) for 5 min to lyse the red blood cells. Following two washes with RPMI medium, cells were pelleted and then resuspended in PBS. Cells were then counted, and viability was analyzed by trypan blue exclusion. Recipient Rag1 KO mice were then inoculated via the tail vein with 2 × 108 viable mononuclear cells. Mice were left for 6 weeks and then tested for the success of reconstitution by staining colonic tissue sections with isolated lymphoid follicles for the presence of T lymphocytes using the marker CD3 and for the presence of B lymphocytes using the marker B220.
Statistical analysis.
Statistical significance was calculated by using either a two-tailed Student t test or the Mann-Whitney t test as indicated, with assistance from GraphPad Prism software (version 4.00; GraphPad Software, San Diego, CA) (www.graphpad.com). A P value of 0.05 was considered significant. The results were expressed as means and standard errors of the means or as means and standard deviations as indicated below.
RESULTS
Depletion of mucus-containing goblet cells correlates with peak bacterial colonization during C. rodentium infection.
Previous studies have indicated that C. rodentium infection causes goblet cell depletion within the colons of infected mice (38); however, the timing of this pathology and how the changes are related to C. rodentium colonization have not been addressed. To begin, we examined the time course and spatial characteristics of C. rodentium colonization in C57BL/6 mice (Fig. 1). When total pathogen burdens were assessed, C. rodentium was found to heavily colonize the colon by day 6 p.i., with the number of bacteria peaking at day 10 p.i. By day 14 p.i, the number of C. rodentium cells began to drop, and the infection essentially cleared by day 21 p.i., with only a few thousand bacteria remaining (Fig. 1 and data not shown). In addition, consistent with recent studies of C. rodentium infection dynamics by other groups (76), we found that bioluminescent C. rodentium cells were predominantly localized in the cecum and distal half of the colon, and little infection was detected in the proximal colon at days 6 and 10 p.i. (not shown).
FIG. 1.

C. rodentium infection peaks at day 10 p.i. in C57BL/6 mice. Each symbol indicates the mean for three independent infections, each with three mice per time point. The error bars indicate standard deviations.
We next examined whether the localization of C. rodentium influenced colonic pathology and the reported goblet cell depletion by comparing tissue sections taken from the distal and proximal colons of uninfected and infected mice at days 6 and 10 p.i. Using the PAS staining technique to stain goblet cell mucins, only minimal changes in PAS staining were observed in any region of the cecum and colon at day 6 pi (data not shown), and there was little overt change in PAS staining of goblet cells in the cecum (data not shown) or in the proximal colon (Fig. 2A and B) at day 10 p.i. However, a dramatic reduction in PAS staining was observed in tissue sections of samples taken from the distal colon at day 10 p.i. (Fig. 2C and D). While a small number of crypts still contained phenotypically mature mucin-filled goblet cells, characterized by a swollen (PAS-stained) apical region and a tapered basolateral compartment containing the nucleus, in most colonic crypts there were almost none of these cells (Fig. 2E). However, examination under higher magnification revealed that these tissues contained many cells that exhibited weak PAS staining in their apical compartments (Fig. 2D). Compared to the cells that stained strongly with PAS stain (Fig. 2E), these weakly stained cells were smaller and exhibited a more columnar morphology (Fig. 2F), reminiscent of the “hypotrophic” goblet cell phenotype observed in the interleukin-10 (IL-10)-deficient mouse model of spontaneous colitis (41). When we determined the number of PAS-positive cells per 100 epithelial cells at day 10 p.i., we observed a modest but statistically significant decrease in the size of the total PAS-positive population (P < 0.036) compared to uninfected controls. However, when we determined the number of cells possessing the phenotypically mature goblet cell morphology that stained strongly with PAS stain (Fig. 2F), we found that there was a substantially greater reduction in the size of this population (P < 0.002) (Table 2). As the infection progressed, the hypotrophic goblet cell phenotype was still predominant at day 14 p.i., but the number of large mucin-filled goblet cells began to rebound again by day 21 p.i. (data not shown). Thus, our data indicate that C. rodentium infection leads to a reduction in PAS staining, potentially due to a decrease in mucin glycoprotein content of goblet cells, rather than an actual loss of the goblet cell lineage. As a result, we observed only a modest decrease in the total number of goblet cells, and the dramatic reduction in the large goblet cells that appeared to be mature explained the previously described goblet cell depletion.
FIG. 2.

Depletion of mucus-containing goblet cells occurs in the distal colon and is pronounced when the C. rodentium burdens peak. (A to D) PAS staining of formalin-fixed paraffin-embedded tissue sections obtained from the proximal (A and B) and distal (C and D) colons of both naïve (uninfected) mice (A and C) and mice at day 10 p.i (B and D). There was a reduction in overall PAS staining within many crypts in distal colons at day 10 p.i., although scattered PAS-positive cells were still evident (arrows in panel D). Bars = 100 μm. DIS, distal colon; PROX, proximal colon. (E and F) Representative images of PAS-positive cells at day 10 p.i. exhibiting a large mucin-filled goblet cell morphology (arrows in panel E) or a mucin-depleted columnar morphology (arrows in panel F). The images are representative of at least three independent infections with two or three mice per time point.
TABLE 2.
Goblet cell enumeration in the distal colon of C57BL/6 mice following C. rodentium infectiona
| Days p.i. | Total no. of PAS-positive cells/100 epithelial cells (avg ± SD) | No. of phenotypically mature goblet cells/100 epithelial cells (avg ± SD) |
|---|---|---|
| Uninfected control | 32.9 ± 7.9 | 17.9 ± 2.8 |
| 6 | 28.1 ± 7.3 | 14.0 ± 4.6 |
| 10 | 21.6 ± 7.3b | 7.7 ± 3.2b |
C. rodentium infection results in depletion of phenotypically mature goblet cells in the distal colon during times corresponding to peak infection. Enumeration was performed using PAS- and hematoxylin-stained tissue sections of distal colons by determining the number of distinctly PAS-positive cells per 100 epithelial cells, differentiating the cells that had a mature goblet phenotype. The data are values from three independent infections, with each infection group containing three mice per time point.
P < 0.05, as determined by the Mann-Whitney t test.
Goblet cell-specific gene expression is reduced during infection.
While infection led to a decrease in overall PAS staining and a reduction in the number of phenotypically mature goblet cells, it remained unclear whether these effects solely reflected a reduction in mucin expression and production, which directly impacts goblet cell morphology (70), or whether they were also accompanied by altered production of additional goblet cell-specific mediators. To assess this, the expression of the major goblet cell mucin Muc2, as well as Tff3, was assayed by semiquantitative RT-PCR. As assessed by densitometry of end point PCR products shown in Fig. 3A, there were slight but nonsignificant reductions in expression of Muc2 and Tff3 mRNA by day 6 p.i., but there were notably larger and significant decreases in expression of Muc2 and especially Tff3 mRNA by day 10 p.i. (Fig. 3A, graph). To determine whether C. rodentium infection impacted Muc2 and Tff3 protein expression, we performed immunoperoxidase staining for Muc2 and Tff3 of formalin-fixed paraffin-embedded sections of distal colons from control and infected mice. In control mice, Muc2 protein expression and Tff3 protein expression were abundant in goblet cells, although the expression patterns were distinct. While Muc2-positive cells were observed from the base to the luminal surface of colonic crypts (Fig. 3B, upper left panel), Tff3-positive goblet cells were confined predominantly to the upper half of the crypts (Fig. 3B, lower left panel), in agreement with previous reports (52). However, consistent with our results showing that infection causes down-regulation of Muc2 and Tff3 mRNA, we observed a dramatic reduction in staining for both the Muc2 and Tff3 proteins at day 10 p.i. throughout crypts having the depleted goblet cell phenotype (Fig. 3B, upper and lower right panels, respectively), although there was still faint Muc2 staining in scattered crypt cells. Thus, our data show that infection leads not only to reduced mucin (Muc2) expression and production, resulting in decreased PAS staining, but also to reduced production of the goblet cell-specific mediator Tff3; taken together, the results suggest that goblet cell function is profoundly altered during infection. Considering that this pathology was most pronounced in regions of the colon heavily infected by C. rodentium, we next addressed whether direct infection might play a role in this modulation of goblet cell function.
FIG. 3.

C. rodentium infection results in reduction of Muc2 and Tff3 gene expression. (A) Representative RT-PCR analysis of Muc2 and Tff3 gene expression in distal colonic tissues of naïve C57BL/6 mice or mice at days 6 and 10 p.i. β-Actin was used as a loading control. Differences in levels of expression relative to the levels in naïve (uninfected) mice after normalization to β-actin were determined by densitometric analysis, as shown in the graph on the right. The bars indicate the means for three independently infected mice per time point. The error bars indicate the standard errors of the means. Asterisk, P < 0.05; ns, not significant (P > 0.05), as determined by Student's t test. (B) Immunoperoxidase staining for Muc2 (upper panels) and Tff3 (lower panels) protein (brown) in serial sections of distal colons from either naïve mice (left panels) or mice at day 10 p.i. (right panels). In control mice, Muc2-positive cells are present from the crypt base to the luminal surface, whereas Tff3 expression is restricted to the top half of the crypts (arrows). The arrowheads indicate cells that are coexpressing the Muc2 and Tff3 proteins. Note the lack of staining in intact crypts at day 10 p.i. (arrows in left panels). Bar = 50 μm.
C. rodentium directly interacts with goblet cells in vivo.
It remains unclear whether C. rodentium is able to infect cells other than enterocytes in vivo, such as goblet cells. To address this question, we first assessed whether C. rodentium cells in the infected colons were close to or in contact with goblet cells. Here, we focused on day 6 p.i., when colonization in the distal colon was established but phenotypically distinct goblet cells were still relatively abundant (Table 2). To identify any potential C. rodentium-goblet cell interactions, we a performed a dual immunostaining analysis to look for colocalization of Muc2-positive goblet cells and the locus of enterocyte effacement-encoded virulence factor Tir, which is expressed on the apical surface of cells only following direct infection by C. rodentium (13). As shown in Fig. 4A, Tir staining was found on the surface epithelium and progressed down some crypts, where Muc2-positive goblet cells also resided. To confirm the colocalization of Tir with Muc2-positive cells, we examined tissues at a higher magnification and found positive Tir staining on the apical surface of enterocytes, as well as on cells that were positive for Muc2 (Fig. 4B). Next, we examined whether C. rodentium cells were adherent to colonic goblet cells using TEM. TEM analysis showed that C. rodentium cells were adherent to the apical plasma membrane of goblet cells (Fig. 4C and D). Although goblet cells have a morphology and function that are very distinct from the morphology and function of their absorptive enterocytic counterparts, they still contain intact, albeit shortened and less dense, microvilli (58). Notably, however, C. rodentium-associated goblet cells also exhibited evidence of microvillar effacement, a hallmark of A/E lesion formation (Fig. 4C). Interestingly, we also occasionally observed bacteria within the apical granule mass of goblet cells, suggesting that C. rodentium may be internalized by some goblet cells (Fig. 4C). In contrast, when mice were infected with ΔescN C. rodentium, which lacks a functional T3SS, no C. rodentium cells were found to be adherent to goblet cells, nor was any effacement of goblet cell (or enterocyte) microvilli observed in these mice (Fig. 4E). To determine the frequency at which goblet cells were infected, we determined the proportions of Muc2-positive (goblet) cells and Muc2-negative (enterocyte, nongoblet) cells that were positive for Tir staining on the surface epithelium, where most C. rodentium cells were localized. Our results showed that there was a moderate but significant difference between the proportion of infected Muc2-positive cells and the proportion of infected Muc2-negative cells, in that fewer than 50% of Muc2-positive cells were positive for Tir staining, whereas just over 60% of Muc2-negative cells were positive for Tir staining (Fig. 4F). These results indicate that along with colonocytes, goblet cells are subject to direct infection by A/E pathogens in vivo, although Muc2-negative cells are more frequently observed to be infected.
FIG. 4.

C. rodentium directly infects colonic goblet cells in vivo. (A) Immunofluorescence staining for the C. rodentium translocated effector Tir (red), goblet cell-specific Muc2 (green), and DNA (blue) in distal colonic tissues at day 6 p.i. Tir staining is present in Muc2-positive cells (arrowheads) and Muc2-negative cells within the surface epithelium, progressing down the length of some crypts (arrow). (B) Magnified image of Tir (red) staining (arrows) on the apical surface of cells staining strongly for Muc2 (green) in the apical compartment and exhibiting distinct goblet cell morphology. GA, goblet cell apical compartment; GN, goblet cell nucleus; E, enterocyte; L, lumen. Original magnification, ×1,000. (C to E) TEM micrographs of distal colons of C57BL/6 mice taken at day 7 (C) and day 11 (D) following infection with wild-type C. rodentium and at day 7 following infection with ΔescN C. rodentium (E). C. rodentium is in direct contact with goblet cells in panels C and D (black arrows). In panel C, effacement of microvilli (black arrows) on a goblet cell and internalization of bacteria into the apical granule mass of the goblet cell (white arrow) can be seen. In panel E, no infection of goblet cells and intact microvilli (arrow) can be seen following infection with ΔescN C. rodentium. (F) Graph showing the proportions of Muc2-positive (goblet) cells and Muc2-negative (nongoblet) cells of the surface epithelia that were positive for Tir staining at day 6 p.i. The bars indicate the averages for three mice from two independent infections. The error bars indicate the standard errors of the means. Asterisk, P < 0.05, as determined by Student's t test.
C. rodentium predominantly associates with crypts that do not exhibit goblet cell depletion.
Our next goal was to examine the interactions of C. rodentium with colonic crypts at day 10 p.i. in order to determine whether C. rodentium could directly mediate the loss of mature goblet cells. If this were the case, we would expect to find C. rodentium associated with hypotrophic goblet cells, which we tested by immunofluorescently staining for both Tir and Muc2. Strikingly, only minimal Tir staining was associated with crypts exhibiting hypotrophic goblet cells, and no C. rodentium cells were identified in proximity to these cells (Fig. 5A). To clarify the relationship between C. rodentium infection and goblet cell depletion, we identified individual colonic crypts that were infected by C. rodentium (i.e., Tir positive) and, as described in Materials and Methods, differentiated between crypts that exhibited a weak overall signal for Muc2, indicative of the goblet cell depletion phenotype, and crypts that displayed a strong signal for Muc2, indicating that goblet cell depletion did not occur. Interestingly, we found that the proportion of crypts that were infected and exhibited goblet cell depletion was more than threefold lower than the proportion of crypts that did not display an overt loss of goblet cells (Fig. 5B). This trend was even more dramatic at day 14 p.i., when in virtually all crypts Muc2 expression was markedly reduced and there was little if any associated Tir staining (data not shown). These results suggest that there is a surprising inverse relationship between C. rodentium infection of crypts and goblet cell depletion. It should be noted that we did observe one or two crypts per colonic section that were heavily invaded by C. rodentium and were strongly Tir positive from the surface epithelium to the crypt base, yet were also depleted of Muc2-positive cells. However, unlike the typical goblet cell-depleted crypts, these crypts invariably looked atrophic and/or necrotic, suggesting that the loss of Muc2 staining in these crypts was due to destruction of the crypt epithelium rather than to modulation of goblet cell function. These data therefore demonstrate that aside from occasional crypt destruction, the widespread goblet cell depletion seen in colonic crypts during C. rodentium infection is independent of direct bacterial contact or infection of the altered goblet cells.
FIG. 5.

C. rodentium associates with crypts that are strongly positive for Muc2 at day 10 p.i. (A) Dual immunofluorescence staining for C. rodentium Tir (red) and Muc2 (green) in a distal colon at day 10 p.i. Tir staining (white arrow) is associated with crypts strongly positive for Muc2 (white arrowhead), but little if any Tir staining (yellow arrow) is associated with crypts that are weakly positive for Muc2 (yellow arrowhead). Note the frequent colocalization of Tir with Muc2-postive cells and luminal Muc2. (B) Proportions of crypts exhibiting depletion of Muc2-positive goblet cells that are positive for Tir compared to crypts that are not depleted of Muc2-postive goblet cells. The bars indicate the averages for 30 crypts counted within colonic sections from two individual mice, and the error bars indicate standard deviations.
Rag1 KO mice do not suffer goblet cell depletion during C. rodentium infection.
The studies described above suggested that goblet cell depletion is not a result of direct infection, since it is frequently observed in uninfected crypts. When alternative mechanisms were considered, the adaptive immune response was shown to modulate goblet cell function during infection by intestinal helminths (35, 48). Moreover, as previous studies have found that a robust adaptive immune response to C. rodentium infection occurs by day 10 p.i. (26), we tested whether the host adaptive immune response mediated the observed reduction in the number of mature goblet cells and Muc2 and Tff3 expression. Thus, Rag1 KO mice (lacking mature T and B lymphocytes) were infected, and their goblet cell responses were analyzed at day 10 p.i. As observed with C57BL/6 mice, mucosal thickening associated with epithelial hyperplasia was observed in Rag1 KO mice, consistent with previous reports (66). However, compared with infected C57BL/6 mice (Fig. 6B), no decrease in the number of mature goblet cells was observed in the Rag1 KO mice at day 10 p.i., as assessed by PAS staining (Fig. 6D and 6E); in fact, there was a modest trend toward increased numbers of PAS-positive cells at day 10 p.i. compared to uninfected controls (Fig. 6E). Moreover, the majority of goblet cells in the Rag1 KO mice, at both the base and surface of the crypts, had a highly differentiated appearance and contained full mucin-filled apical granule masses that were stained strongly with PAS stain (Fig. 6D). However, as observed for immunocompetent mice in the studies mentioned above, we occasionally observed heavily infected and atrophic crypts that exhibited reduced PAS staining, (Fig. 6D). The maintenance of overall goblet cell function within the colons of infected Rag1 KO mice was further confirmed by immunostaining for the Muc2 and Tff3 proteins. High levels of both proteins were detected in infected Rag1 KO mice at day 10 p.i., and the staining patterns were similar to those observed in uninfected colons (Fig. 7A). In addition, there was not a significant decrease in Muc2 or Tff3 gene expression at day 10 p.i. compared with C57BL/6 mice (Fig. 7B). To determine how the preservation of mature goblet cells is related to the number of bacteria, we quantified the bacterial burdens at days 6 and 10 p.i. Consistent with previous reports (66), Rag1 KO mice had greater bacterial burdens than C57BL/6 mice at day 10 p.i. (Fig. 6F), which indicates that the maintenance of goblet cell number and function in the Rag1 KO mice was not due to reduced numbers of bacteria in these mice and confirms that direct C. rodentium infection plays a minor role in the overall goblet cell depletion observed in C57BL/6 mice. These results strongly suggested that T and/or B lymphocytes were involved in the loss of mature goblet cells during C. rodentium infection.
FIG. 6.

C. rodentium infection does not result in goblet cell depletion phenotype in Rag1 KO mice. (A to D) Histology of PAS- and hematoxylin-stained distal colonic sections taken from (A) naïve C57BL/6 mice, (B) C57BL/6 mice at day 10 p.i., (C) naïve Rag1 KO mice, and (D) Rag1 KO mice at day 10 p.i. The mature PAS-positive cell population is still large in Rag1 KO mice at day 10 p.i. Heavily infected mucin-depleted atrophic crypts are indicated by arrowheads. Original magnification, ×100. Bar = 100 μm. (E) Quantitation of goblet cells in distal colons from naïve C57BL/6 and Rag1 KO mice or from infected mice at day 10 p.i. The bars indicate the mean numbers of goblet cells per 100 epithelial cells counted in PAS- and hematoxylin-stained sections of the distal colon. The error bars indicate standard errors of the means for at least three independent infections with one to three mice per time point. Asterisk, P < 0.05. (F) Comparison of numbers of bacteria in whole colons of C57BL/6 mice (filled bars) and Rag1 KO mice (open bars) at day 10 p.i. The bars indicate the means from three independent infections, each with two or three mice per time point. The error bars indicate the standard deviations. Asterisk, P < 0.05, as determined by the Mann-Whitney t test.
FIG. 7.

Muc2 and Tff3 are abundantly expressed in Rag1 KO mice following C. rodentium infection. (A) Immunohistochemical staining for Muc2 (upper panels) and Tff3 (lower panels) in the distal colons of naïve mice (right panels) and infected mice (left panels). Note the abundance of Tff3 and Muc2 in crypts that are evidently undergoing hyperplasia, with staining patterns similar to those of naïve mice (arrows). Bars = 50 μm. Original magnification, ×200. (B) Quantitative RT-PCR analysis of Muc2 and Tff3 gene expression in distal colons from naïve and infected C57BL/6 and Rag1 KO mice. Each bar indicates the mean relative expression for four mice from three independent infections compared to naïve mice of the same strain, which were assigned an arbitrary expression value of 1.00. Genes encoding Muc2 and Tff3 were both expressed at significantly higher levels in Rag1 KO mice than in C57BL/6 mice at day 10 p.i. All samples were normalized to β-actin. The error bars indicate the standard errors of the means. Asterisk, P < 0.05, as determined by the Mann-Whitney t test.
Proinflammatory cytokine expression in C57BL/6 and Rag1 KO mice during C. rodentium infection.
Because proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), have been implicated in causing goblet cell depletion in the small intestine in a Salmonella enterica serovar Typhimurium ligated ileal loop model (1), we compared TNF-α gene expression in C57BL/6 mice and TNF-α gene expression in Rag1 KO mice to begin to address which host molecules are responsible for the loss of mature goblet cells in the colon during infection. As shown in Fig. 8, TNF-α expression was induced in both strains at day 10 p.i. but was induced to a greater extent in C57BL/6 mice; however, the difference did not reach statistical significance (P = 0.0727). These results indicated that cytokines besides TNF-α are probably involved in the observed changes in colonic goblet cells during C. rodentium infection. We have previously shown that gamma interferon expression is lower in Rag1 KO mice than in C57BL/6 mice during infection (66), but recent studies have shown that IL-17A (IL-17), a proinflammatory T-cell-derived cytokine that has been reported to directly affect intestinal epithelial function (54), is also upregulated in mice with C. rodentium-induced colitis (42). Therefore, we compared IL-17 mRNA levels in C57BL/6 and Rag1 KO mice. As expected, we found that in infected C57BL/6 mice, IL-17 mRNA levels were significantly increased and were more than 100-fold greater than the levels in uninfected mice; in contrast, IL-17 expression in Rag1 KO mice did not increase to levels greater than those in uninfected C57BL/6 control mice (Fig. 8), despite the greater numbers of bacteria in the Rag1 KO mice (Fig. 6F). These results, coupled with those of previous studies (66), show that effector T-cell cytokines are highly expressed at times when goblet cell depletion is apparent and may play a direct or indirect role in functional modulation of goblet cells during infection.
FIG. 8.

Cytokine gene expression during C. rodentium infection: quantitative RT-PCR analysis of TNF-α and IL-17 expression in distal colons taken from C57BL/6 and Rag1 KO at day 10 following C. rodentium infection. Induction of TNF-α was moderately increased in both strains at day 10 p.i.; the levels in C57BL/6 mice were increased, but the difference was not significant (P = 0727). However, IL-17 gene expression was significantly increased in infected C57BL/6 mice compared to the minimal IL-17 expression observed in infected Rag1 KO mice. All samples were normalized to β-actin. The bars indicate the means for eight C57BL/6 mice and four Rag1 KO mice from three independent infections. The error bars indicate the standard errors of the means. Asterisk, P < 0.05, as determined by the Mann-Whitney t test.
Adoptive transfer of T and B lymphocytes rescues the goblet cell depletion phenotype in Rag1 KO mice.
While the results described above indicated that a functional adaptive immune system was important for meditating the observed goblet cell depletion phenotype, we tested this hypothesis directly by reconstituting Rag1 KO mice with T and B lymphocytes isolated from spleens and MLNs of C57BL/6 mice (using PBS as a negative control) and subsequently challenging them with C. rodentium. At day 10 p.i., PBS-treated (nonreconstituted) mice or mice reconstituted with T and B lymphocytes were euthanized, and the distal colons were used for histological assessment of goblet cells via PAS staining, as well as the presence of infiltrating lymphocytes via CD3 (T-cell) and B220 (B-cell) staining. Consistent with the hypothesis that T and/or B cells mediate the goblet cell depletion phenotype, we observed reduced overall PAS staining within crypts of the reconstituted mice compared with crypts of nonreconstituted mice (Fig. 9A and B). Examination of the T- and B-lymphocyte populations within these tissues revealed that while B220-positive cells were concentrated mainly in isolated lymphoid follicles found only in reconstituted mice (data not shown), the goblet cell depletion seen in the reconstituted mice was associated with a prominent population of CD3-positive cells in the mucosa and submucosa of the reconstituted mice (Fig. 9C) but not the nonreconstituted mice (Fig. 9D). Moreover, the reduced PAS staining and the large CD3-positive cell population in distal colons of infected reconstituted mice at day 10 p.i. were also accompanied by a reduction in Muc2 mRNA expression and a significant reduction in Tff3 mRNA expression compared to infected nonreconstituted mice (Fig. 9E). Lastly, similar to the results for infected C57BL/6 mice, we observed significantly greater expression of IL-17 mRNA and a slight, nonsignificant increase in TNF-α expression in reconstituted mice compared to infected nonreconstituted mice at day 10 p.i. (Fig. 9F). Together, these results show that the adaptive immune response, presumably through the actions of T lymphocytes, plays a central role in regulating goblet cell gene expression, with downstream effects on goblet cell function and morphology during infection by a noninvasive enteric pathogen.
FIG. 9.

Adaptive transfer of T and B cells into Rag1 KO mice restored the goblet cell depletion phenotype during C. rodentium infection. (A and B) PAS staining of distal colons removed on day 10 p.i. from Rag1 KO mice that were reconstituted with T and B lymphocytes from naïve C57BL/6 mice (Recon) (A) or with PBS (nonreconstituted control) (B). The arrows indicate intact crypts exhibiting reduced PAS staining and numerous hypotrophic goblet cells. The asterisks indicate crypts magnified in the insets. Note that reduced PAS staining in PBS-treated mice (B) was associated mainly with crypts that were severely disrupted due to bacterial overload (arrowheads), which was not observed in infected reconstituted mice. Original magnification, ×100. Bar = 100 μm. (C and D) Anti-CD3 staining showing greater numbers of infiltrating CD3-positive cells in the lamina propria and submucosa of reconstituted Rag1 KO mice (arrows) (C) than in the lamina propria and submucosa of PBS-treated mice (D). Original magnification, ×200. Bar = 50 μm. (E and F) Quantitative RT-PCR analysis of expression of genes encoding Muc2 and Tff3 (E) or TNF-α and IL-17 (F) in distal colons of reconstituted or nonreconstituted (PBS-treated) mice at day 10 p.i. All samples were normalized to β-actin. The bars indicate the means for seven reconstituted mice and three nonreconstituted mice from two independent infections. The error bars indicate the standard errors of the means. Asterisk, P < 0.05, as determined by the Mann-Whitney t test.
DISCUSSION
While goblet cell depletion during C. rodentium infection has been reported previously (38), our studies are the first studies to directly characterize this pathology and specifically address the underlying mechanisms. We show here that during C. rodentium-induced colitis, there is a significant reduction in the size of the mature goblet cell population in the distal colon during periods corresponding to heavy pathogen burden, and this is associated with reduced expression of the goblet cell-specific genes encoding Muc2 and Tff3 at the mRNA level and, histologically, at the protein level. We also demonstrate that in addition to colonocytes, goblet cells represent a target of C. rodentium infection, as the bacteria directly interact with and infect goblet cells. However, we show that only a portion of goblet cells in the murine colon are infected, and it is likely the host adaptive immune system that mediates the majority of the goblet cell depletion that occurs during C. rodentium infection.
The relationship between the histological changes in the goblet cell population and the alterations in goblet cell-specific gene expression seen in this model was intriguing and provided insight into how the immune system modulates goblet cells during infection. While infection did lead to a significant reduction in the total number of PAS-stained mucin-containing cells relative to other non-carbohydrate-producing cells, this in itself is unlikely to account for the dramatic reduction in Muc2 and Tff3 immunostaining that was observed. Rather, the prevalence of hypotrophic PAS-positive cells may reflect an altered state of goblet cell function that occurs in the distal colon during infection. As initially suggested by Makkink et al. for another model of colitis showing goblet cell depletion (41), the reduced Muc2 protein expression may have been directly responsible for the hypotrophic goblet cell phenotype that we observed, since Muc2 is the main morphological determinant of goblet cell morphology (41, 70). In fact, mice genetically deficient in this mucin lack phenotypically distinct goblet cells, yet they are strongly reactive for other goblet cell markers, like Tff3 (70). However, in addition to reduced Muc2 levels, we also found a marked reduction in Tff3 protein levels in C57BL/6 mice but not in Rag1 KO mice. Because Tff3 is thought to be expressed primarily by mature goblet cells (51), this suggests that the modulation of goblet cell function may reflect immune system-mediated impairment of the ability of immature goblet cells to fully differentiate into mature goblet cells.
The biological consequences of the functional modulation of goblet cells are currently unclear. In some respects it is paradoxical that the host immune system reduces expression of Muc2 and Tff3, considering the protective roles that these goblet cell-derived proteins play in the intestine. Muc2 was recently shown to be important in maintaining overall mucosal homeostasis, ultimately suppressing spontaneous tumor growth (70), as well as colitis development (68). In vitro studies have reported that intestinal mucins prevent EPEC attachment (40, 56) to epithelial cells, as well as bacterial translocation (17) across epithelial cell monolayers. Moreover, in mice lacking Tff3 the ability to heal colonic injury induced by the cytotoxic agent dextran sodium sulfate is impaired, and the mice suffer an exaggerated and fatal colitis as a result (45). Thus, down-regulating both these genes could compromise the host defense when an animal is challenged by an enteric bacterial pathogen.
On the other hand, the impact of the loss of these goblet cell-derived factors may be minimal or even beneficial to the host during infection with enteric bacteria. For example, similar to commensal species (57), enteropathogenic bacteria such as Yersinia enterocolitica have been demonstrated to use carbohydrate-laden mucins as a food source (43), and S. enterica serovar Typhimurium is thought to bind to intestinal mucins to facilitate colonization (73). Thus, reducing mucin production might be important for reducing energy sources for the pathogenic bacteria, as well as for reducing potential anchoring sites required for initial colonization. Indeed, as C. rodentium constitutes approximately 90% of the bacterial flora at the peak of infection (38), reducing a potential nutrient source, such as mucins, may inhibit pathogen growth. In this regard, the robust mature goblet cell population observed in infected Rag1 KO mice may facilitate the increased bacterial burdens that are observed within the colons of these mice and perhaps even the previously described impaired clearance of the pathogen (66).
The mechanisms underlying the immune system-driven loss of mature Muc2- and Tff3-expressing goblet cells are currently unclear; however, goblet cell depletion has also been observed in human colonic tissues in association with hyperproliferation of colonic crypts, in a manner dependent upon activation of T cells within the lamina propria (15). While infection-induced alterations in the turnover of the colonic epithelium may be involved in the observed goblet cell depletion, we observed that both C57BL/6 mice and Rag1 KO mice showed evidence of colonic hyperplasia during infection, consistent with previous reports by workers in our lab (66) and other groups (55). We also found that the numbers of epithelial cells within elongated crypts were not significantly different in the two mouse strains following infection (unpublished observations). Indeed, the relationship between crypt hyperplasia and goblet cell depletion phenotypes observed in other models of intestinal inflammation appears to be complex: for example, the immune system-mediated pathology that occurs during murine helminth infections results in both crypt and goblet cell hyperplasia (2, 34, 35). Moreover, in SAMP1/YitFc mice which develop spontaneous Crohn's disease-like ileitis (72), inflammation-induced crypt elongation is associated with the expansion of secretory lineages, including Paneth and goblet cells (72). Taken together, these studies suggest that the mechanisms underlying the depletion of mature goblet cells during C. rodentium infection may involve processes that are independent of, but coordinated with, the induction of crypt hyperplasia.
The concept of immunomodulation of goblet cells during enteric bacterial infection is intriguing in light of the role that the immune system plays in modulating goblet cell function when it is faced with other intestinal challenges. For example, the robust Th2 response that typically accompanies intestinal nematode infections induces goblet cell hyperplasia (34) and goblet cell-specific gene expression, which are thought to contribute to host defense (3, 34). In contrast, we observed loss of the mature goblet cell phenotype. Given that Th1 responses (26) and, more recently, Th17 responses (42) have been linked to C. rodentium infection, it is possible that these polarized T-helper-type responses are specifically responsible for mediating the loss of the goblet cell phenotype. In this regard, while we observed increased IL-17 expression during infection in immunocompetent mice compared to Rag1 KO mice, we have in previous studies observed a similar trend for gamma interferon expression (66). Therefore, further studies are needed to address which T-cell subsets and potentially which cytokine(s) are specifically responsible for the modulation of goblet cell function during C. rodentium infection.
In addition, our report of direct infection of goblet cells in vivo by C. rodentium reflects a novel and intriguing host-pathogen interaction in the intestine that may have consequences for local colonization by this pathogen. Given that C. rodentium, like EPEC and EHEC, can subvert enterocyte function, it is tempting to speculate that these pathogens can also subvert the function of intestinal goblet cells; however, this possibility has yet to be explored in detail. Furthermore, the evidence of possible bacterial internalization within goblet cells is intriguing, although its significance and frequency of occurrence remain unclear. While it seems counterintuitive that cells as specialized for secretion as goblet cells could be involved in uptake of luminal contents, it is interesting to note that rodent colonic goblet cells have been observed to internalize their apical membrane along with experimentally injected luminal cationic ferritin via endocytosis (9). Clearly, the interactions between enteric pathogens like C. rodentium and goblet cells reflect a dynamic host-bacterium interaction, with the goblet cells exposed to A/E effectors and C. rodentium directly exposed to proteins secreted by the goblet cells.
In conclusion, we demonstrate here that although goblet cells are infected by C. rodentium, they are also subject to functional modulation by the host immune system during in vivo infection by this A/E bacterial pathogen. As the host can utilize goblet cells for protection against an array of challenges, understanding how the host modulates goblet cells during A/E and other bacterial challenges should help us determine what role these important cell types play during infectious colitis and during maladaptive responses against normal microflora, such as those observed during human inflammatory bowel disease.
Acknowledgments
We gratefully acknowledge Jan Dekker for providing the anti-murine Muc2 antisera, Daniel Podolsky for providing the anti-Tff3 antisera, and Wanyin Deng for providing the anti-C. rodentium Tir antisera. We also thank Mehran Ghoreishi for help with the reconstitution experiments and Sharon Edwards for help with the immunohistochemistry.
This study was supported by operating grants from the Canadian Institutes for Health Research and the Crohn's and Colitis Foundation of Canada to B.A.V. B.A.V. is the Children with Intestinal and Liver Disorders (CHILD) Foundation Research Scholar, a Michael Smith Foundation for Health Research Scholar, and the Canada Research Chair in Pediatric Gastroenterology. K.S.B.B. was supported by an MSFHR graduate scholarship. J.A.G. is a CAG/CIHR/AstraZeneca and MSFHR Postdoctoral Fellow.
Editor: J. F. Urban, Jr.
Footnotes
Published ahead of print on 5 November 2007.
REFERENCES
- 1.Arnold, J. W., G. R. Klimpel, and D. W. Niesel. 1993. Tumor necrosis factor (TNFα) regulates intestinal mucus production during salmonellosis. Cell. Immunol. 151336-344. [DOI] [PubMed] [Google Scholar]
- 2.Artis, D., C. S. Potten, K. J. Else, F. D. Finkelman, and R. K. Grencis. 1999. Trichuris muris: host intestinal epithelial cell hyperproliferation during chronic infection is regulated by interferon-gamma. Exp. Parasitol. 92144-153. [DOI] [PubMed] [Google Scholar]
- 3.Artis, D., M. L. Wang, S. A. Keilbaugh, W. He, M. Brenes, G. P. Swain, P. A. Knight, D. D. Donaldson, M. A. Lazar, H. R. P. Miller, G. A. Schad, P. Scott, and G. D. Wu. 2004. RELMβ/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract Proc. Natl. Acad. Sci. USA 10113596-13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle, E. C., N. F. Brown, and B. B. Finlay. 2006. Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell. Microbiol. 81946-1957. [DOI] [PubMed] [Google Scholar]
- 5.Brand, S., F. Beigel, T. Olszak, K. Zitzmann, S. T. Eichhorst, J.-M. Otte, J. Diebold, H. Diepolder, B. Adler, C. J. Auernhammer, B. Goke, and J. Dambacher. 2005. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am. J. Physiol. Gastrointest. Liver Physiol. 289G960-G968. [DOI] [PubMed] [Google Scholar]
- 6.Celli, J., W. Deng, and B. B. Finlay. 2000. Enteropathogenic Escherichia coli (EPEC) attachment to epithelial cells: exploiting the host cell cytoskeleton from the outside. Cell. Microbiol. 21-9. [DOI] [PubMed] [Google Scholar]
- 7.Clarke, S. C., R. D. Haigh, P. P. E. Freestone, and P. H. Williams. 2003. Virulence of enteropathogenic Escherichia coli, a global pathogen. Clin. Microbiol. Rev. 16365-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cliffe, L. J., N. E. Humphreys, T. E. Lane, C. S. Potten, C. Booth, and R. K. Grencis. 2005. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science 3081463-1465. [DOI] [PubMed] [Google Scholar]
- 9.Colony, P. C., and R. D. Specian. 1987. Endocytosis and vesicular traffic in fetal and adult colonic goblet cells. Anat. Rec. 218365-372. [DOI] [PubMed] [Google Scholar]
- 10.Dean, P., and B. Kenny. 2004. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol. Microbiol. 54665-675. [DOI] [PubMed] [Google Scholar]
- 11.Deng, W., Y. Li, B. A. Vallance, and B. B. Finlay. 2001. Locus of enterocyte effacement from Citrobacter rodentium: sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Infect. Immun. 696323-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng, W., J. L. Puente, S. Gruenheid, Y. Li, B. A. Vallance, A. Vazquez, J. Barba, J. A. Ibarra, P. O'Donnell, P. Metalnikov, K. Ashman, S. Lee, D. Goode, T. Pawson, and B. B. Finlay. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. USA 1013597-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng, W., B. A. Vallance, Y. Li, J. L. Puente, and B. B. Finlay. 2003. Citrobacter rodentium translocated intimin receptor (Tir) is an essential virulence factor needed for actin condensation, intestinal colonization and colonic hyperplasia in mice. Mol. Microbiol. 4895-115. [DOI] [PubMed] [Google Scholar]
- 14.Deplancke, B., and H. R. Gaskins. 2001. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am. J. Clin. Nutr. 731131S-1141. [DOI] [PubMed] [Google Scholar]
- 15.Evans, C. M., A. D. Phillips, J. A. Walker-Smith, and T. T. MacDonald. 1992. Activation of lamina propria T cells induces crypt epithelial proliferation and goblet cell depletion in cultured human fetal colon. Gut 33230-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frankel, G., A. D. Phillips, I. Rosenshine, G. Dougan, J. B. Kaper, and S. Knutton. 1998. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol. Microbiol. 30911-921. [DOI] [PubMed] [Google Scholar]
- 17.Gork, A. S., N. Usui, E. Ceriati, R. A. Drongowski, M. D. Epstein, A. G. Coran, and C. M. Harmon. 1999. The effect of mucin on bacterial translocation in I-407 fetal and Caco-2 adult enterocyte cultured cell lines. Pediatr. Surg. Int. 15155-159. [DOI] [PubMed] [Google Scholar]
- 18.Gruenheid, S., I. Sekirov, N. A. Thomas, W. Deng, P. O'Donnell, D. Goode, Y. Li, E. A. Frey, N. F. Brown, P. Metalnikov, T. Pawson, K. Ashman, and B. B. Finlay. 2004. Identification and characterization of NleA, a non-LEE-encoded type III translocated virulence factor of enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 511233-1249. [DOI] [PubMed] [Google Scholar]
- 19.Guttman, J. A., Y. Li, M. E. Wickham, W. Deng, A. W. Vogl, and B. B. Finlay. 2006. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell. Microbiol. 8634-645. [DOI] [PubMed] [Google Scholar]
- 20.Guttman, J. A., F. N. Samji, Y. Li, W. Deng, A. Lin, and B. B. Finlay. 2007. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell. Microbiol. 9131-141. [DOI] [PubMed] [Google Scholar]
- 21.Guttman, J. A., F. N. Samji, Y. Li, A. W. Vogl, and B. B. Finlay. 2006. Evidence that tight junctions are disrupted due to intimate bacterial contact and not inflammation during attaching and effacing pathogen infection in vivo. Infect. Immun. 746075-6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Handa, Y., M. Suzuki, K. Ohya, H. Iwai, N. Ishijima, A. J. Koleske, Y. Fukui, and C. Sasakawa. 2007. Shigella IpgB1 promotes bacterial entry through the ELMO-Dock180 machinery. Nat. Cell Biol. 9121-128. [DOI] [PubMed] [Google Scholar]
- 23.Happel, K. I., E. A. Lockhart, C. M. Mason, E. Porretta, E. Keoshkerian, A. R. Odden, S. Nelson, and A. J. Ramsay. 2005. Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect. Immun. 735782-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heller, F., P. Florian, C. Bojarski, J. Richter, M. Christ, B. Hillenbrand, J. Mankertz, A. Gitter, N. Bürgel, M. Fromm, M. Zeitz, I. Fuss, W. Strober, and J. D. Schulzke. 2005. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129550-564. [DOI] [PubMed] [Google Scholar]
- 25.Higgins, L. M., G. Frankel, I. Connerton, N. S. Goncalves, G. Dougan, and T. T. MacDonald. 1999. Role of bacterial intimin in colonic hyperplasia and inflammation. Science 285588-591. [DOI] [PubMed] [Google Scholar]
- 26.Higgins, L. M., G. Frankel, G. Douce, G. Dougan, and T. T. MacDonald. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect. Immun. 673031-3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinz, M., H. Schwegler, C. E. Chwieralski, G. Laube, R. Linke, W. Pohle, and W. Hoffmann. 2004. Trefoil factor family (TFF) expression in the mouse brain and pituitary: changes in the developing cerebellum. Peptides 25827-832. [DOI] [PubMed] [Google Scholar]
- 28.Kaper, J. B. 1998. Enterohemorrhagic Escherichia coli. Curr. Opin. Microbiol. 1103-108. [DOI] [PubMed] [Google Scholar]
- 29.Karam, S. M. 1999. Lineage commitment and maturation of epithelial cells in the gut. Front. Biosci. 4D286-D298. [DOI] [PubMed] [Google Scholar]
- 30.Katz, J. P., N. Perreault, B. G. Goldstein, C. S. Lee, P. A. Labosky, V. W. Yang, and K. H. Kaestner. 2002. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 1292619-S2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly, M., E. Hart, R. Mundy, O. Marches, S. Wiles, L. Badea, S. Luck, M. Tauschek, G. Frankel, R. M. Robins-Browne, and E. L. Hartland. 2006. Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infect. Immun. 742328-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenny, B., S. Ellis, A. D. Leard, J. Warawa, H. Mellor, and M. A. Jepson. 2002. Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol. Microbiol. 441095-1107. [DOI] [PubMed] [Google Scholar]
- 33.Khan, M. A., C. Ma, L. A. Knodler, Y. Valdez, C. M. Rosenberger, W. Deng, B. B. Finlay, and B. A. Vallance. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect. Immun. 742522-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan, W. I., P. Blennerhasset, C. Ma, K. I. Matthaei, and S. M. Collins. 2001. Stat6 dependent goblet cell hyperplasia during intestinal nematode infection. Parasite Immunol. 2339-42. [DOI] [PubMed] [Google Scholar]
- 35.Khan, W. I., and S. M. Collins. 2004. Immune-mediated alteration in gut physiology and its role in host defence in nematode infection. Parasite Immunol. 26319-326. [DOI] [PubMed] [Google Scholar]
- 36.Kindon, H., C. Pothoulakis, L. Thim, K. Lynch-Devaney, and D. Podolsky. 1995. Trefoil peptide protection of intestinal epithelial barrier function: cooperative interaction with mucin glycoprotein. Gastroenterology 109516-523. [DOI] [PubMed] [Google Scholar]
- 37.Lambert, M. E., P. F. Schofield, A. G. Ironside, and B. K. Mandal. 1979. Campylobacter colitis. Br. Med. J. i857-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luperchio, S. A., and D. B. Schauer. 2001. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 3333-340. [DOI] [PubMed] [Google Scholar]
- 39.Ma, C., M. E. Wickham, J. A. Guttman, W. Deng, J. Walker, K. L. Madsen, K. Jacobson, W. A. Vogl, B. B. Finlay, and B. A. Vallance. 2006. Citrobacter rodentium infection causes both mitochondrial dysfunction and intestinal epithelial barrier disruption in vivo: role of mitochondrial associated protein (Map). Cell. Microbiol. 81669-1686. [DOI] [PubMed] [Google Scholar]
- 40.Mack, D. R., S. Michail, S. Wei, L. McDougall, and M. A. Hollingsworth. 1999. Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 276G941-G950. [DOI] [PubMed] [Google Scholar]
- 41.Makkink, M. K., N. M. J. Schwerbrock, M. Mähler, J. A. Boshuizen, I. B. Renes, M. Cornberg, H. J. Hedrich, A. W. C. Einerhand, H. A. Büller, S. Wagner, M. L. Enss, and J. Dekker. 2002. Fate of goblet cells in experimental colitis. Dig. Dis. Sci. 472286-2297. [DOI] [PubMed] [Google Scholar]
- 42.Mangan, P. R., L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006. Transforming growth factor-β induces development of the TH17 lineage. Nature 441231-234. [DOI] [PubMed] [Google Scholar]
- 43.Mantle, M., and C. Rombough. 1993. Growth in and breakdown of purified rabbit small intestinal mucin by Yersinia enterocolitica. Infect. Immun. 614131-4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maresca, M., D. Miller, S. Quitard, P. Dean, and B. Kenny. 2005. Enteropathogenic Escherichia coli (EPEC) effector-mediated suppression of antimicrobial nitric oxide production in a small intestinal epithelial model system. Cell. Microbiol. 71749-1762. [DOI] [PubMed] [Google Scholar]
- 45.Mashimo, H., D.-C. Wu, D. K. Podolsky, and M. C. Fishman. 1996. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274262-265. [DOI] [PubMed] [Google Scholar]
- 46.Moncada, D. M., S. J. Kammanadiminti, and K. Chadee. 2003. Mucin and Toll-like receptors in host defense against intestinal parasites. Trends Parasitol. 19305-311. [DOI] [PubMed] [Google Scholar]
- 47.Nagai, T., A. Abe, and C. Sasakawa. 2005. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J. Biol. Chem. 2802998-3011. [DOI] [PubMed] [Google Scholar]
- 48.Nair, M. G., K. J. Guild, and D. Artis. 2006. Novel effector molecules in type 2 inflammation: lessons drawn from helminth infection and allergy. J. Immunol. 1771393-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nataro, J. P., and J. B. Kaper. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11142-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nougayrede, J.-P., P. J. Fernandes, and M. S. Donnenberg. 2003. Adhesion of enteropathogenic Escherichia coli to host cells. Cell. Microbiol. 5359-372. [DOI] [PubMed] [Google Scholar]
- 51.Podolsky, D. K., K. Lynch-Devaney, J. L. Stow, P. Oates, B. Murgue, M. DeBeaumont, B. E. Sands, and Y. R. Mahida. 1993. Identification of human intestinal trefoil factor. Goblet cell-specific expression of a peptide targeted for apical secretion. J. Biol. Chem. 2686694-6702. [PubMed] [Google Scholar]
- 52.Renes, I. B., M. Verburg, D. J. Van Nispen, J. A. Taminiau, H. A. Buller, J. Dekker, and A. W. Einerhand. 2002. Epithelial proliferation, cell death, and gene expression in experimental colitis: alterations in carbonic anhydrase I, mucin MUC2, and trefoil factor 3 expression. Int. J. Colorectal Dis. 17317-326. [DOI] [PubMed] [Google Scholar]
- 53.Sachdev, H. P., V. Chadha, V. Malhotra, A. Verghese, and R. K. Puri. 1993. Rectal histopathology in endemic Shigella and Salmonella diarrhea. J. Pediatr. Gastroenterol. Nutr. 1633-38. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz, S., J. F. Beaulieu, and F. M. Ruemmele. 2005. Interleukin-17 is a potent immuno-modulator and regulator of normal human intestinal epithelial cell growth. Biochem. Biophys. Res. Commun. 337505-509. [DOI] [PubMed] [Google Scholar]
- 55.Simmons, C. P., S. Clare, M. Ghaem-Maghami, T. K. Uren, J. Rankin, A. Huett, R. Goldin, D. J. Lewis, T. T. MacDonald, R. A. Strugnell, G. Frankel, and G. Dougan. 2003. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 715077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith, C. J., J. B. Kaper, and D. R. Mack. 1995. Intestinal mucin inhibits adhesion of human enteropathogenic Escherichia coli to HEp-2 cells. J. Pediatr. Gastroenterol. Nutr. 21269-276. [DOI] [PubMed] [Google Scholar]
- 57.Sonnenburg, J. L., J. Xu, D. D. Leip, C.-H. Chen, B. P. Westover, J. Weatherford, J. D. Buhler, and J. I. Gordon. 2005. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 3071955-1959. [DOI] [PubMed] [Google Scholar]
- 58.Specian, R. D., and M. R. Neutra. 1981. The surface topography of the colonic crypt in rabbit and monkey. Am. J. Anat. 160461-472. [DOI] [PubMed] [Google Scholar]
- 59.Specian, R. D., and M. G. Oliver. 1991. Functional biology of intestinal goblet cells. Am. J. Physiol. Cell Physiol. 260C183-C193. [DOI] [PubMed] [Google Scholar]
- 60.Steinberg, S. E., J. G. Banwell, J. H. Yardley, G. T. Keusch, and T. R. Hendrix. 1975. Comparison of secretory and histological effects of shigella and cholera enterotoxins in rabbit jejunum. Gastroenterology 68309-317. [PubMed] [Google Scholar]
- 61.Stevens, J. M., E. E. Galyov, and M. P. Stevens. 2006. Actin-dependent movement of bacterial pathogens. Nat. Rev. Microbiol. 491-101. [DOI] [PubMed] [Google Scholar]
- 62.Sugawara, I., H. Yamada, C. Li, S. Mizuno, O. Takeuchi, and S. Akira. 2003. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol. Immunol. 47327-336. [DOI] [PubMed] [Google Scholar]
- 63.Taupin, D., and D. K. Podolsky. 2003. Trefoil factors: initiators of mucosal healing. Nat. Rev. Mol. Cell Biol. 4721-732. [DOI] [PubMed] [Google Scholar]
- 64.Taupin, D. R., K. Kinoshita, and D. K. Podolsky. 2000. Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc. Natl. Acad. Sci. USA 97799-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vallance, B. A., W. Deng, M. De Grado, C. Chan, K. Jacobson, and B. B. Finlay. 2002. Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen Citrobacter rodentium in infected mice. Infect. Immun. 706424-6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vallance, B. A., W. Deng, L. A. Knodler, and B. B. Finlay. 2002. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 702070-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vallance, B. A., and B. B. Finlay. 2000. Exploitation of host cells by enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 978799-8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van der Sluis, M., B. A. E. De Koning, A. C. J. M. De Bruijn, A. Velcich, J. P. P. Meijerink, J. B. Van Goudoever, H. A. Buller, J. Dekker, I. Van Seuningen, I. B. Renes, and A. W. C. Einerhand. 2006. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131117-129. [DOI] [PubMed] [Google Scholar]
- 69.Van Klinken, B. J.-W., A. W. C. Einerhand, L. A. Duits, M. K. Makkink, K. M. A. J. Tytgat, I. B. Renes, M. Verburg, H. A. Buller, and J. Dekker. 1999. Gastrointestinal expression and partial cDNA cloning of murine Muc2. Am. J. Physiol. Gastrointest Liver Physiol. 276G115-G124. [DOI] [PubMed] [Google Scholar]
- 70.Velcich, A., W. Yang, J. Heyer, A. Fragale, C. Nicholas, S. Viani, R. Kucherlapati, M. Lipkin, K. Yang, and L. Augenlicht. 2002. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2951726-1729. [DOI] [PubMed] [Google Scholar]
- 71.Reference deleted.
- 72.Vidrich, A., J. M. Buzan, S. Barnes, B. K. Reuter, K. Skaar, C. Ilo, F. Cominelli, T. Pizarro, and S. M. Cohn. 2005. Altered epithelial cell lineage allocation and global expansion of the crypt epithelial stem cell population are associated with ileitis in SAMP1/YitFc mice. Am. J. Pathol. 1661055-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vimal, D., M. Khullar, S. Gupta, and N. Ganguly. 2000. Intestinal mucins: the binding sites for Salmonella typhimurium. Mol. Cell. Biochem. 204107-117. [DOI] [PubMed] [Google Scholar]
- 74.Wales, A. D., M. J. Woodward, and G. R. Pearson. 2005. Attaching-effacing bacteria in animals. J. Comp. Pathol. 1321-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang, F., W. V. Graham, Y. Wang, E. D. Witkowski, B. T. Schwarz, and J. R. Turner. 2005. Interferon-γ and tumor necrosis factor-α synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 166409-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wiles, S., K. M. Pickard, K. Peng, T. T. MacDonald, and G. Frankel. 2006. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect. Immun. 745391-5396. [DOI] [PMC free article] [PubMed] [Google Scholar]