

Abstract
Francisella tularensis can cause severe disseminated disease after respiratory infection. The identification of factors involved in mortality or recovery following induction of tularemia in the mouse will improve our understanding of the natural history of this disease and facilitate future evaluation of vaccine candidate preparations. BALB/c mice were infected intranasally with the live vaccine strain (LVS) of F. tularensis subsp. holarctica and euthanized at different stages of disease to analyze the induction of immune molecules, gross anatomical features of organs, bacterial burdens, and progression of the histopathological changes in lung and spleen. Tissue-specific interleukin-6 (IL-6), macrophage inflammatory protein 2, and monocyte chemotactic protein 1 were immune markers of mortality, while anti-LVS immunoglobulin M and IL-1β were associated with survival. Moribund mice had enlarged spleens and lungs, while surviving mice had even more prominent splenomegaly and normal-appearing lungs. Histopathology of the spleens of severely ill mice was characterized by disrupted lymphoid follicles and fragmented nuclei, while the spleens of survivors appeared healthy but with increased numbers of megakaryocytes and erythrocytes. Histopathology of the lungs of severely ill mice indicated severe pneumonia. Lungs of survivors at early time points showed increased inflammation, while at late times they appeared healthy with peribronchial lymphoid aggregates. Our results suggest that host immune factors are able to affect bacterial dissemination after respiratory tularemia, provide new insights regarding the pathological characteristics of pulmonary tularemia leading to systemic disease, and potentially identify immune markers associated with recovery from the disease.
Francisella tularensis is a gram-negative facultative intracellular bacterium that causes tularemia. Different clinical presentations of tularemia exist, including ulceroglandular, glandular, oropharyngeal, typhoidal, and pneumonic. Approximately 200 cases of tularemia are reported in the United States every year. Pneumonia presents in 10% to 20% of cases as a primary event or a complication of ulceroglandular or typhoidal tularemia (22). Pneumonic tularemia can also occur with other forms, like glandular, oculoglandular, and oropharyngeal, as a result of secondary bacteremic spread to the lungs. Both primary pneumonic tularemia and pneumonia complicating the typhoidal form can occur 2 days to several months after infection. If left untreated, respiratory tularemia can progress to respiratory failure, liver, kidney, and splenic involvement, meningitis, septic shock, and death in up to 30 to 60% of cases. The number of tularemia cases reported worldwide is relatively low, but the risk of the use of F. tularensis as a biological weapon has encouraged research on its pathogenesis, as well as the development of effective vaccines and therapeutics against the disease.
Respiratory tularemia has been studied in murine models after inoculation with either the live vaccine strain (LVS) of F. tularensis subsp. holarctica or type A F. tularensis strains via the intranasal route (42) or the aerosol route (11). F. tularensis spreads from the lungs to organs, such as the liver and the spleen, with several pathological consequences, including severe inflammation and areas of necrosis (11). More recently, disseminated tularemia with the involvement of the spleen and liver has been described in an oral mouse model after initial infection of the mesenteric lymph nodes (25).
Research has also been carried out to evaluate the factors involved in both innate and adaptive immune responses to F. tularensis infection. Examples of these reports include studies of the involvement of Toll-like receptors 2 and 4 in tularemia induced in the mouse via the respiratory or the intradermal route (6, 9, 10, 28), analysis of proinflammatory gene expression following murine intraperitoneal or intradermal infection (8), and research on the contributions of B and T lymphocytes to protection and response in experimental tularemia (5, 14, 15). It is still unclear what exactly triggers the host to develop severe symptoms due to dissemination and fatal disease and what mechanisms are involved in initial protection prior to the initiation of acquired immune responses.
Less effort has been made to understand what factors are involved in bacterial clearance following inhalation of high F. tularensis doses and the long-term consequences tularemia might have on vital organs following spontaneous recovery. In this work, studies were conducted using the respiratory mouse model of tularemia, induced by the LVS of F. tularensis, and the features of pulmonary disease followed by dissemination to peripheral organs were investigated, focusing on mice with terminal tularemia and mice that fully recovered from disease. Our results will improve the understanding of the natural history of the disease and increase our knowledge of potential protective immunity against the disease, facilitating our search for improved vaccine strategies.
MATERIALS AND METHODS
Preparation of inoculum.
F. tularensis subsp. holarctica LVS (ATCC 29684) was obtained from the CDC, Fort Collins, CO. LVS was grown on chocolate agar plates (Remel, Lenexa, KS) for 72 h at 37°C to form a layer, and colonies were scraped off and resuspended in sterile phosphate-buffered saline (PBS) to an optical density at 600 nm (OD600) of 0.3. CFU were determined by plating out 100 μl from 10-fold dilutions on chocolate agar plates after incubation at 37°C for 72 h. Bacterial suspensions were prepared, and CFU counts were confirmed for each experiment.
Mice and LVS respiratory infection.
Female BALB/c mice, 7 to 10 weeks old, were obtained from Jackson Laboratories (Bar Harbor, ME) and given pelleted food and water ad libitum. All experimental procedures were in compliance with the guidelines of the Institutional Animal Care and Use Committee at the Boston University School of Medicine. The mice were anesthetized with ketamine HCl (Fort Dodge Animal Health, Fort Dodge, IA) and xylazine (Lloyd Laboratories, Shenandoah, IA) and challenged intranasally with 104 CFU of LVS in 20 μl of PBS under biological safety level 2 conditions. Mock-inoculated mice received 20 μl of PBS. All animals were closely observed until they had completely awakened from anesthesia.
Survival, symptoms, and tissue processing.
The mice were monitored daily for signs of disease. Typical signs included ruffled fur, hunched posture, decreased responsiveness to stimuli, eye discharge, and weight loss. Animal survival was recorded for up to 50 days postinfection, and in some experiments, death was considered an endpoint. In selected experiments, mice were humanely euthanized at 24, 48, 72, and 96 h postinfection or when severe disease was observed at day 7 postinfection (moribund). Animals recovered from disease signs and euthanized at days 7, 15, and 50 postinfection (survivors) were also included for analysis. Lungs and spleens were harvested aseptically and weighed, and their gross anatomy was observed and photographed (with a Canon EOS-20D digital camera). To determine bacterial loads, organs were homogenized in sterile PBS according to their size (1 ml of normal saline for each 70 mg of spleen or 150 mg of lung), and CFU per tissue weight was calculated by culture of lysates on chocolate agar plates after incubation at 37°C for 72 h. Similarly, blood was collected from the submandibular vein, diluted appropriately, and plated out for CFU counts. Results were expressed as log10 mean CFU/milligram of tissue or milliliter of blood.
Cytokine, chemokine, and antibody measurement.
Tissues were homogenized in PBS according to their weight and size (as described above) and centrifuged for 20 min at 1,500 × g, and the supernatants were collected, immediately placed on ice to minimize proteolysis, and stored at −80°C. Serum was obtained from blood by incubating samples for 10 min at room temperature and centrifuging them for 20 min at 1,500 × g. Upon use, samples were thawed at room temperature, diluted appropriately, and analyzed. Levels of monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein 2 (MIP-2), interleukin-6 (IL-6), IL-1β, and gamma interferon (IFN-γ) were measured in lung and spleen supernatants by using ELISA BD OptEIA (San Diego, CA) or R&D systems (Minneapolis, MN) kits, and plates were read at OD450 on an ELx800 enzyme-linked immunosorbent assay reader (Bio-Tek Instruments, Inc., Winooski, VT). Results were expressed as mean picograms/milliliter based on the standard curves provided by the manufacturers. To analyze LVS-specific immunoglobulin G (IgG) and IgM antibody levels, Immulon II microtiter plate wells were coated with 100 μl of whole bacteria (106 CFU/well) in PBS, incubated at 37°C for 3 h, and then stored overnight at 4°C. Supernatants were diluted in 0.05% PBS-Tween 20, added to the bacterium-coated wells, and incubated for 1 h at 37°C. Alkaline phosphatase-conjugated anti-mouse IgG or IgM (100 μl; Sigma, St. Louis, MO) diluted in PBS-Tween 20 was added, and the plates were incubated for 1 h at 37°C and washed. Color was developed with 100 μl 1-step p-nitrophenylphosphate (Pierce, Rockford, IL) and measured at OD405 on the ELx800 enzyme-linked immunosorbent assay reader. Colorimetric values were converted to nanograms/milliliter, based on the standard curves for IgG or IgM generated by using known concentrations of IgG and IgM on plates coated with goat anti-mouse IgG and IgM F(ab′)2-specific antibodies (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) as described previously (27). Levels of IL-6 and anti-LVS IgM in mouse sera were also determined.
Histopathology.
Lungs and spleens were removed aseptically. Formalin-fixed lungs and spleens were embedded in paraffin, and 5-μm sections were stained with standard hematoxylin and eosin and analyzed by light microscopy. The lungs were inflated with 10% buffered formalin through the trachea to preserve the tissue architecture. Histopathological analysis of tissues from moribund mice was conducted at either day 6 or 7 after intranasal infection, while for survivors, it was done either 7 days (early survivors) or 50 days (late survivors) after infection.
Statistical analysis.
Comparisons of groups were performed by the Mann-Whitney U nonparametrical test using GraphPad Prism version 4.02 (San Diego, CA). P values of ≤0.05 were considered significant, while P values of ≤0.01 indicated high significance.
RESULTS
Outcome of respiratory tularemia in the mouse model.
Respiratory tularemia followed by dissemination into distant organs, including the spleen, was consistently induced by intranasal inoculation of mice with 104 CFU of LVS. Infected mice showed signs of moderate disease (ruffled fur, hunched posture, or decreased responsiveness to stimuli) by 3 to 4 days following inoculation. At 5 to 8 days postinfection, approximately 70% of the mice developed severe disease and/or died (Fig. 1A), and weight loss was observed, in addition to the development of clinical symptoms (Fig. 1B). In initial experiments, mice were infected and closely observed for survival and disease signs. This led us to determine the distinction between mice reaching the terminal disease stage (moribund) and mice recovering from disease (survivors). A moribund animal was defined as one showing a deteriorating condition with the disease signs listed above, with the addition of severe eye discharge and the complete inability to respond to external stimuli. The frequency and accuracy of clinical-sign observation also revealed that at this terminal stage mice died within a few hours. A smaller percentage of the mice never reached the moribund stage, despite being symptomatic at earlier time points (days 3 to 4). These animals overcame the disease, gradually regained weight, and became asymptomatic (Fig. 1A and B). Recovery from the symptomatic stage was observed as early as 7 days postinoculation. In subsequent experiments, moribund and surviving mice were humanely sacrificed for measurement of parameters. For moribund mice, all analyses were performed at day 7 and for survivors at 7, 15, and 50 days postinoculation.
FIG. 1.
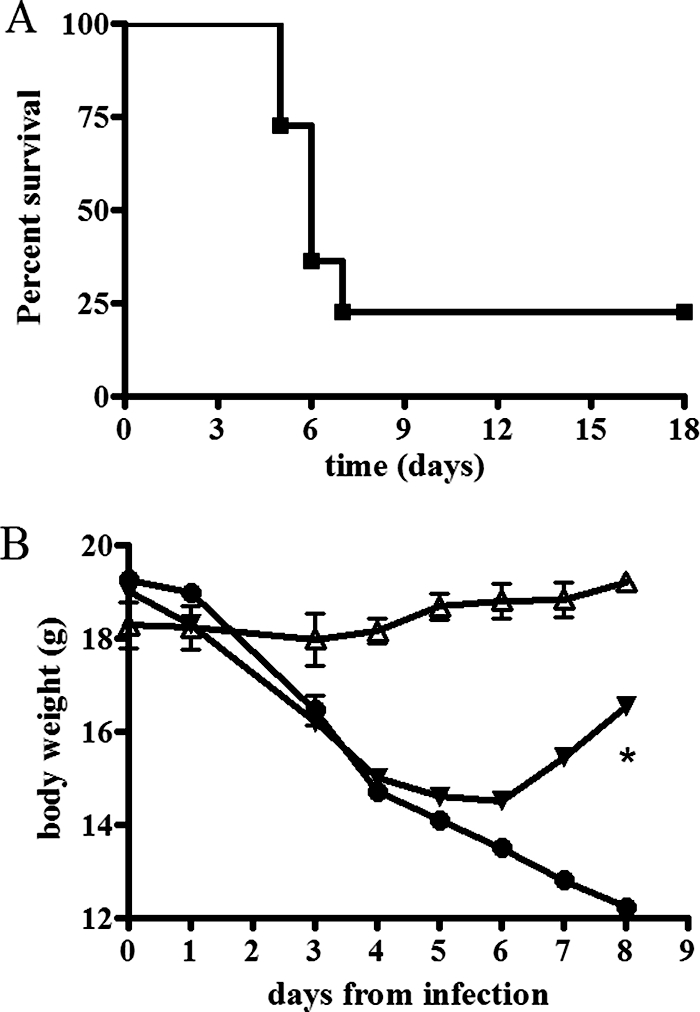
Survival and weight loss curves following pulmonary infection with F. tularensis LVS. Mice were inoculated via the intranasal route with 104 CFU of the LVS strain, and survival and body weight were monitored over time. (A) All mice showed disease signs by day 3 or 4, and approximately 75% became moribund and died between days 5 and 8, while a small percentage (∼25%) overcame the illness and recovered. In this representative experiment, a total of 22 mice were included, 5 of which were survivors. (B) Moribund mice showed a body weight loss of approximately 25% (n = 12; filled circles) compared to uninfected controls (n = 4; open triangles). Survivors also lost body weight concurrently with the appearance of clinical signs but regained it upon recovery (n = 3; filled inverted triangles). Differences in body weight between moribund mice and survivors were statistically significant at day 8 postinfection (*, P < 0.05). The error bars represent standard deviations.
Immunoglobulin, cytokine, and chemokine levels in tissues from moribund mice versus mice that had recovered from respiratory infection.
In supernatants of lungs and spleens obtained from moribund animals, IgM levels (36.2 ± 30.4 ng/ml in spleens and 10.4 ± 6.6 ng/ml in lungs) were similar to those of negative controls (not detectable in spleens and 37.14 ± 16.6 ng/ml in lungs), while in mice that recovered and survived, they reached average levels of 575.2 ± 283.3 ng/ml in spleen lysates and 335.9 ± 88.5 ng/ml in lung lysates (day 7), peaking at day 15 (816.8 ± 337.4 ng/ml for spleens and 845.7 ± 134.6 ng/ml for lungs), and returned to baseline levels at later time points (day 50) (Fig. 2A and B). Differences in antibody levels between tissues of moribund mice and survivors were statistically significant (P < 0.05 and P < 0.01). In mouse serum, IgM levels reflected the trend observed in target tissue lysates, although no significant differences were detected between moribund mice and survivors (data not shown). Anti-LVS IgG levels were detected only at later time points (day 50) in tissue supernatants of mice surviving disease (data not shown). Levels of MIP-2, MCP-1, and IL-6 were elevated above baseline in both lungs and spleens of moribund mice, while in tissues of animals surviving tularemic disease, the levels of these immune mediators were extremely low and similar to those observed in uninfected controls (Fig. 2C, D, E, and F and 3A and B). Differences in the levels of MCP-1 and IL-6 in the lysates of lungs from moribund mice compared to mice that survived infection were highly significant (P < 0.01). Levels of IL-6 in serum were also higher in moribund animals than in either infected mice that survived or uninfected controls, but the difference was not found to be statistically significant (data not shown). IL-1β production was detected in lungs and spleens of moribund mice, but it was significantly higher in mice that recovered and survived infection at day 7 (P < 0.05 and P < 0.01) (Fig. 3C and D). In mice surviving tularemia at days 15 and 50 postinfection, levels of IL-1β returned to baseline (Fig. 3C and D). In both lung and spleen tissue lysates, IFN-γ levels were elevated in both moribund mice and survivors on day 7, while no detectable amounts were measured in mice that survived at days 15 and 50 or in uninfected controls (Fig. 3E and F).
FIG. 2.

Antibody and chemokine responses in lungs and spleens of mice infected with LVS. Following intranasal infection with LVS, mice (three to six animals per group) were humanely sacrificed at a terminal disease stage or at different time points after recovering from symptoms. The results are shown in light gray for spleens and dark gray for lungs. Organs from mice injected with PBS were used as negative controls. Data are presented as the quantity (in ng or pg) of antibody or chemokine per milliliter at different stages of disease, and the error bars represent standard deviations. Differences between moribund mice and survivors were either significant (*, P < 0.05) or highly significant (**, P < 0.01). ND, not detectable.
FIG. 3.

Cytokine levels in lungs and spleens of mice infected with LVS. Following intranasal infection with LVS, supernatants from spleens (light-gray histograms) and lungs (dark-gray histograms) from moribund and survivor mice (three to six animals per group) were collected and analyzed for cytokine production. Organs from mice injected with PBS were used as negative controls. The data are reported as amounts (in pg) of cytokine per milliliter at different stages of disease, and the error bars represent standard deviations. Differences between moribund mice and survivors were either significant (*, P < 0.05) or highly significant (**, P < 0.01). ND, not detectable.
Organ enlargement and bacterial burdens in pulmonary and splenic tissues of moribund versus surviving mice.
Upon dissection, spleens from moribund mice were pale, brittle, visibly enlarged, and approximately twofold greater in weight than those of control mice (Fig. 4A and Table 1). In contrast, spleens from mice fully surviving infection showed slight discoloration and tissue changes, but their splenomegaly was even more pronounced, up to threefold larger than those of uninfected mice (Fig. 4C and Table 1). In fully recovered mice, tissue gross anatomy was observed at 7, 15, and 50 days postinfection, and the splenomegaly persisted up to the latest time point. Tissue discoloration in the enlarged spleens of survivors appeared less evident than that observed in moribund mice (Fig. 4C). The lungs of moribund mice were enlarged and greater in size (Table 2), while the tissue and dimensions of lungs in survivors appeared normal (Fig. 4B and D).
FIG. 4.

Changes in lung and spleen gross anatomy after infection with LVS. (A) Spleens collected from representative moribund mice appeared enlarged and pale (right) compared to those of uninfected controls (left). (B) Lungs from representative moribund mice did not collapse and showed some color variation (right) compared to those from uninfected controls (left). (C) Splenomegaly in representative survivors at early (day 7; center) and late (day 50; right) time points in comparison to normal spleens from uninfected control mice (left). (D) Lung tissue from survivors at both early (day 7; center) and late (day 50; right) times appeared normal in size and color, similar to those of uninfected controls (left). Bars = 5 mm.
TABLE 1.
Spleen characteristics at different stages of tularemia
| Mice | Organ wt (mg)a | Gross anatomy | Histopathology |
|---|---|---|---|
| Uninfected controls (n = 4) | 72.5 ± 5 | Normal | Normal |
| Moribund (n = 5) | 120 ± 12.2b | Pale, friable, visibly enlarged | Lymphocyte depletion, necrosis |
| Survivors | |||
| Day 7 (n = 6) | 185 ± 48.4c | No color variation, visibly enlarged | Erythrocytes in sinusoids, megakaryocytes |
| Day 50 (n = 3) | 186.6 ± 30.5c | No color variation, visibly enlarged | Erythrocytes in sinusoids, megakaryocytes |
Average weights ± standard deviations.
P < 0.05 compared to uninfected controls; Mann-Whitney U test.
P < 0.01 compared to uninfected controls; Mann-Whitney U test.
TABLE 2.
Lung features at different disease stages of tularemia
| Mice | Organ wt (mg)a | Gross anatomy | Histopathology |
|---|---|---|---|
| Uninfected controls (n = 4) | 175 ± 17.3 | Normal | Normal |
| Moribund (n = 5) | 308 ± 43.8b | Color variation, visibly enlarged | Diffuse edema, neutrophils and necrosis |
| Survivors | |||
| Day 7 (n = 6) | 226.6 ± 64.5 | Normal | Neutrophil and some lymphocyte infiltration, no fibrin deposits or edema |
| Day 50 (n = 3) | 246.6 ± 64.2 | Normal | Prominent lymphoid aggregates |
Average weights ± standard deviations.
P < 0.05 compared to uninfected controls; Mann-Whitney U test.
In selected experiments, animals were sacrificed at 24, 48, 72, and 96 h to determine changes of organ weight and bacterial burdens at early time points in both tissue lysates and blood (Fig. 5). A significant enlargement of spleens, compared to those from uninfected animals, was observed as early as 96 h postinfection (P < 0.05) (Fig. 5A). Compared to uninfected controls or moribund mice, spleen enlargement in mice that survived infection was highly significant (P < 0.01 and P < 0.05, respectively) (Fig. 5A). In splenic tissue, LVS CFU were detectable at 48 h postinfection and increased daily, reaching a mean of 4.4 × 106 ± 5.6 × 106 CFU/mg of tissue in moribund mice (Fig. 5C). Bacteria were not detected in any of the spleens from mice that survived past day 7 (Fig. 5C), but upon analysis of blood collected from all mice, LVS was detected over time, providing evidence that an infection had occurred (Fig. 5E). Bacteremia was detected from 48 h and persisted up to 96 h postinfection (Fig. 5E), and when approximately 75% of the mice became moribund, they showed a mean of 9 × 103 ± 5.2 ×103 CFU/ml of blood, while in those mice recovering from disease (∼25%), LVS was already cleared (Fig. 5E). Only moribund mice presented significantly enlarged lung tissue compared to controls (P < 0.05), and a significant difference in weight was also seen between lungs from moribund mice and lungs from mice that recovered from infection (P < 0.05) (Fig. 5B). Bacteria were found as early as 24 h postinfection in lung tissue, peaking at 2.2 × 106 ± 7.7 × 105 CFU/mg of tissue at the moribund stage. A lower number of bacteria (2.9 × 102 ± 1.4 ×102 CFU/mg of tissue) were found in lungs of recovered mice 7 days postinfection, but 15 and 50 days later, the infection was cleared (Fig. 5D).
FIG. 5.

Analysis of organ weight and enumeration of bacterial loads in tissue and blood after induction of tularemia. Following intranasal infection, mice (n = 3 to 6) were sacrificed at different time intervals. Lungs and spleens were dissected, weighed, and homogenized, and samples were serially diluted to determine LVS CFU by culture. The data are reported as weight per milligram and CFU per milligram of either spleen (A and C) or lung (B and D) tissue over time, and the error bars represent standard deviations (SD). (E) In a separate experiment, bacterial loads were also evaluated in blood from all mice (n = 25) collected at different time intervals (24 to 96 h). Also shown are CFU values detected in the blood of ∼75% of the mice that became moribund at day 7 (n = 18) and the ∼25% of survivor mice at days 7, 15, and 50 (n = 7). The data are expressed as CFU per milliliter, and the error bars represent SD. Differences in organ weight (mg) were either significant (*, P < 0.05) or highly significant (**, P < 0.01) in comparison to uninfected controls. “Survivor” data in panels A and B include organ weights determined at days 7, 15, and 50 postinfection (no differences were observed between the time points).
Histopathology of pulmonary and splenic tissue in moribund mice versus survivors.
Histolopathological analysis of spleens from moribund mice demonstrated depletion of lymphocytes and destruction of the follicular architecture. Nuclear fragments within the follicles and necrotic foci were also detected in spleen tissue (Fig. 6B). In contrast, the spleens of survivors appeared to be normal, with no alterations of the follicular architecture, but the splenic sinusoids were congested and a high number of megakaryocytes were observed (Fig. 6C). The elevated numbers of megakaryocytes were observed in the splenic tissues of survivors at both early and late time points (Fig. 6D).
FIG. 6.

Hematoxylin and eosin staining of formalin-fixed splenic tissue. Spleens were removed at different stages of disease. (A) Spleen from an uninfected control with no pathological changes. (B) Spleen from a representative moribund animal sacrificed at day 7 with lymphoid follicle disruption. The inset shows infiltration of fragmented nuclei, some neutrophils, and the absence of megakaryocytes. (C and D) Spleens from survivors sacrificed at early (day 7) (C) and late (day 50) (D) time points showed normal lymphoid follicle architecture. The insets demonstrate the presence of prominent megakaryocytes, as indicated by the arrows. The sections were photographed with a 10× objective, with a 50× objective for the insets.
Lungs of terminally ill mice had evidence of severe pneumonia, characterized by diffuse peribronchial and perivascular edema, moderate to massive infiltration of neutrophils, and necrosis (Fig. 7B). In the adjacent alveoli, vasculitis, defined by the presence of neutrophils in the arterial wall, fibrin deposits, and nuclear fragments, was noted. The lungs of asymptomatic mice presented with severe infiltration of neutrophils and lymphocytes, but no fibrin deposits or edema of the arterial walls was observed at early time points (Fig. 7C). The lungs of mice surviving infection after 50 days appeared healthy and showed only prominent peribronchial lymphoid aggregates (Fig. 7D).
FIG. 7.

Hematoxylin and eosin staining of formalin-fixed pulmonary tissue. (A) No alterations were observed in the lung tissue of uninfected control mice. (B) Seven days after infection, moribund mice presented with pneumonia characterized by diffuse edema, neutrophil infiltration, and fibrin deposits. The inset shows a large area of diffuse edema. (C) Survivors sacrificed at day 7 postinfection presented with severe cellular infiltration but no fibrin deposits or edema. Neutrophil infiltration is evident in the inset. (D) Survivors sacrificed at day 50 no longer showed pathological changes in the lung, and lymphoid aggregates were evidenced, as shown by the arrows. The sections were photographed with a 10× objective, with a 50× objective for the insets.
DISCUSSION
Animal models of tularemia have principally been developed in the mouse and have proved to be very helpful tools in elucidating several aspects of disease pathogenesis, including the characterization of innate and adaptive immunity to F. tularensis, in testing the efficacy of antibiotics and other therapies (30, 32, 33, 39) and vaccine candidates against the disease (7, 13, 34). Various methods of inducing disease have been used, and the respiratory route is of particular interest (11, 42) due to the severity of pneumonic tularemia and the impact that aerosol spread might have in a bioterrorist attack. Despite the extensive use of the mouse model for the study of tularemia, little is known about bacterial clearance following inhalation of F. tularensis in contrast to the factors that contribute to mortality. Also, the long-term sequelae of tularemia in vital organs following spontaneous recovery, especially after dissemination, are still unclear. We defined factors that discriminate between the outcomes of (i) death of mice due to disseminated disease and (ii) recovery by overcoming clinical symptoms and surviving infection, even after prolonged periods of time. This was accomplished by observing signs of disease, characterizing anatomical and histopathological parameters, and analyzing markers of immunity following infection of mice via the intranasal route with the LVS strain of F. tularensis. We acknowledge that LVS is an alternative to the use of much more virulent type A Francisella isolates (35, 40). This attenuated strain offers certain advantages over the use of the type A strain, and for the purpose of this study, it proved to be an ideal model strain, as it did not induce 100% death and allowed the study of correlates of mortality versus survival. The interpretation of these results was done with the drawbacks of the use of LVS in mind, and future, similar studies using type A strains are planned.
In our mouse model, anti-LVS IgM was the first marker to obviously distinguish between death and survival, and we speculate that it may be associated with infection clearance. Anti-LVS IgM levels were raised in survivors at early time points and returned to baseline in survivors at late time points. IgM may play a direct role in survival, as Cole et al. (8) observed a robust response in mice surviving challenge with LVS after administration of LVS lipopolysaccharide 2 days prior to infection. One factor that should be kept in mind is that this phenomenon could be unique to LVS, as several groups have shown that upon transfer of specific antibodies, there is protection against F. tularensis subsp. holarctica but none against F. tularensis subsp. tularensis strains (1, 19, 38).
In moribund mice, but not survivors, MIP-2 and MCP-1 were clear indicators of critical illness, as shown by their elevated levels in both lung and spleen supernatants. MIP-2 is a potent chemoattractant for neutrophils and has previously been found to be involved in the recruitment of polymorphonuclear leukocytes in experimental bacterial pneumonia caused by K. pneumoniae and Escherichia coli (37), in mouse sepsis models (41), and in herpes simplex virus type 1 and Pseudomonas aeruginosa corneal-infection models (24, 44). The elevated levels of MIP-2 in lung and spleen tissues at a terminal stage of pneumonic tularemia support the histological findings characterized by neutrophil infiltration. MCP-1 is associated with the recruitment of macrophages, the main cell type infected by F. tularensis, and increased levels of the chemokine in sick mice reflect this type of response. Levels of the IL-6 cytokine were elevated in moribund animals, while in survivors, these levels were similar to those detected in uninfected control mice. In mouse serum, a similar trend was observed, but the difference between moribund mice and survivors was not found to be significant. We speculate that there may be an opposite response in other tissue sites (i.e., liver or kidney), probably due to stimulation of different cell populations, and that the contributions of these other organs might even out IL-6 levels in the serum. IL-6 is a cytokine with a well-established role in inflammatory processes and is an important marker of sepsis (29). This finding suggests that mice showing severe symptoms underwent organ failure, while the absence of an IL-6 response in survivors suggests that the septic process was aborted. A recent study reported the controversial role of IL-6 as a predictor of death outcome due to sepsis induced by cecal ligation and puncture, as a high percentage of mice exhibited low levels of the cytokine at the moribund stage (43). Interestingly, levels of the proinflammatory cytokine IL-1β, induced following in vitro stimulation of human monocytes and macrophages with LVS (4, 20), were found to be raised above baseline in tissue supernatants of moribund mice but were even higher in day 7 survivors. This finding makes IL-1β an important immune marker for the identification of the recovery stage and could be of remarkable value to help clarify mechanisms of infection control in moribund mice. Raised levels of IFN-γ were observed in moribund mice and in survivors at the early stage. IFN-γ is known to play a pivotal role in protective host responses to F. tularensis, and tissues from experimentally infected mice have previously been shown either to have increased levels of the cytokine in lung lysates (16, 28) or to express it in different tissues (8, 23). The fact that we still observed raised levels of IFN-γ in mice recovering from infection indicates that this molecule is also involved in the control of tularemia.
The clinical and pathological characteristics of tularemia in animal spleens have been described and include enlargement, severe tissue pallor, and splenic granulomatous lesions (2, 3, 11). In a study conducted in 1988, Anthony et al. described how LVS caused splenomegaly in C57BL/6 mice at 5 and 8 days after intravenous infection (2). In our study, the spleens of moribund mice were enlarged, pale (not the normal dark-red to purple color), and friable, and elevated bacterial counts were detected. These findings are in accordance with what has been observed in previous aerosol studies conducted in mice, where splenomegaly was associated with elevated LVS counts (11). In this report, histological analysis of splenic tissue from moribund mice reflected the compromised appearance of the tissue gross anatomy: we found necrotic lesions and follicle pattern disruption due to reduction of small lymphocytes. Spleens of mice that recovered from disease symptoms appeared healthy but visibly enlarged, and the splenomegaly was even greater than for moribund animals, up to threefold greater than in uninfected controls. We also noted that in survivors, the enlargement persisted over time: mice sacrificed at 7, 15, and 50 days postinfection all presented with prominent splenomegaly, despite the absence of LVS CFU in the tissue. Morphologically, the spleens of survivors appeared healthier than those of severely ill mice and contained high numbers of megakaryocytes (extramedullary hemopoiesis), as observed in survivors sacrificed at both early (day 7) and late (day 50) time points. In mice, increased extramedullary hemopoiesis can occur in both the spleen and liver as a compensatory response to deficient bone marrow cells. In human diseases like myelofibrosis, splenomegaly has been observed in some patients (17, 21). Extramedullary hemopoiesis has rarely been reported in direct association with infection, although Murray et al. observed megakaryocytes in spleens from IFN-γ−/− mice infected with bacillus Calmette-Guérin via the intraperitoneal and intravenous routes (31). In other experimental models, a correlation between extramedullary hemopoiesis and splenomegaly was also seen following administration of CpG oligodeoxynucleotides and IL-13 in mice (26, 36). Our findings indicate that the extramedullary hemopoiesis evident in all tularemia survivors could contribute to splenomegaly and its persistence over time.
Compared to uninfected controls, moribund mice had increased lung size and weight, discolored foci, and bacterial counts greater than 106 CFU/mg of tissue. The yield of bacterial loads in lungs over time was comparable to those in previous reports (11, 16, 18), but with this model, we emphasize how different disease outcomes (mortality versus survival) could occur even though all of the mice became infected at early time points (bacteremia at 48 to 96 h). Histology of lungs revealed severe pneumonia characterized by diffuse inflammation with neutrophil and fibrin infiltration, and these observations are in accordance with previous studies (11, 42). Histologically, the lungs of survivors at an early time point (day 7) showed characteristics of severe inflammation that were absent in survivors at a later time point (day 50). This event, as well as the occurrence of bacteremia over a period of time, demonstrates that all animals became infected, as indicated by detection of bacteria in blood and organs at early time points. We speculate that in some animals (survivors), lung immunity was able to control bacterial replication, preventing dissemination and leading to recovery (bacteria and inflammation were still present in the lungs of survivors sacrificed 7 days after infection). At later time points, LVS colonies were no longer detectable in murine tissue and blood, and in addition, lung histology appeared normal and healthy with areas of lymphoid aggregates. Conlan et al. (12) have previously described the presence of these areas in lungs from mice immunized with LVS via the aerosol route and speculated that they could represent either residual infection foci or specific anti-Francisella T cells that might potentially be recalled by reinfection with more virulent strains. Our group is currently investigating the nature of this type of response, which could play an important role in overcoming disease.
In our study, we identified immune markers associated with death or survival following induction of pulmonary murine tularemia. We also characterized important differences in the gross anatomy and histopathological changes of target organs between moribund mice and survivors. Our findings will help to clarify the natural history of Francisella tularemia and eventually aid our understanding of whether characteristics of survival after natural infection differ from those of survival due to the protective effect of vaccine candidates after bacterial challenge.
Acknowledgments
We thank Frank Gibson III for comments and suggestions throughout the study, Xiu Ping Liu for technical advice, and Robina Folland and Richard Trevino for administrative support.
This work was supported by National Institutes of Health Grant/National Institute of Allergy and Infectious Diseases U19 AI056543.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 19 November 2007.
REFERENCES
- 1.Allen, W. 1962. Immunity against tularemia: passive protection of mice by transfer of immune tissues. J. Exp. Med. 115411-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anthony, L. S., E. Skamene, and P. A. Kongshavn. 1988. Influence of genetic background on host resistance to experimental murine tularemia. Infect. Immun. 562089-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baskerville, A., P. Hambleton, and A. B. Dowsett. 1978. The pathology of untreated and antibiotic-treated experimental tularaemia in monkeys. Br. J. Exp. Pathol. 59615-623. [PMC free article] [PubMed] [Google Scholar]
- 4.Bolger, C. E., C. A. Forestal, J. K. Italo, J. L. Benach, and M. B. Furie. 2005. The live vaccine strain of Francisella tularensis replicates in human and murine macrophages but induces only the human cells to secrete proinflammatory cytokines. J. Leukoc. Biol. 77893-897. [DOI] [PubMed] [Google Scholar]
- 5.Bosio, C. M., and K. L. Elkins. 2001. Susceptibility to secondary Francisella tularensis live vaccine strain infection in B-cell-deficient mice is associated with neutrophilia but not with defects in specific T-cell-mediated immunity. Infect. Immun. 69194-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, W., R. KuoLee, H. Shen, M. Busa, and J. W. Conlan. 2005. Toll-like receptor 4 (TLR4) plays a relatively minor role in murine defense against primary intradermal infection with Francisella tularensis LVS. Immunol. Lett. 97151-154. [DOI] [PubMed] [Google Scholar]
- 7.Chen, W., H. Shen, A. Webb, R. KuoLee, and J. W. Conlan. 2003. Tularemia in BALB/c and C57BL/6 mice vaccinated with Francisella tularensis LVS and challenged intradermally, or by aerosol with virulent isolates of the pathogen: protection varies depending on pathogen virulence, route of exposure, and host genetic background. Vaccine 213690-3700. [DOI] [PubMed] [Google Scholar]
- 8.Cole, L. E., K. L. Elkins, S. M. Michalek, N. Qureshi, L. J. Eaton, P. Rallabhandi, N. Cuesta, and S. N. Vogel. 2006. Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol. 1766888-6899. [DOI] [PubMed] [Google Scholar]
- 9.Cole, L. E., K. A. Shirey, E. Barry, A. Santiago, P. Rallabhandi, K. L. Elkins, A. C. Puche, S. M. Michalek, and S. N. Vogel. 2007. Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect. Immun. 754127-4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collazo, C. M., A. Sher, A. I. Meierovics, and K. L. Elkins. 2006. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microb. Infect. 8779-790. [DOI] [PubMed] [Google Scholar]
- 11.Conlan, J. W., W. Chen, H. Shen, A. Webb, and R. KuoLee. 2003. Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb. Pathog. 34239-248. [DOI] [PubMed] [Google Scholar]
- 12.Conlan, J. W., H. Shen, R. KuoLee, X. Zhao, and W. Chen. 2005. Aerosol-, but not intradermal-immunization with the live vaccine strain of Francisella tularensis protects mice against subsequent aerosol challenge with a highly virulent type A strain of the pathogen by an alphabeta T cell- and interferon gamma-dependent mechanism. Vaccine 232477-2485. [DOI] [PubMed] [Google Scholar]
- 13.Conlan, J. W., H. Shen, A. Webb, and M. B. Perry. 2002. Mice vaccinated with the O-antigen of Francisella tularensis LVS lipopolysaccharide conjugated to bovine serum albumin develop varying degrees of protective immunity against systemic or aerosol challenge with virulent type A and type B strains of the pathogen. Vaccine 203465-3470. [DOI] [PubMed] [Google Scholar]
- 14.Cowley, S. C., E. Hamilton, J. A. Frelinger, J. Su, J. Forman, and K. L. Elkins. 2005. CD4−CD8− T cells control intracellular bacterial infections both in vitro and in vivo. J. Exp. Med. 202309-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Culkin, S. J., T. Rhinehart-Jones, and K. L. Elkins. 1997. A novel role for B cells in early protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 1583277-3284. [PubMed] [Google Scholar]
- 16.Duckett, N. S., S. Olmos, D. M. Durrent, and D. W. Metzger. 2005. Intranasal interleukin-12 treatment for protection against respiratory infection with the Francisella tularensis live vaccine strain. Infect. Immun. 732306-2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elsayes, K. M., V. R. Narra, G. Mukundan, J. S. J. Lewis, C. O. Menias, and J. P. Heiken. 2005. MR imaging of the spleen: spectrum of abnormalities. Radiographics 25967-982. [DOI] [PubMed] [Google Scholar]
- 18.Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 592922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fulop, M., P. Mastroeni, M. Green, and R. W. Titball. 2001. Role of antibody to lipopolysaccharide in protection against low- and high-virulence strains of Francisella tularensis. Vaccine 194465-4472. [DOI] [PubMed] [Google Scholar]
- 20.Gavrilin, M. A., I. J. Bouskl, N. L. Knatz, M. D. Duncan, M. W. Hall, J. S. Gunn, and M. D. Wewers. 2006. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1β processing and release. Proc. Natl. Acad. Sci. USA 103141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Georgiades, C. S., E. G. Neyman, I. R. Francis, M. B. Sneider, and E. K. Fishman. 2002. Typical and atypical presentations of extramedullary hemopoiesis. Am. J. Roent. 1791239-1243. [DOI] [PubMed] [Google Scholar]
- 22.Gill, V., and B. A. Cunha. 1997. Tularemia pneumonia. Semin. Respir. Infect. 1261-67. [PubMed] [Google Scholar]
- 23.Golovliov, I., G. Sandstrom, M. Ericsson, A. Sjostedt, and A. Tarnvik. 1995. Cytokine expression in the liver during the early phase of murine tularemia. Infect. Immun. 63538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan, S., N. Cole, E. B. Hume, L. Garthwaite, T. C. Conibear, D. H. Miles, Y. Aliwaga, M. B. Krockenberger, and M. D. Willcox. 2007. The role of CXC chemokine receptor 2 in Pseudomonas aeruginosa corneal infection. 2007. J. Leukoc. Biol. 81315-318. [DOI] [PubMed] [Google Scholar]
- 25.KuoLee, R., X. Zhao, J. Austin, G. Harris, J. W. Conlan, and W. Chen. 2007. Mouse model of oral infection with virulent type A Francisella tularensis. Infect. Immun. 751651-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai, Y. H., J. M. Heslan, S. Poppema, J. F. Elliott, and T. R. Mosmann. 1996. Continuous administration of IL-13 to mice induces extramedullary hemopoiesis and monocytosis. J. Immunol. 1563166-3173. [PubMed] [Google Scholar]
- 27.Mackinnon, F. G., Y. Ho, M. S. Blake, F. Michon, A. Chandraker, M. H. Sayegh, and L. M. Wetzler. 1999. The role of B/T costimulatory signals in the immunopotentiating activity of neisserial porin. J. Infect. Dis. 180755-761. [DOI] [PubMed] [Google Scholar]
- 28.Malik, M., C. S. Bakshi, B. Sahay, A. Shah, S. A. Lotz, and T. J. Sellati. 2006. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 743657-3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martins, G. A., M. DaGloria DaCosta Cavarlho, and C. Rocha Gattass. 2003. Sepsis: a follow-up of cytokine production in different phases of septic patients. Int. J. Mol. Med. 11585-591. [PubMed] [Google Scholar]
- 30.Mason, W. L., H. T. Eigelsbach, S. F. Little, and J. H. Bates. 1980. Treatment of tularemia, including pulmonary tularemia, with gentamicin. Am. Rev. Respir. Dis. 12139-45. [DOI] [PubMed] [Google Scholar]
- 31.Murray, P. J., R. A. Young, and G. Q. Daley. 1998. Hematopoietic remodeling in interferon-gamma-deficient mice infected with mycobacteria. Blood 912914-2924. [PubMed] [Google Scholar]
- 32.Pammit, M. A., V. N. Budhavarapu, E. K. Raulie, K. E. Klose, J. M. Teale, and B. P. Arulanandam. 2004. Intranasal interleukin-12 treatment promotes antimicrobial clearance and survival in pulmonary Francisella tularensis subsp. novicida infection. Antimicrob. Agents Chemother. 484513-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell, P., S. M. Eley, M. J. Fulop, D. L. Bell, and R. W. Titball. 1998. The efficacy of ciprofloxacin and doxycycline against experimental tularaemia. J. Antimicrob. Chemother. 41461-465. [DOI] [PubMed] [Google Scholar]
- 34.Shen, H., W. Chen, and J. W. Conlan. 2004. Mice sublethally infected with Francisella novicida U112 develop only marginal protective immunity against systemic or aerosol challenge with virulent type A or B strains of F. tularensis. Microb. Pathog. 37107-110. [DOI] [PubMed] [Google Scholar]
- 35.Shen, H., W. Chen, and J. W. Conlan. 2004. Susceptibility of various mouse strains to systemically- or aerosol-initiated tularemia by virulent type A Francisella tularensis before and after immunization with the attenuated live vaccine strain of the pathogen. Vaccine 222116-2121. [DOI] [PubMed] [Google Scholar]
- 36.Sparwasser, T., L. Hultner, E. S. Koch, A. Luz, G. B. Lipford, and H. Wagner. 1999. Immunostimulatory CpG-oligodeoxynucleotides cause extramedullary murine hemopoiesis. J. Immunol. 1622368-2374. [PubMed] [Google Scholar]
- 37.Standiford, T. J., S. L. Kunkel, M. J. Greenberger, L. L. Laichalk, and R. M. Strieter. 1996. Expression and regulation of chemokines in bacterial pneumonia. J. Leukoc. Biol. 5924-28. [DOI] [PubMed] [Google Scholar]
- 38.Stenmark, S., H. Lindgren, A. Tarnvik, and A. Sjostedt. 2003. Specific antibodies contribute to the host protection against strains of Francisella tularensis subspecies holarctica. Microb. Pathog. 3573-80. [DOI] [PubMed] [Google Scholar]
- 39.Steward, J., T. Piercy, M. S. Lever, A. J. Simpson, and T. J. Brooks. 2006. Treatment of murine pneumonic Francisella tularensis infection with gatifloxacin, moxifloxacin or ciprofloxacin. Int. J. Antimicrob. Agents 27439-443. [DOI] [PubMed] [Google Scholar]
- 40.Twine, S. M., H. Shen, J. F. Kelly, W. Chen, A. Sjostedt, and J. W. Conlan. 2006. Virulence comparison in mice of distinct isolates of type A Francisella tularensis. Microb. Pathog. 40133-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walley, K. R., N. W. Lukacs, T. J. Standiford, R. M. Strieter, and S. L. Kunkel. 1997. Elevated levels of macrophage inflammatory protein 2 in severe murine peritonitis increase neutrophil recruitment and mortality. Infect. Immun. 653847-3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu, T. H., J. A. Hutt, K. A. Garrison, L. S. Berliba, Y. Jhou, and C. R. Lyons. 2005. Intranasal vaccination induces protective immunity against infection with virulent Francisella tularensis biovar A. Infect. Immun. 732644-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao, H., J. Siddiqui, and D. G. Remick. 2006. Mechanisms of mortality in early and late sepsis. Infect. Immun. 745227-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan, X. T., T. M. Tumpey, S. L. Kunkel, J. E. Oakes, and R. N. Lausch. 1998. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Investig. Ophthalmol. Vis. Sci. 391854-1862. [PubMed] [Google Scholar]