

Abstract
Protein aggregation is a phenomenon observed in all organisms and has often been linked with cell disorders. In addition, several groups have reported a virtual absence of protein aggregates in healthy cells. In contrast to previous studies and the expected outcome, we observed aggregated proteins in aerobic exponentially growing and “healthy” Escherichia coli cells. We observed overrepresentation of “aberrant proteins,” as well as substrates of the major conserved chaperone DnaK (Hsp70) and the protease ClpXP (a serine protease), in the aggregates. In addition, the protein aggregates appeared to interact with chaperones known to be involved in the aggregate repair pathway, including ClpB, GroEL, GroES, and DnaK. Finally, we showed that the levels of reactive oxygen species and unfolded or misfolded proteins determine the levels of protein aggregates. Our results led us to speculate that protein aggregates may function as a temporary “trash organelle” for cellular detoxification.
Formation or accumulation of aggregated proteins, which are present in all organisms, has become an important area of intensive research, mainly due to observations that there is a link with many disorders, including aging or neurodegenerative diseases (16).
Major substrates for aggregation are unfolded or misfolded proteins. Indeed, misfolded proteins inappropriately expose hydrophobic surfaces normally buried in the protein's interior, leading to nonnative conformations able to interact and form aggregates. In cells, protein folding may fail because of amino acid misincorporation. Moreover, DnaK/Hsp70 and GroEL/Hsp60 chaperone protein substrates, as well as partially degraded proteins, are more prone to unfolding and thus to aggregation (15).
A common theory is that protein aggregation may be the consequence of a failed quality control mechanism normally charged with repairing or removing the misfolded or unfolded proteins. Indeed, to maintain functional proteins, the cells contain chaperones or proteases that assist proper protein folding and disaggregation of aggregates (17). For instance, Hsp104/ClpB has the capacity to rescue unfolded or misfolded proteins from an aggregated state in cooperation with the Hsp70/DnaK chaperone system (9). However, as recently shown in Huntington's disease, protein aggregates could serve as “temporary storage zones” within cells in order to maintain their function and integrity for prolonged periods of time (1, 13).
A common opinion is that in healthy cells, the levels of formation and accumulation of protein aggregates are extremely low, although protein unfolding occurs constantly. Indeed, several groups have observed a virtual absence of protein aggregates in healthy cells (2, 5, 15), although it is possible that insufficient material was used for detection.
Here we isolated and characterized protein aggregates in exponentially growing “healthy” Escherichia coli cells using classical procedures to extract E. coli insoluble cell fractions and protein aggregates. Our results led us to speculate that these aggregates function as a “trash organelle” that may act as a temporary storage zone for cellular detoxification.
MATERIALS AND METHODS
Bacterial strains and media.
Strains (E. coli MG1655 and mutants) were grown aerobically in liquid Luria-Bertani medium in a rotary shaker at 37°C and 200 rpm. Overnight cultures were diluted 100-fold in Luria-Bertani medium and, except for cultures of PsodA strains, were allowed to grow until the optical density at 600 nm (OD600) was 0.5 without antibiotics. Deletion mutants were constructed by P1 transduction in parental strain MG1655 and selection of kanamycin resistance for ΔdnaK::kan (JW0013), ΔclpX::kan (JW0428), and ΔclpB::kan (JW2573) (3). The ΔdnaK::kan and other kanamycin resistance genes were removed as described previously (7). The hpx mutant was a kind gift from F. Barras. The skx strain (katE::Tn10 katG::Tn10 sodA49 sodB::MudPR3 zij::mini-kanΩ) and plasmid PsodA pDT1-19 (pBR322 carrying the lacI gene and the sodA gene under Ptac control) were provided by D. Touati.
Protein preparation.
Exponentially growing cells were harvested from 10-liter cultures and then washed twice with phosphate buffer (pH 7, 0.05 M) at 4°C by centrifugation at 5,500 × g for 20 min at 4°C. The pellet was resuspended in phosphate buffer and lysed by four cycles with a French press, and all samples were treated with 2 mg/ml DNase and 0.5 mg/ml RNase. Following determination of the OD600, we obtained pellets after centrifugation at 18,000 × g for 4 min (corresponding to the maximum decrease in absorbance) and for 26 min with no detectable variation in absorbance; these pellets were designated LP and SP, respectively. The supernatant (SN) was collected after both centrifugation steps were performed. Next, protein aggregates were extracted from LP and SP as previously described (5), with a minor modification (the percentage of Triton X-100 was changed). Briefly, the pellets were resuspended in buffer A (50 mM Tris, 150 mM NaCl [pH 8]) with 1% Triton X-100 and incubated at 4°C for 3 h. This procedure was repeated with 0.5% Triton X-100. The pellets were then washed with buffer A and centrifuged for 30 min at 18,000 × g to obtain pellets designated ILP and ISP. Next, all samples were dried by speed vacuuming and then resolubilized in rehydration buffer before they were subjected to isoelectric focusing. The protein concentration of the SN was determined with a bicinchoninic acid protein assay kit (Pierce).
Two-dimensional (2D) SDS-PAGE.
Protein samples (100 μg) in rehydration buffer containing 7 M urea, 2 M thiourea, 4% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 100 mM dithiothreitol, 0.2% (vol/vol) ampholyte 3-10 (Bio-Rad), and 0.01% (wt/vol) bromophenol blue were adsorbed onto 17-cm immobilized pH gradient strips (linear pH 3 to 10 gradient). After isoelectric focusing, the strips were (i) subjected to equilibration for 20 min in 60 mM Tris base containing 2.3% (wt/vol) sodium dodecyl sulfate (SDS), 10% (vol/vol) glycerol, 5% (vol/vol) β-mercaptoethanol, and 0.1% (wt/vol) bromophenol blue or (ii) first derivatized with 2,4-dinitrophenylhydrazone (6). Separation based on molecular weight was performed with a 9% acrylamide gel using the Protean II XL SDS-polyacrylamide gel electrophoresis (PAGE) multicell slab gel system (Bio-Rad). Proteins were stained with silver nitrate for mass spectrometry (MS) analysis (Amersham).
Identification of proteins by MS.
Excised silver-stained spots were destained using a ProteoSilver destainer kit (Sigma) and were digested with sequencing grade modified Trypsin Gold (Promega, Madison, WI) as described previously (14). Proteomic analysis was performed by liquid chromatography (LC) nano-electrospray ionization (nano-ESI) MS/MS. The nano high-performance liquid chromatography instrument used was a Finnigan Surveyor system (Thermo Electron, San Jose, CA) equipped with a Spark micro AS autosampler, a Rheodyne 10-port switching valve, and a nano dynamic (NSI) probe assembly on an ion trap spectrometer (Finnigan LCQ-DECAXP). The peptide mixture was dissolved in 5% formic acid in water, and about two-thirds of the total volume was injected onto a Finnigan ProteomeX 2.0 workstation. Peptides were separated on a reversed-phase PicoFrit column (5-μm BioBasic C18; pore size, 300 Å; 75 μm by 10 cm; 15-μm tip; New Objective, Woburn, MA). The peptides were ionized with a capillary temperature of 160°C and a spray voltage of 2.2 kV. Xcalibur data acquisition software allowed one full-scan mass spectrum followed by three MS/MS spectra of the three most intense peaks using the dynamic exclusion features of the Data Dependant program set at a repeat count of 2 within 0.5 min and an exclusion duration of 1 min. The TurboSequest algorithm created Dta files in the Bioworks 3.1 software package (Thermo Electron Corporation), which were processed for protein identification using the nonredundant NCBI E. coli database (September 2005 release; E. coli K-12; 4,220 sequences), in which proteins appear under unique names and accession numbers. Protein identities were therefore unambiguously assigned. The spectra were analyzed for average peptide peaks (range, m/z 400 to 4,000) with a signal-to-noise ratio threshold of 1,000. The search parameter criteria used were as follows: two missed cleavage sites allowed, variable methionine oxidation, cysteine carbamidomethylation, no fixed modification, and maximum precursor and fragment errors of 1.5 and 1.0 Da. The best score protein identification was maximum coverage with at least three unique peptides (minimum duplicated experiments), evaluated by calculation of a cross-correlation number (Xcorr) versus charge state chosen to be Xcorr > 1.7 for singly charged ions, Xcorr > 2.5 for doubly charged ions, and Xcorr > 3.5 for triply charged ions.
RESULTS
Characterization of E. coli pellets.
The size of protein aggregates remaining in a supernatant is a function of the centrifugation time and the acceleration rate. With this in mind, we first aimed to separate soluble protein from protein aggregates in a French press crude extract (CE) obtained from an exponentially growing culture of E. coli cells. For this purpose, we used the protocol described by Dougan et al. (9), with minor modifications (see Material and Methods). We observed that whereas after 4 min of centrifugation at 18,000 × g of the CE we were able to obtain a limpid supernatant, some protein (5.3% ± 1.1%) continued to sediment between 4 and 30 min. We therefore decided to recover large particles that formed a pellet (LP) after 4 min of centrifugation, small particles that formed a pellet (SP) after between 4 and 30 min of centrifugation, and the SN after 30 min of centrifugation (Fig. 1). No intact cells were found in the fractions by microscopy, indicating that the extracts contained only cell debris. LP and SP were then subjected to a previously described protocol to extract Triton X-100-insoluble protein aggregates (insoluble pellets) (5). We observed persistence of Triton X-100-insoluble pellets in both LP and SP, which were designated ILP and ISP respectively (Fig. 2). Next, we evaluated the relative amounts of LP, SP, ILP, and ISP. We observed that LP and SP accounted for a few percent of the total protein present in the CE, whereas ILP and ISP accounted for less than 1% of the total protein present in the CE (Fig. 2). Moreover, when E. coli cultures were grown at 30°C, we still detected Triton X-100-insoluble protein aggregates, but to a lesser extent (Fig. 2), suggesting that protein aggregation in exponentially growing cells is a general phenomenon. Finally, because protein aggregates formed after heat shock have been shown to rapidly deconstruct (9), we wondered if the protein aggregates observed here also deconstructed over time. We observed that when de novo protein synthesis was stopped by addition of chloramphenicol (2.5 mg/ml), no variation in the quantity of proteins was detected in any of the four pellets (LP, SP, ILP, or ISP) over time in the wild-type strain (see Fig. S1 in the supplemental material). Therefore, in contrast to previous observations that no protein aggregates were obtained when only a small number of cells (equivalent to a 10-ml culture at an OD600 of 0.5) were used (2, 5, 15), with enough loaded material (equivalent to a 500-ml culture at an OD600 of 0.5) we detected Triton X-100-insoluble protein aggregates in exponentially growing E. coli cells.
FIG. 1.
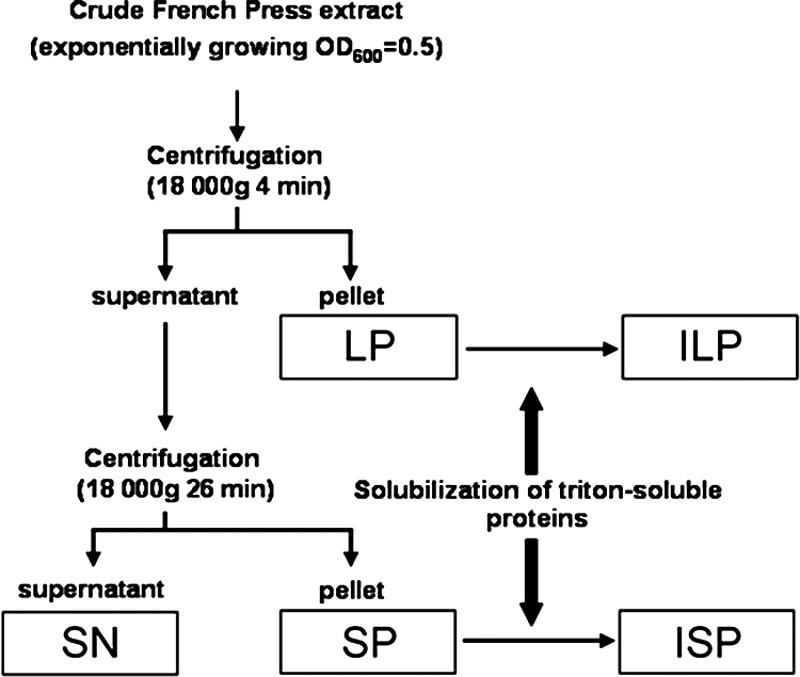
Schematic diagram of extract clarification, showing the separation of the pellets and SN, as described in Materials and Methods.
FIG. 2.
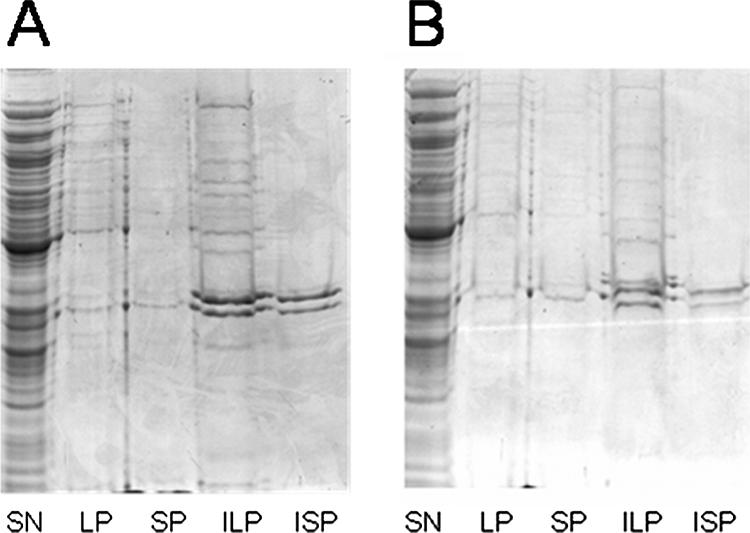
Quantity of protein in the pellet and Triton X-100-insoluble pellet depends on the cell growth temperature: Coomassie blue-stained SDS-PAGE gels showing proteins from SN, LP, ILP, SP, and ISP fractions of cells grown at 37°C (A) and 30°C (B). For all fractions except ILP and ISP, samples were prepared from equal amounts of cells (based on the OD600). For ILP and ISP, fivefold more cells were loaded.
2D gel electrophoresis analysis.
To characterize their protein contents, all four pellet fractions and the SN were subjected to 2D gel electrophoresis, followed by silver nitrate staining. As shown in Fig. 2, when the same quantities of proteins were loaded on the gels, we visualized about 450, 300, 200, 150, and >700 spots for LP, SP, ILP, ISP, and SN, respectively. When the spots observed for the different fractions (LP, SP, ILP, ISP, and SN) were compared, we detected more than 50 spots that were present in the pellets but absent in the SN. We considered the SN devoid of protein aggregates and believed that it contained the majority of the spots, and thus we used it as the control for the comparative study.
Overrepresentation of “aberrant” proteins in aggregates. (i) Spot analysis.
To characterize the specificity of each pellet, some spots were selected for further identification by MS. We analyzed a total of 115 spots (see Table S1 in the supplemental material). In many cases, (i) we detected more than one protein signature in any given spot, and (ii) the same protein could be found in different spots on the same 2D gel (see Table S1 in the supplemental material). Spot overlap analysis revealed a total of 204 protein signatures corresponding to 95 different identified proteins. For each of the 204 protein signatures we were able to estimate an experimental isoelectric point (pI) and experimental molecular weight (Mw). In our study, an “aberrant” protein was defined as (i) a protein with a pI shift greater than 0.3, (ii) a protein whose Mw was decreased by more than 20% (designated a protein degradation product [PDP]), or (iii) a protein whose Mw was increased by more than 20% (designated a cross-linked protein). Indeed, more than 96% of the proteins retrieved from the Expasy proteome or SN proteins had standard deviations of ±0.3 for pI and ±20% for Mw, confirming the usefulness of these parameters in identifying “aberrant” proteins (see Fig. S2 in the supplemental material). Among the 204 protein signatures present in LP, SP, ILP, and ISP (see Table S1 in the supplemental material), we found 99 aberrant protein signatures, which we divided into 62 proteins with shifts in the isoelectric point, 18 PDPs, and 19 cross-linked protein signatures. Finally, spot analysis of the LP and SP proteins (composed of both Triton X-100-soluble and Triton X-100-insoluble proteins) not included in ILP and ISP (containing only Triton X-100-insoluble proteins), respectively, indicated that the Triton X-100-soluble proteins present in LP or SP included large numbers of aberrant proteins. Ten of the spots analyzed from pellets that contained only aberrant proteins could not be detected in the SN. In addition, we detected aberrant protein signatures in only 2 of the 11 SN spots containing at least one aberrant protein also found in pellets (see Table S1 in the supplemental material). As detection by MS depends on the amount of protein present in a spot, we concluded that the levels of aberrant proteins were higher in pellet spots than in SN spots.
(ii) Migration front analysis.
We noted a large difference between fractions in the apparent concentration of the material migrating with the front (Fig. 3). In order to identify this material in each 2D gel, we divided the area corresponding to the migration front into 17 gel pieces (10 by 4 mm), each corresponding to 0.4 pI unit. These pieces were then individually analyzed using an LC nano-ESI MS/MS spectrometer (see Table S2 in the supplemental material). We observed that, with the exception of a few proteins, protein signatures detected in ILP and ISP were also present in LP and SP, respectively (see Table S2 in the supplemental material), suggesting that our LC nano-ESI MS/MS analysis was quite efficient. We detected 70 protein signatures in the migration front, including seven PDP proteins and 34 ribosomal proteins. Interestingly, only one PDP protein, OmpA, was found in the SN, indicating that PDP proteins are specifically found in pellet fractions. Moreover, ribosomal proteins were not expected in the pellet fractions. Indeed, using Svedberg units, we were able to calculate that in our experimental procedure more than 1 and 10 h were needed to spin down polysome (1000S) and ribosomal particles (70S), respectively. Moreover, as indicated in Materials and Methods, RNase was added before pellet preparation, allowing us to reasonably assume that polysomes were absent. Of the 34 ribosomal proteins identified in pellets, only 3 were absent in the SN. In addition, we observed many ribosomal proteins with correct theoretical isoelectric points and also ribosomal proteins displaying several deviations, resulting in many isoelectric protein signatures in pellets (Fig. 4). This suggests that the ribosomal proteins present in the migration fronts of pellets were in fact aberrant proteins. This was rare or absent for the SN migration front (Fig. 4) and the other proteins detected in the migration front (data not shown).
FIG. 3.

Silver-stained 2D gel analysis of proteins in pellets and SN: protein patterns for LP, SP, ILP, ISP, and SN obtained after 2D gel electrophoresis and visualization by silver staining. Experiments were repeated three times with cells from three different cultures.
FIG. 4.

Schematic distribution of ribosomal proteins in migration fronts. The migration fronts of LP, SP, and SN were divided into 17 equal gel pieces and analyzed by LC nano-ESI MS/MS. ILP and ISP data are not shown as the distributions were the same as those of LP and SP except for one protein. The 34 different ribosomal proteins identified were classified by increasing pI (acidic to basic) from 1 to 34. Analysis of repartition of these proteins with isoelectric points (pI 3 to 10) revealed proteins identified (filled circle) or not identified (open circles) at their theoretical isoelectric points and proteins identified with incorrect isoelectric points (asterisks).
Taken together, the spot and migration front analyses demonstrated that aberrant proteins were more abundant in pellets than in SN, both in terms of number and in terms of quantity.
Overrepresentation of cytosolic proteins and abnormal ribosomal proteins in pellets.
We identified all classes of proteins, except periplasmic proteins, in pellet fractions. After pooling all the protein signatures from all pellets, we identified 159 different proteins (see Tables S1 and S2 in the supplemental material). A closer examination showed that 44% were cytosolic proteins and 22% were ribosomal proteins, as predicted by the ecocyc website (http://ecocyc.org/). Finding a cytosolic or ribosomal protein in a pellet would occur only if the protein was either present inside a protein aggregate or associated with a protein aggregate. Consequently, our results show that the Triton X-100-insoluble protein aggregates were not artifactual membrane-associated protein aggregates as expected but were composed primarily of cytosolic and ribosomal proteins. Moreover, we showed that LP and SP were composed of both Triton X-100-insoluble protein aggregates and Triton X-100-soluble proteins. Although we expected to find membrane-associated proteins among the Triton X-100-soluble proteins, we wanted to test the possibility that Triton X-100-soluble aggregates would also be found. To investigate this, we analyzed the compositions of the spots present in LP and SP but absent in ILP and ISP, respectively. We found that in the Triton X-100-soluble protein fractions there was overrepresentation of cytosolic proteins, suggesting that these proteins were in fact aggregated or associated with protein aggregates. This was especially the case for several chaperones with known involvement in the aggregate repair pathway, including ClpB, GroEL, GroES, and DnaK (4). These results show that cytosolic and abnormal ribosomal proteins, normally not expected in pellets of exponentially growing cells, were in fact Triton X-100-soluble or Triton X-100-insoluble protein aggregates and aggregate-associated proteins.
Overrepresentation of ClpXP and/or DnaK substrates in protein aggregates.
Because DnaK and GroEL substrates are more prone to aggregation, we investigated the possible increase in the number of these proteins in the pellets. Moreover, as we had already detected numerous PDPs, we wondered if we would also see an increase in the number of proteins known to be degraded by ClpXP protease. Previous studies have shown that about 2 to 3%, 9 to 18%, and 10 to 15% of the E. coli proteins are ClpXP (10), DnaK (8, 9, 18), and GroEL (11) substrates, respectively. Of the 95 different protein spots identified (see Table S1 in the supplemental material), 38% were DnaK substrates and 12% were ClpXP substrates, whereas only 8% were GroEL substrates. Together, these results show that there was overrepresentation of DnaK and/or ClpXP substrates among the aggregated proteins and that there was no overrepresentation of GroEL substrates.
Quantities of protein in pellets depend on the levels of ROS and unfolded or misfolded proteins in healthy E. coli cells.
Having shown that all four pellet fractions contain proteins previously shown to be more prone to aggregation (aberrant or DnaK substrates), we wanted to know if pellets of mutants involved in defense against oxidative stress or heat shock have larger quantities of aggregate proteins. As shown in Fig. 5, when SDS-PAGE analysis was carried out with equal amounts of the wild-type and mutant pellets, we found that, as expected for mutants involved in defense against protein aggregation (like ΔclpB or ΔdnaK mutants) or proteolysis (like ΔclpX mutants), the quantities of protein in the pellets and/or Triton X-100-insoluble pellets were greater than the quantities in wild-type pellets. Previously, other groups have shown that ΔdnaK mutant cells accumulate protein aggregates at physiological growth temperatures (9). Interestingly, protein aggregation was only slightly increased in ΔclpB or ΔclpX mutant cells, compared to the large increase in aggregation seen in ΔdnaK mutant cells. Moreover, the quantities of protein in pellet fractions increased for mutants involved in defense against oxidative stress, like skx mutants (deficient in cytosolic superoxide dismutase and catalases) and hpx mutants (deficient in catalase and alkyl hydroperoxidase), compared to the levels observed in wild-type cells. Interestingly, mutants involved in defense against oxidative stress appeared to have a different aggregation pattern. Overproduction of SodA resulted in a decrease in the amount of protein found in the pellet fraction of wild-type cells; this phenomenon was most dramatic in the hpx mutant (Fig. 5). Taken together, these results indicate that the amount of protein in pellets is directly or indirectly dependent on the level of reactive oxygen species (ROS) and unfolded or misfolded proteins.
FIG. 5.

Quantities of protein in the pellet and Triton X-100-insoluble pellet depend on the levels of ROS and unfolded or misfolded proteins: Coomassie blue-stained SDS-PAGE gels showing proteins from LP, ILP, SP, and ISP in cells with different genetic backgrounds. For each fraction, samples were prepared from equal amounts of cells of different strains (based on the OD600).
DISCUSSION
In this study, we isolated and characterized Triton X-100-soluble protein aggregates and Triton X-100-insoluble protein aggregates in healthy E. coli cells. We found that proteins known to be most prone to aggregation are overrepresented in protein aggregates. In addition, we observed the presence of several chaperones involved in protein aggregate deconstruction in these fractions, including DnaK, GroEL, GroES, and ClpB (4). Furthermore, we demonstrated that the quantity of protein aggregates is determined by the levels of ROS and unfolded or misfolded proteins.
Existence of protein aggregates in healthy E. coli cells.
The analyses of pellets by MS clearly indicated a large presence of cytosolic proteins or ribosomal proteins in pellet fractions. In contrast to the common opinion that protein aggregates are virtually absent in healthy cells (2, 5, 15), our results indicate the existence of protein aggregates associated with chaperones involved in disaggregation. Moreover, we were unable to detect differences in composition between LP and SP, suggesting that various protein aggregate sizes were present in the bacterial population. In addition, we report that the time of centrifugation of CE is an important parameter and may sometimes lead to misinterpretation of results. Indeed, as referred to in the Results, a limpid supernatant was obtained after 4 min of centrifugation; however, this supernatant likely contained protein aggregates (SP).
Overrepresentation of potentially unfolded or misfolded proteins in protein aggregates.
We observed that more than 80% of the protein signatures identified in protein aggregates showed a predisposition to aggregate based on previous findings. These proteins included PDPs, isoelectric point shift or cross-linked proteins, and substrates of the major conserved chaperone DnaK and protease ClpXP. Moreover, these proteins were more abundant in the aggregates than in the SN. Surprisingly, GroEL substrates were not overrepresented in pellets, suggesting that even though they are prone to aggregation, these proteins do not always aggregate in healthy E. coli cells.
Dynamic formation of protein aggregates.
We separated large and small aggregate proteins associated with chaperones and showed that there was an increased presence of ClpXP substrates (12% of the proteins in pellets) or DnaK substrates (38%), suggesting that there is dynamic aggregate formation and evolution with time. Indeed, chaperones, including DnaK, GroEL, GroES, HtpG, ClpB, and Tig, involved in protein disaggregation following heat shock were all detected in insoluble cell fractions with their correct pI and Mw. Judging from the peptide coverage, the results suggest that PDP proteins result from several partial degradation steps and not abortive de novo synthesis (see Fig. S3 in the supplemental material). Nine of these proteins (FusA, HtpG, GroEL, OmpA, ProS, RpsA, TufB, YaeT, and YleA) showed several stages of pI and/or Mw modification, and three of them (TufB, RpsA, and OmpA) were found at the migration front, suggesting that there are several steps of degradation.
Protein aggregates: a “trash organelle”?
The presence of protein aggregates overrepresenting PDPs, cross-linked or aberrant proteins, and chaperones involved in disaggregation suggests that the aggregates have a role as a “trash organelle.” Aggregates could be temporary or definitive storage zones in the protein degradation pathway. Indeed, the change in aggregate protein quantities with time leads us to propose that the flow of newly aggregated proteins is equivalent to that of newly disaggregated proteins. Consequently, a flow equal to zero would correspond to a definitive storage zone, whereas a flow not equal to zero would correspond to a temporary storage zone. However, considering the observations that (i) the formation of protein aggregates during heat shock is a reversible process involving chaperones and proteases (9, 12), (ii) in our conditions the levels of abnormal protein and ROS controlling the level of oxidized protein determine the amount of aggregated protein, and (iii) inside protein aggregates PDP proteins result from several partial degradation steps, we suggest that protein aggregation is a temporal storage disposal method allowing maintenance of cellular function and integrity and not the natural consequence of a failed quality control mechanism for repairing or removing misfolded or unfolded proteins.
Supplementary Material
Acknowledgments
We thank M. Chippaux, B. Py, A. Galinier, A. Magalon, V. Geli, B. Ezraty, D. Byrne, and A. Ducret of IBSM, Marseille, France, and A. Mogk of ZMBH, Universität Heidelberg, Heidelberg, Germany, for helpful comments on the manuscript. We thank S. Lignon and R. Lebrun for their technical assistance with the LC nano-ESI MS/MS analysis.
This work was supported by ACI Jeunes Chercheurs C.C., by ANR grant ANR-05-BLAN-SPV005511, and by two fellowships from the Ministère de l'Education Nationale.
Footnotes
Published ahead of print on 26 November 2007.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Arrasate, M., S. Mitra, E. S. Schweitzer, M. R. Segal, and S. Finkbeiner. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431805-810. [DOI] [PubMed] [Google Scholar]
- 2.Baars, L., A. J. Ytterberg, D. Drew, S. Wagner, C. Thilo, K. J. van Wijk, and J. W. de Gier. 2006. Defining the role of the Escherichia coli chaperone SecB using comparative proteomics. J. Biol. Chem. 28110024-10034. [DOI] [PubMed] [Google Scholar]
- 3.Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 21-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Zvi, A. P., and P. Goloubinoff. 2001. Review: mechanisms of disaggregation and refolding of stable protein aggregates by molecular chaperones. J. Struct. Biol. 13584-93. [DOI] [PubMed] [Google Scholar]
- 5.Chapman, E., G. W. Farr, R. Usaite, K. Furtak, W. A. Fenton, T. K. Chaudhuri, E. R. Hondorp, R. G. Matthews, S. G. Wolf, J. R. Yates, M. Pypaert, and A. L. Horwich. 2006. Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc. Natl. Acad. Sci. USA 10315800-15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad, C. C., J. Choi, C. A. Malakowsky, J. M. Talent, R. Dai, P. Marshall, and R. W. Gracy. 2001. Identification of protein carbonyls after two-dimensional electrophoresis. Proteomics 1829-834. [DOI] [PubMed] [Google Scholar]
- 7.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deuerling, E., H. Patzelt, S. Vorderwulbecke, T. Rauch, G. Kramer, E. Schaffitzel, A. Mogk, A. Schulze-Specking, H. Langen, and B. Bukau. 2003. Trigger factor and DnaK possess overlapping substrate pools and binding specificities. Mol. Microbiol. 471317-1328. [DOI] [PubMed] [Google Scholar]
- 9.Dougan, D. A., A. Mogk, and B. Bukau. 2002. Protein folding and degradation in bacteria: to degrade or not to degrade? That is the question. Cell. Mol. Life Sci. 591607-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flynn, J. M., S. B. Neher, Y. I. Kim, R. T. Sauer, and T. A. Baker. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11671-683. [DOI] [PubMed] [Google Scholar]
- 11.Houry, W. A., D. Frishman, C. Eckerskorn, F. Lottspeich, and F. U. Hartl. 1999. Identification of in vivo substrates of the chaperonin GroEL. Nature 402147-154. [DOI] [PubMed] [Google Scholar]
- 12.Rosen, R., D. Biran, E. Gur, D. Becher, M. Hecker, and E. Z. Ron. 2002. Protein aggregation in Escherichia coli: role of proteases. FEMS Microbiol. Lett. 2079-12. [DOI] [PubMed] [Google Scholar]
- 13.Rujano, M. A., F. Bosveld, F. A. Salomons, F. Dijk, M. A. van Waarde, J. J. van der Want, R. A. de Vos, E. R. Brunt, O. C. Sibon, and H. H. Kampinga. 2006. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 4e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68850-858. [DOI] [PubMed] [Google Scholar]
- 15.Tomoyasu, T., A. Mogk, H. Langen, P. Goloubinoff, and B. Bukau. 2001. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol. Microbiol. 40397-413. [DOI] [PubMed] [Google Scholar]
- 16.Tran, P. B., and R. J. Miller. 1999. Aggregates in neurodegenerative disease: crowds and power? Trends Neurosci. 22194-197. [DOI] [PubMed] [Google Scholar]
- 17.Wickner, S., M. R. Maurizi, and S. Gottesman. 1999. Posttranslational quality control: folding, refolding, and degrading proteins. Science 2861888-1893. [DOI] [PubMed] [Google Scholar]
- 18.Winter, J., K. Linke, A. Jatzek, and U. Jakob. 2005. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol. Cell 17381-392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.