

Abstract
Mycobacterium avium subsp. paratuberculosis infection of cattle takes place through the intestinal mucosa. To identify M. avium subsp. paratuberculosis genes associated with the invasion of bovine epithelial cells in vitro, we screened a library of transposon mutants. Several mutants of M. avium subsp. paratuberculosis were identified which invaded Madin-Darby bovine kidney (MDBK) epithelial cells less efficiently than wild-type (wt) M. avium subsp. paratuberculosis. The ΔOx mutant had the transposon located in the MAP3464 gene, a putative oxidoreductase gene whose expression is upregulated upon bacterial contact with MDBK cells. Complete restoration of invasion comparable to that for the wt bacterium was achieved by introducing a copy of the complete oxidoreductase operon into the ΔOx mutant. Immunoprecipitation and Western blot analysis indicated that wt M. avium subsp. paratuberculosis activates Cdc42 and RhoA pathways of internalization 15 and 60 min after infection of the host cell, respectively. The ΔOx mutant, however, failed to activate the Cdc42 pathway. To determine whether an M. avium subsp. paratuberculosis protein delivered to the host cell mediates the entry of the wt bacterium by activation of the Cdc42 pathway, affinity precipitation of active Cdc42 from MDBK-infected cells followed by mass spectrometry was carried out. We identified a 17-amino-acid bacterial peptide associated with the Cdc42 of cells infected with wt M. avium subsp. paratuberculosis but not with the ΔOx mutant. The sequence of the peptide matches MAP3985c, a hypothetical protein, possibly functioning as a putative Cdc42 effector. These findings reveal a novel signaling pathway activated during M. avium subsp. paratuberculosis entry that links the product of MAP3464 gene to activation of Cdc42 in the host cell.
Mycobacterium avium subsp. paratuberculosis causes Johne's disease, a chronic debilitating intestinal disease of cattle and other ruminants. M. avium subsp. paratuberculosis infects the small intestine and ultimately causes granulomatous inflammation. The inflammation leads to diarrhea, poor absorption of nutrients, severe weight loss, and the eventual death of the infected animal. The U.S. dairy industry has reported annual losses of $1.5 billion due to the disease (14). It has also been estimated that 22% of the dairy herds in the United States are infected with M. avium subsp. paratuberculosis (9). Infection is acquired during the early phase of a calf's life by the ingestion of feed or water contaminated with M. avium subsp. paratuberculosis or by drinking milk from an infected animal (3, 5).
It has been suggested that after oral ingestion, M. avium subsp. paratuberculosis enters the intestinal wall, preferably through M cells present in the follicle-associated epithelium covering the continuous Peyer's patches in the distal ileum (13). After crossing the mucosal cell wall, the bacterium enters and survives within submucosal macrophages. Although the mechanism of entry in the mucosa is important in establishing M. avium subsp. paratuberculosis infection, most of the bacterial moieties involved in the interaction with intestinal cells are unknown. A previous report demonstrated that the invasion of M cells in the intestine is mediated by the formation of a fibronectin bridge between a fibronectin attachment protein (FAP) present on the M. avium subsp. paratuberculosis surface and β1 integrin receptors on the surfaces of M cells (20). Because disruption of the fibronectin binding fails to completely eliminate invasion, it is likely that several other virulence factors are involved in the interaction between M. avium subsp. paratuberculosis and the intestinal cells. For instance, a 35-kDa protein exposed on the outer layer of M. avium subsp. paratuberculosis has been associated with this organism's invasion of bovine epithelial cells (4).
Intracellular pathogens gain access to nonphagocytic eukaryotic cells via a “zipper” (receptor-mediated entry) or a “trigger” mechanism (1). The first mechanism is initiated by specific contacts between bacterial ligands (adhesins) and host cell surface receptors. The progressive sliding of the bacterium on the host cell membrane ends with the inclusion of the invading bacterium. Unlike the zipper mechanism, the “trigger mechanism” is initiated by the action of pathogen effector proteins delivered into the host cytosol by specialized protein secretion systems and involves dramatic cytoskeletal rearrangements. These alterations consist of membrane extensions in the form of filopodia and lamellipodia at the site of contact of the bacteria with the host eukaryotic cell surface. M. avium subsp. paratuberculosis invades bovine epithelial cells, but the signal transduction pathways that lead to cytoskeletal rearrangements are currently unknown. Other intracellular pathogens have been shown to trigger host cell cytoskeleton rearrangement, such as the formation of filopodia and lamellipodia through activation of small GTPases of the Rho subfamily (Rac1, Rho, and Cdc42) (21, 24, 25). For instance, invasion by Mycobacterium avium subsp. avium leads to activation of Rho and Cdc42 small GTPases (6, 19). Once activated, Cdc42 binds to a protein similar to neuronal Wiskott-Aldrich syndrome protein (N-WASP). Phosphorylation of N-WASP further activates N-WASP in cooperation with Cdc42 and its subsequent binding to Arp2/3 complex, a protein complex that has been shown to catalyze the nucleation of actin filaments, filopodium formation, and engulfment of the bacteria (18).
Since binding and invasion of the epithelial mucosal cells is the first step toward the establishment of infection, we investigated the ability of M. avium subsp. paratuberculosis to invade Madin-Darby bovine kidney (MDBK) epithelial cells. Previous work showed that M. avium subsp. paratuberculosis can establish infection and replicate within MDBK cells and demonstrated that the efficiency of infection peaks 4 h after exposure to the wild-type (wt) bacterium (16). In the current study, we screened an M. avium subsp. paratuberculosis transposon library for mutants attenuated in their ability to invade MDBK cells. We identified an attenuated mutant (the ΔOx mutant) with the transposon integrated in the MAP3464 gene, a 1.2-kb open reading frame (ORF) homologous to the Mycobacterium tuberculosis Rv3359 gene, which encodes a possible oxidoreductase.
MATERIALS AND METHODS
Cell line.
A bovine epithelial cell line (MDBK) was purchased from the American Type Culture Collection (Manassas, VA) and maintained on Dulbecco's modified Eagle medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Gemini Bio-products, Woodland, CA) as described before (16).
Bacteria.
Mycobacterium avium subsp. paratuberculosis ATCC 19698, a bovine clinical isolate from an animal with Johne's disease, was grown at 37°C on either modified Middlebrook 7H9 broth or 7H11 agar (Difco Laboratories, Sparks, MD) supplemented with 2 mg/liter of mycobactin J (Allied Monitor, Fayette, MO), 10% (vol/vol) oleic acid-albumin-dextrose-catalase (Hardy Diagnostics, Santa Maria, CA), and 0.05% Tween 80 (Sigma, St. Louis, MO). For the invasion assay, individual colonies were selected and resuspended in Hanks' balanced salt solution (HBSS) to give turbidity equivalent to a 0.5 McFarland standard. To minimize clumping, the bacterial suspension was passed through a 26-gauge needle 10 times and then allowed to settle for 5 min. Only the top fraction of the suspension containing dispersed bacteria was used for the assays.
Construction of an M. avium subsp. paratuberculosis transposon library.
To identify genes associated with invasion, a transposon library was constructed by transformation of M. avium subsp. paratuberculosis strain 19698 competent cells with the temperature-sensitive plasmid pTNGJC-KAN, a plasmid containing the transposon Tn5367 harboring a kanamycin-resistant gene, as previously described (27). Briefly, M. avium subsp. paratuberculosis competent cells were centrifuged at 3,000 × g for 15 min at 4°C and washed in a solution of 10% glycerol and 0.1% Tween 80. Washing and centrifugation were repeated three times, and the pellet was resuspended in 1 ml of 10% glycerol (Sigma). Bacteria were electroporated with plasmid pTNGJC-KAN by use of a GenePulser X cell (Bio-Rad, Hercules, CA), plated onto 7H11 agar plates with 400 μg/ml of kanamycin (Sigma), and incubated at 30°C for 3 weeks. Twenty colonies were randomly selected and screened by PCR for the presence of the kanamycin gene by use of the primers 5′-TAATGTCGGGCAATCAGGTG-3′ (forward) and 5′-TGTTCAACAGGCCAGCCA-3′ (reverse). PCR cycling was as follows: 35 cycles of 95°C for 30 s, 57°C for 1 min, and 72°C for 1 min. Prior to the first cycle, a temperature of 95°C was held for 5 min, and at the end of the last cycle, a temperature of 72°C was maintained for 10 min. The confirmed kanamycin-resistant colonies were then grown in 7H9 broth containing kanamycin at a nonpermissive temperature (40°C) for an additional week. The suspensions were diluted and placed onto 7H11 agar with kanamycin at 37°C. Colonies were picked randomly and collected in individual wells of a 96-well tissue culture plate, generating a library of 1,980 individual transposon mutants. Because pTNGJC-KAN contains a temperature-sensitive mycobacterial origin of replication, the shift in temperature eliminated the plasmid, and all surviving kanamycin-resistant cells necessarily contained the transposon in random positions of the bacterial chromosome.
Invasion assay.
Approximately 600 transposon mutants were individually screened for the ability to enter MDBK cells, as previously described (11). Briefly, 105 MDBK cells/ml growing in 24-well plates were infected with individual mutants (multiplicity of infection [MOI], 100) and incubated at 37°C in a 5% CO2 incubator for 4 h. The cell monolayers were then washed three times with HBSS to remove extracellular bacteria and then treated with 1 ml of tissue culture medium supplemented with 200 μg/ml of amikacin (Sigma) for 2 h at 37°C. Following treatment, the monolayers were washed twice with HBSS, and the viable intracellular bacteria were released by incubation with 0.5 ml of 0.1% Triton X-100 (Sigma) in sterile water for 10 min. After the addition of 0.5 ml of Middlebrook 7H9 broth, samples were disrupted by vigorous pipetting. Lysates were collected and viable bacteria were quantified by plating for CFU onto Middlebrook 7H11 agar containing mycobactin J. The percentages of invasion of mutants and wt bacterium were calculated as the percentages of the inoculated bacteria that were recovered from the cell lysate.
Identification of transposon-inactivated mutants.
Chromosomal DNA of the low-invasion mutants was extracted and purified as previously described (15). The isolation of DNA sequences flanking transposon insertions was carried out using a nonspecific nested suppression PCR method previously described by Tamme et al. (23). Briefly, a first round of PCR was conducted at a low annealing temperature by use of 1.6 μM of a transposon-specific primer (5′CCA TCA TCG GAA GAC CTC-3′) and a polymerase lacking exonuclease activity (FideliTag; USB, Cleveland, OH) to generate a mixture of products, including fragments of the desired flanking sequence. PCR was carried out from 100 ng of genomic DNA in a total volume of 30 μl in 3% (vol/vol) dimethyl sulfoxide and 15 μl of FideliTag PCR master mix containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 3 mM MgCl2, 0.4 mM deoxynucleoside triphosphates, and FideliTag. PCR cycling conditions were as follows: 35 cycles of 95°C for 30 s, 50°C for 1 min, and 72°C for 4 min. Prior to the first cycle, a temperature of 95°C was held for 5 min, and at the end of the last cycle a temperature of 72°C was maintained for 10 min. Only 1 μl of the first-round amplification was then used as a template for the second-round PCR (nested PCR) using a primer that is 6 nucleotides (GACCCC) longer at the 3′ end than the primer used in the first round of PCR and a polymerase exhibiting exonuclease activity (Pfu DNA polymerase; Stratagene, La Jolla, CA). Reactions were carried out in a total volume of 50 μl in 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2, 0.4 mM deoxynucleoside triphosphates, 5% (vol/vol) dimethyl sulfoxide, 1.6 μM of the primer, and 2.5 U of Pfu DNA polymerase. The PCR cycling was the same as for the first PCR, except the annealing cycle was 30 s at 56°C. The PCR products of the second amplification were run in a 1% agarose gel, and each PCR band that appeared in the gel was excised and purified using a DNA gel extraction kit (Stratagene) before cloning into the pCR2.1 TOPO vector (Invitrogen). Samples were sequenced using the M13 reverse primer at the Center for Gene Research and Biotechnology, Oregon State University, Corvallis, OR. Database searches and sequence comparisons were performed using the BLAST server from the National Center for Biotechnology Information. To determine a function for the identified genes, we searched for M. tuberculosis homologues with TubercuList (http://genolist.pasteur.fr/TubercuList/) and BLAST.
Amplification of MAP3464 and 16S rRNA genes from genomic DNA.
MAP3464 and 16S rRNA genes were amplified from genomic DNA by use of specific primers and the GC-rich PCR system following the manufacturer's instructions (Roche, Penzberg, Germany). The primers for the amplification of the MAP3464 gene were 5′-TTTGAATTCATGCCCACCCCTCCGGACGT-3′ (forward) and 5′-TCGAAGCTTTTAAGTTGCGGCGCTGGTGTG-3′ (reverse). For the amplification of the 16S rRNA, primers 5′-CGAACGGGTGAGTAACACG-3′ (forward) and 5′-TGCACACAGGCCACAAGGGA-3′ (reverse) were used. PCR amplification was carried out at 35 cycles of 95°C for 30 s, 62°C for 30 s, and 72°C for 2 min.
RNA extraction, cDNA synthesis, and reverse transcription-PCR (RT-PCR).
To determine whether the MAP3464 gene was upregulated when the bacterium was exposed to the host cell, MDBK cells were incubated with wt M. avium subsp. paratuberculosis in HBSS at 37°C for 15, 30, and 60 min. The supernatant was collected and bacteria were pelleted at 3,500 × g for RNA preparation, as previously described (10). Total RNA from bacteria incubated at 37°C in HBSS for 60 min was also prepared as a control. Bacterial pellets were mixed with 1 ml of TRIzol reagent (Invitrogen), and RNA was isolated by rapid mechanical agitation in a bead beater. To remove cellular debris, cells were centrifuged at 13,000 × g for 5 min at 4°C. The supernatant was removed and added to a 2-ml phase lock gel heavy (Eppendorf, Hamburg, Germany) containing 300 μl of chloroform-isoamyl alcohol (24:1). Samples were centrifuged for 10 min at 4°C, and the aqueous layer was collected, extracted with the same volume of phenol-chloroform, and precipitated with isopropanol. The pellet was then washed with 75% ethanol and dried at room temperature for 10 min. RNA samples were treated with DNase I (Clontech, Palo Alto, CA) for 1 h at 37°C, followed by precipitation with ethanol. Total RNA was quantified by measuring absorbance at 260 nm, and quality was determined by measuring the 260/280-nm absorbance ratio. Ratios of ≥1.8 were considered acceptable. RNA was then electrophoresed on a 1% denaturing agarose gel to confirm quality.
Total RNA was reverse transcribed using the SuperScript first-strand synthesis system for RT-PCR following the manufacturer's instructions (Invitrogen). Total RNA (1.5 μg) was incubated with 1 μl of a 10 mM concentration of deoxynucleotide triphosphate mix and 1 μl of random hexamers at 65°C for 5 min. Samples were then mixed with 2 μl of 10× RT buffer, 4 μl of 25 mM MgCl2, 2 μl of 0.1 M dithiothreitol, 1 μl of RNase out recombinant RNase inhibitor (40 U/μl) and incubated at 25°C for 2 min. Finally, 1 μl of SuperScript II reverse transcriptase (50 U/μl) was added to each tube, and the samples were incubated at 42°C for 50 min. The reaction was terminated at 70°C for 15 min. The tube was placed on ice and centrifuged briefly, and 1 μl of RNase H was added to each sample and incubated for 20 min at 37°C. For RT-PCR, the MAP3464 and 16S RNA genes were amplified from 1 μl of cDNA as described above. Ten microliters of the PCR products was separated on a 1% agarose gel and visualized by staining with ethidium bromide. The 16S RNA gene was used as a constitutively expressed control to ensure that the same amount of RNA was added to each reaction mixture.
Quantitative real-time RT-PCR.
Quantitative fluorogenic amplification of cDNA was performed using an iCycler real-time detection system and SYBR green technology (Bio-Rad, Hercules, CA) as previously described (27). Briefly, PCRs were carried out in 50-μl reaction mixtures consisting of 25 μl of IQ SYBR green supermix, 1 μl of each primer (10 μM), 1 μl of cDNA, and 23 μl of water. Each PCR amplification consisted of denaturation at 95°C for 30 s, annealing at 62°C for 30 s, and extension at 72°C for 2 min. Real-time PCR efficiency was determined using a dilution series of cDNA template with a fixed concentration of primers. Slopes calculated by the LightCycler software were used with the following formula to calculate efficiency (E): E = 10(1/slope). Cycle threshold (CT) is defined as the fraction cycle number at which the fluorescence reaches 10 times the standard deviation of the baseline and was quantified as described in the user bulletin for the ABI PRISMS 7700 sequence detection system. Since the 16S RNA gene is constitutively expressed, target genes from both control and experimental groups were normalized to the expression level of the 16S RNA gene. To determine the change (n-fold) in gene expression, the following formula was used: change (n-fold) = 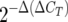 , where ΔCT is CT (target) − CT (16S) and Δ(ΔCT) is ΔCT (experimental) − ΔCT (control). Standard deviations were calculated for the samples.
, where ΔCT is CT (target) − CT (16S) and Δ(ΔCT) is ΔCT (experimental) − ΔCT (control). Standard deviations were calculated for the samples.
Complementation of the ΔOx mutant.
To complement the ΔOx mutant, the MAP3464, MAP3465, and MAP3466 genes were PCR amplified using wt genomic DNA as a template. The sequences of the upstream and downstream primers were as follows: MAP3464 forward, 5′-TTT GGA TCC ATG CCA CCC TCC GGA CGT-3′; MAP3464 reverse, 5′-TTT GGA TCC TTA AGT TGC GGC GCT GGT GTG-3′; MAP3465 forward, 5′-TTT GAA TTC ACA CCA GCG CCG CAA CTT AAT-3′; MAP3465 reverse, 5′-TTT GAA TTC GGC CGG CCT CAG CGT TTC AG-3′; MAP3466 forward, 5′-AAA GTT AAC CTG GGT GTG ATG ACT CGC TCC-3′; and MAP3466 reverse, 5′-AAA GTT AAC TCA GCG CCG CAA CCG GAT GCG-3′. The MAP3464, MAP3465, and MAP3466 PCR products were cloned into the pGEM vector (Promega) and verified by sequencing using the T7 promoter. Subsequently, the PCR products were digested with BamHI, EcoRI, and HpaI, respectively, prior to cloning into pMAV261-AprII, a vector containing the hsp60 promoter and an apramycin-resistant gene. Plasmids pMAV261-MAP3464 and pMAV261-MAP3464-65-66 were electroporated into ΔOx competent cells as described before. The transformants were then plated onto 7H11 agar plates containing 200 μg/ml of apramycin and kanamycin. To confirm transformation, the MAP3464 gene was identified in the complemented mutant clones by PCR amplification as described before.
Cell surface biotinylation.
Biotinylation of bacterial surface proteins for Western blotting was performed using a protein biotinylation system (Amersham, Little Chalfont, Buckinghamshire, United Kingdom). Briefly, three 125-cm2 flasks of 90 to 95% confluent MDBK cells were infected at 37°C for 1 h with wt M. avium subsp. paratuberculosis or with the ΔOx mutant. The supernatant was collected and extracellular bacteria were pelleted at 3,500 × g for 20 min and resuspended in 40 mM bicarbonate buffer, pH 8.6, at a concentration of 1 McFarland standard. Three milliliters of the suspension was surface labeled by incubation with 40 μl of biotinamidohexanoic acid N-hydroxysuccinimide ester (20 mg/ml) (Sigma) in dimethylformamide for 1 h at 4°C. Unreacted biotinylation reagent was removed by three successive washes in ice-cold phosphate-buffered saline buffer, and biotinylated bacteria was subsequently lysed with 200 μl of ice-cold lysis buffer (250 mM NaCl, 25 mM Tris [pH 7.5], 5 mM EDTA [pH 8], 1% NP-40, and a protease inhibitory cocktail) by rapid mechanical agitation in a bead beater. wt and ΔOx bacteria grown in 7H9 medium biotinylated and lysed in the same manner were used as controls. Cell lysates were centrifuged at 10,000 × g for 2 min at 4°C, and protein concentration in the clarified supernatant was measured using the Bradford assay (Bio-Rad). Total proteins (0.5 μg) were analyzed by electrophoresis on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Bio-Rad) at 120 V for 2 h and transferred to a nitrocellulose membrane in a trans-blot semidry apparatus (Bio-Rad). Protein-containing membranes were blocked overnight in 5% (wt/vol) dry milk in Tris-buffered saline (TBS). After several washes in TBS with 0.1% Tween (TBST), the membrane was probed with 1:1,500 (vol/vol) of horseradish peroxidase (HRP)-conjugated streptavidin for 1 h. The membranes were washed again with TBST and proteins were visualized with an enhanced chemiluminescence detection kit (Amersham).
Immunoprecipitation and Western blotting.
MDBK cells growing in a six-well plate were infected with wt M. avium subsp. paratuberculosis or with the ΔOx mutant at 37°C for 15, 30, 60, or 120 min (MOI, 1:100). Uninfected MDBK cells were used as the control. Monolayers were then washed with HBSS and lysed with 1 ml of sterile water in the presence of 0.05 M phenylmethylsulfonyl fluoride in isopropanol. Cells were gently scraped off the plates, passed through a 21-gauge needle 10 times, and centrifuged at 3,500 × g for 10 min at 4°C. The protein concentration in the supernatant was measured using the Bradford assay (Bio-Rad). Equal amounts of cell lysates were incubated for 1 h at 4°C with 10 μl of α-phosphotyrosine or α-phosphothreonine monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Immunocomplexes were captured by adding 30 μl Protein A/G Plus-agarose immunoprecipitation beads (Santa Cruz Biotechnology) and gently rocking at 4°C for 30 min. Beads were washed several times with phosphate-buffered saline, and the captured proteins were resolved on a 12% SDS-PAGE gel at 120 V for 2 h and transferred to a nitrocellulose membrane using a semidry transfer apparatus (Bio-Rad). Protein-containing membranes were blocked overnight with 5% dry milk in TBS. After several washes in TBST, the membranes were probed with α-Cdc42, α-RhoA, and α-Rac antibodies (Santa Cruz Biotechnology) in TBS. Blots were washed with TBST and incubated with HRP-conjugated secondary antibody (Amersham) in TBS for 1 h. Proteins were visualized with an enhanced chemiluminescence detection kit (Amersham) followed by autoradiography.
Affinity precipitation of active Cdc42, in-gel tryptic digestion, and mass spectrometry.
Affinity precipitation of active Cdc42 from MDBK-infected cells was carried out using the EZ-Detect Cdc42 activation kit according to the manufacturer's instructions (Pierce, Rockford, IL). Briefly, MDBK cells growing in 125-ml flasks were infected with wt M. avium subsp. paratuberculosis or with the ΔOx mutant for 30 min at 37°C. After infection, the cells were rinsed once with ice-cold TBS and lysed with a cold lysis/binding/wash buffer containing 1 mM phenylmethylsulfonyl fluoride. Cells were scraped from the plate and clarified by centrifugation at 10,000 × g for 15 min in a microcentrifuge. Equal amounts of cell lysates were incubated in a spin column with 20 μg of the p21-binding domain (PDB) of activated kinase 1 (Pak1) in the presence of a SwellGel-immobilized glutathione disc (GST-Pak1-PDB) at 4°C for 1 h. After incubation, the mixture was centrifuged at 7,200 × g for 30 s to remove unbound proteins. The resin was washed three times with lysis/binding/wash buffer, and the sample was eluted in 50 μl of 2× SDS sample buffer without β-mercaptoethanol after boiling at 95°C for 5 min. Samples were centrifuged at 7,200 × g for 2 min and analyzed on a 12% SDS-PAGE gel. As a positive control, cell lysates from uninfected cells were treated with 0.1 mM GTPγ at 30°C for 15 min to activate endogenous Cdc42.
Coomassie dye-stained 1-D gel electrophoresis-separated proteins were digested using an in-gel tryptic digestion kit following the manufacturer's instructions (Pierce, Rockford, IL). Protein bands were excised from the one-dimensional (1-D) acrylamide gel and destained at 37°C for 1 h with 200 μl of 50 mM ammonium bicarbonate in a 50% (vol/vol) acetonitrile solution (destaining solution). Reduction and alkylation of the cystine residues from proteins were performed by adding 50 mM Tris[2-carboxyethyl]phosphine and 100 mM iodoacetamide, respectively. Samples were washed twice by incubation with the destaining solution at 37°C for 30 min, dehydrated by incubation with 50 μl of 100% acetonitrile for 15 min at room temperature, and dried at room temperature for 10 min. For peptide extraction, protein bands were digested with 20 ng of sequencing-grade modified trypsin and 25 μl of 25 mM ammonium bicarbonate at 37°C for 4 h. The digestion mixture was removed and placed in a clean tube for liquid chromatographic separation and mass spectrometry using a Waters nanoAcquity high-pressure liquid chromatography instrument connected to a Waters Q-Tof Ultima global mass spectrometer (Milford, MA). Briefly, 5 μl of each sample was loaded onto a Waters symmetry C18 trap at 4 μl/min. The peptides were then eluted from the trap onto a Waters Atlantis analytical column (10 cm by 75 μm) at 350 nl/min. Peptide “parent ions” were monitored as they eluted from the analytical column with 0.5-s survey scans from m/z 400 to 2,000. Up to three parent ions per scan that had sufficient intensity and had two, three, or four positive charges were chosen for mass spectrometry. The raw data were processed with MassLynx 4.0 to produce pkl files, a set of smoothed and centroided parent ion masses with the associated fragment ion masses. The pkl files were searched with Mascot 2.0 (Matrix Science Ltd, London, United Kingdom) database-searching software and the Swiss protein database, using mass tolerances of 0.2 for the parent and fragment masses. Peaks Studio (Bioinformatics Solutions Inc., Ontario, Canada) was also used to search the data, using mass tolerances of 0.1.
Statistics.
Each experiment was repeated at least three times, and the results were expressed as means ± standard deviations of the means. The significance of the difference between the experimental and control group was analyzed by Student's t test. P values of <0.05 were considered significant.
RESULTS
Mycobacterium avium subsp. paratuberculosis transposon mutants attenuated in their ability to enter MDBK cells.
To identify M. avium subsp. paratuberculosis genes associated with invasion of epithelial cells, a library of transposon mutants containing approximately 2,000 clones was created. Individual screening of 600 mutants for impaired bacterial invasion in MDBK was carried out. Since previous work showed that the efficiency of infection peaks 4 h after infection with the wt bacterium, mutants were allowed to invade MDBK cells for 4 h. As shown in Fig. 1, the screening of the transposon library led to the identification of five mutants (the ΔOx, Δ41, Δ42, Δ32, and Δ37 mutants) with efficiencies of invasion (1.1 to 2.3%) significantly lower than that of the wt bacterium (6.9%). Genomic DNA was isolated from the mutants, and the sequences of the genes associated with invasion were obtained by PCR amplification of the region containing the transposon Tn5367. The DNA sequences of the PCR products encoding the flanking region of the gene inactivated by the transposon were compared with the published M. avium subsp. paratuberculosis genome sequence by use of the BLAST program. All the mutants had one copy of the transposon identified by Southern blotting (data not shown). Sequencing of the five genes showed that NADH-dependent flavin oxidoreductase, nuoL (NADH-ubiquinone oxidoreductase), Mce1D (mycobacterial cell entry), ArcA (arginine deaminase), and trkA (potassium transporter protein) were involved in M. avium subsp. paratuberculosis invasion of MDBK cells. Details regarding the genes are shown in Table 1.
FIG. 1.

Invasion efficiency of wt M. avium subsp. paratuberculosis and transposon mutants 4 h after infection of MDBK cells. MDBK cells were infected at a MOI of 10 and incubated at 37°C for 4 h. The number of intracellular bacteria was calculated as the fraction of the inoculated bacteria that was recovered from the cell lysate. ΔOx mutant, MAP3464; Δ41 mutant, MAP3212; Δ42 mutant, MAP3607; Δ32 mutant, MAP0941; Δ37 mutant, MAP2808. Values represent the means of three experiments ± standard deviations. *, P value of <0.05 compared with the invasion percentage for the wt bacteria.
TABLE 1.
M. avium subsp. paratuberculosis genes identified as playing a role in the invasion of MDBK cells
| Clone | Gene | M. tuberculosis H37Rv homologue | % Amino acid identity-similarity | Protein |
|---|---|---|---|---|
| ΔOx | MAP3464 | Rv3359 | 81-90 | NADH-dependent flavin oxidoreductase |
| Δ41 | MAP3212 | Rv3156 | 69-73 | NADH-ubiquinone oxidoreductase (nuoL) |
| Δ42 | MAP3607 | Rv0172 | 81-90 | Mycobacterial cell entry (Mce1D) |
| Δ32 | MAP0941 | Rv1000 | 63-72 | Arginine deaminase (ArcA) |
| Δ37 | MAP2808 | Rv2691 | 81-90 | Potassium transporter protein (trkA) |
The ΔOx mutant has the transposon inserted into the MAP3464 gene, which codes for a hypothetical protein.
In order to verify the results obtained using the nested suppression PCR, amplification of the MAP3464 gene from the ΔOx mutant or from the wt genomic DNA was carried out using specific primers. As shown in Fig. 2A, the amplification of the MAP3464 gene was achieved from the genomic DNA of the wt bacteria (lanes 2 and 3) but not from the ΔOx mutant (lane 1). As expected, 16S RNA gene amplification was obtained in both cases. Using a nested suppression PCR method, we showed that the transposon was inserted in the MAP3464 gene of the ΔOx mutant, a 1.2-kb ORF homologous to the M. tuberculosis Rv3359 gene, which encodes a possible oxidoreductase. A comparison of the homologous gene regions in the M. avium subsp. paratuberculosis and M. tuberculosis genomes is shown in Fig. 2B. BLAST analysis against other bacterial genomes showed the that MAP3464 gene product is similar to a variety of NADH-flavin oxidoreductases/NADH oxidase enzymes, found mostly in bacteria or fungi, that contain a triose phosphate isomerase-barrel fold domain. The triose phosphate isomerase-barrel fold is a closed barrel structure composed of an eightfold repeat of beta-alpha units, where the eight parallel beta strands on the inside are covered by the eight alpha helices on the outside. The active site is always found at the C-terminal end of this domain. A search in the conserved domain database (rpsblast; NCBI) showed a conserved Old Yellow Enzyme (OYE)-like flavin mononucleotide-binding domain in the MAP3464 gene product. OYE was the first flavin-dependent enzyme identified (22); however, its true physiological role remains elusive to this day. Each monomer of OYE contains flavin mononucleotide as a noncovalently bound cofactor. It uses NADPH as a reducing agent, and as electron acceptors in the catalytic reaction, it uses oxygen, quinones, and α-β-unsaturated aldehydes and ketones. Computation of physical and chemical parameters with ProtParam showed that the MAP3464 gene product has a large number of alanine residues (14.8%), a molecular mass of 43.7 kDa, and a theoretical pI of 9.05. Further sequence analysis, using the Signal IP 3.0 and the PSORT World Wide Web servers for prediction of protein sorting signals and protein localization sites in amino acid sequences, suggested that MAP3464 is a nonsecreted protein synthesized in the bacterial cytoplasm.
FIG. 2.

The ΔOx mutant has the transposon inserted in the MAP3464 gene. (A) PCR from genomic DNA using specific primers for the MAP3464 and 16S RNA genes. Lanes: MW, molecular weight marker; 1, ΔOx mutant; 2 and 3, wt M. avium subsp. paratuberculosis; 4, PCR negative control. (B) Organization of the oxidoreductase operon in M. tuberculosis and M. avium subsp. paratuberculosis genomes.
The MAP3464 gene is located upstream of the MAP3465 (corresponding protein of 93.3 kDa) and MAP3466 (corresponding protein of 87.6 kDa) genes, which are presumed to be coregulated by the same promoter. These genes encode hypothetical proteins highly homologous to the M. tuberculosis Rv1747 gene and other ATP-binding cassette (ABC) transporters. ABC transporters are multidomain transmembrane proteins responsible for the active transport of substances across cellular membranes. ABC transporters are minimally composed of four domains, with two transmembrane domains responsible for binding and transport of substances and two nucleotide-binding domains responsible for coupling the energy of ATP hydrolysis to conformational changes in the transmembrane domains.
The expression of MAP3464 is upregulated upon bacterial contact with MDBK cells.
The expression of MAP3464 in extracellular wt bacteria exposed to MDBK cells was examined by semiquantitative RT-PCR and RT-PCR at 15, 30, and 60 min postinoculation. RNA was extracted and reverse transcribed, and the cDNA was used for the amplification of the genes encoding the MAP3464 protein and 16S RNA. RT-PCR (Fig. 3A) and real-time RT-PCR (Fig. 3B) amplifications demonstrated that the expression of MAP3464 mRNA increases upon 30 min of exposure to MDBK cells compared to what was seen for unexposed bacteria. The levels of expression upon 30 and 60 min of contact were comparable, i.e., sixfold increases compared with what was seen for unexposed bacteria. Furthermore, we exposed the ΔOx mutant to MDBK for 1 h and performed RT-PCR to evaluate MAP3464 gene transcription. As shown in Fig. 3C, while MAP3464 was not expressed in the ΔOx mutant (lanes 3 and 4), it was highly upregulated when the wt bacteria were exposed to MDBK cells for 1 h (lane 2). The 16S RNA was used as a control because its expression is not influenced by the conditions used (Fig. 3A, bottom, and C, bottom).
FIG. 3.

Epithelial cell contact triggers upregulation of MAP3464 gene expression. (A) The expression of the MAP3464 and 16S genes in wt M. avium subsp. paratuberculosis upon contact with MDBK cells was examined by semiquantitative RT-PCR. After 0, 15, 30, and 60 min of infection, RNA was extracted from extracellular bacteria and cDNA was synthesized as described in Materials and Methods (lanes 1, 2, 3, and 4). For RT-PCR, the MAP3464 and 16S RNA genes were amplified from 1 μl of cDNA by use of specific primers. (B) Upregulation of MAP3464 gene expression (n-fold) upon 15, 30, and 60 min of infection with wt M. avium subsp. paratuberculosis as determined by real-time RT-PCR. The data represent the average of three independent experiments ± standard deviation. The levels of MAP3464 gene expression at 30 and 60 min after infection were comparable. (C) The expression of the MAP3464 and 16S genes in wt M. avium subsp. paratuberculosis and the ΔOx mutant upon 1 h of contact with MDBK cells was examined by semiquantitative RT-PCR. The expression of MAP3464 increased in wt M. avium subsp. paratuberculosis after 1 h of infection (lane 2) compared with what was seen for bacteria incubated in HBSS at 37°C for 1 h (lane 1). As shown in lanes 3 and 4, MAP3464 was not expressed in the ΔOx mutant in the tested conditions. The 16S RNA gene was used as a control because it is constitutively expressed. MW, molecular weight marker.
Complementation of the ΔOx mutant.
To examine whether the replacement of the MAP3464 gene was associated with reversion of the attenuated phenotype, MDBK cells were infected with the ΔOx complemented mutant containing a copy of the MAP3464 gene. The presence of the intact MAP3464 gene in the ΔOx mutant resulted in the growth of the complemented strain in a fashion similar to that seen for the mutant (data not shown). This result suggested that the inactivation of the MAP3464 gene might have a polar effect on neighboring genes; consequently, the downstream genes in the operon, the MAP3465 and MAP3466 genes, might be responsible for the observed phenotype. As shown in Fig. 4, when the complete operon was supplied, the invasion phenotype was fully restored for the ΔOx mutant. These data strongly suggest that the MAP3464 gene and the two downstream genes play a role in M. avium subsp. paratuberculosis invasion of MDBK cells.
FIG. 4.

Invasion of MDBK cells by the wt bacterium, the ΔOx mutant, and the complemented ΔOx mutant containing a complete copy of the oxidoreductase operon (Ox Com.). Cells were infected with bacteria at a MOI of 10, and the number of intracellular bacteria was determined at 4 h after infection as described in Materials and Methods. Values are the means of three experiments ± standard errors of the mean. P values were <0.05 for the comparison between the Ox mutant and either wt or the complemented ΔOx mutant.
Differential protein surface staining of wt M. avium subsp. paratuberculosis and the ΔOx mutant.
To visualize surface membrane proteins differentially expressed in wt M. avium subsp. paratuberculosis and the ΔOx mutant, whole bacteria were biotinylated after 1 h of exposure to MDBK cells. Bacterial lysates were separated by gel electrophoresis, and incorporated biotin was detected by Western blotting with streptavidin-conjugated HRP. As shown in Fig. 5, a band corresponding to an approximately 34-kDa surface-exposed protein was identified in the wt bacteria (lanes 1 and 2) but not in the ΔOx mutant (lanes 3 and 4). Exposure to MDBK cells does not seem to be required for the expression of the 34-kDa protein on the surface of wt M. avium subsp. paratuberculosis (lane 1), as this protein was detected in lysates of unexposed bacteria as well (lane 2). The surface membrane proteins that were biotinylated represented low-abundance proteins at the whole-bacterium level. For instance, the 34-kDa protein was too low in abundance to be Coomassie stained, and it was necessary to utilize an antibody to allow its detection.
FIG. 5.

Effect of the oxidoreductase mutation on presentation of proteins in the cell surface. MDBK cells were infected at 37°C with wt M. avium subsp. paratuberculosis or with the ΔOx mutant. Extracellular bacteria were collected after 1 h of infection and surface-labeled using biotinamidohexanoic acid N-hydroxysuccinimide ester (20 mg/ml). Labeled bacteria were lysed, and 0.5-μg portions of the cell lysates were analyzed on a 12% SDS-PAGE gel. Gels were transferred to nitrocellulose membranes and probed with HRP-conjugated streptavidin. Lane 2 shows a biotinylated protein identified in extracellular wt M. avium subsp. paratuberculosis upon contact with MDBK cells that was not present in the ΔOx mutant (lane 4). wt M. avium subsp. paratuberculosis and the ΔOx mutant incubated in HBSS for 1 h at 37°C were used as controls (lanes 1 and 3).
The ΔOx mutant failed to activate the Cdc42 signal transduction pathway.
To define the pathway that M. avium subsp. paratuberculosis uses to enter into epithelial cells and whether the defect in oxidoreductase expression affected the induction of actin polymerization, lysates of MDBK cells infected with wt M. avium subsp. paratuberculosis or with the ΔOx mutant at different time points were immunoprecipitated with α-phosphotyrosine or α-phosphothreonine antibodies followed by Western blot analysis with anti-Cdc42 (Fig. 6A) or anti-RhoA (Fig. 6B) antibodies. As shown in Fig. 6, wt M. avium subsp. paratuberculosis induced the activation of Cdc42 and RhoA pathways of internalization 15 and 60 min after infection, respectively. In contrast, the ΔOx mutant failed to use the Cdc42 pathway but was able to enter into the cells by use of the RhoA transduction pathway. As expected, Cdc42 and RhoA were not activated in uninfected cells. Similar quantities of proteins per gel were confirmed by immunoprecipitation/Western blotting with α-Cdc42 antibody (Fig. 6C).
FIG. 6.

Cdc42 and RhoA transduction signals are activated during M. avium subsp. paratuberculosis infection. Lysates of MDBK cells infected with wt M. avium subsp. paratuberculosis or the ΔOx mutant at different time points were immunoprecipitated (IP) with α-phosphotyrosine (αTyr), α-phosphothreonine (αThr), or α-Cdc42 antibodies. Uninfected cells (U) were used as controls. The α-phosphotyrosine or α-phosphothreonine immunoprecipitates were examined by immunoblotting with α-Cdc42 (A) and α-RhoA (B) antibodies. Similar quantities of proteins were confirmed by immunoprecipitation and immunoblotting with α-Cdc42 antibody (C). α-, anti-.
A peptide with homology to the MAP3985c protein binds Cdc42.
To determine whether an M. avium subsp. paratuberculosis-secreted protein delivered to the host cell mediates entry in a Cdc42-like manner, a pull-down assay of active Cdc42 from MDBK cells infected with wt M. avium subsp. paratuberculosis or with the ΔOx mutant was performed. Cell lysates were prepared 30 min after infection and precipitated with GST-Pak1-PDB fusion protein. The pull-down active Cdc42 was then run on a 1-D polyacrylamide gel under nonreducing conditions. As shown in Fig. 7, two bands of approximately 209 kDa (a prominent band) and 45 kDa (approximately the size of Cdc42) were detected in lysates of cells infected with wt M. avium subsp. paratuberculosis (lane 3) or the ΔOx mutant (lane 4), while very little signal was detected in uninfected cell lysates (lane 2). As a positive control, lysates from uninfected cells were treated with 0.1 mM GTPγ to activate endogenous Cdc42 (lane 1). The two predominant bands from each sample were excised from the gel and digested with trypsin, and the resultant peptides were analyzed by mass spectrometry. A bacterial peptide of 17 amino acids (HYELAPASIEGWPLPAK) with homology to the Escherichia coli (100% identity) and Pseudomonas sp. (44% identity) TraA proteins was associated with the Cdc42 proteins of cells infected with wt M. avium subsp. paratuberculosis but not with those infected with the ΔOx mutant. TraA is an extracellular protein with a proposed function in pilin formation. The fact that only one peptide was found bound to Cdc42 probably means that it represents the motif of the protein that recognizes Cdc42. As an alternative explanation, it is known that as less of a protein is injected onto the mass spectrometer, fewer and fewer peptides are detected. It could be there is just too little of the protein in the sample, so only peptides with very good mass spectrometry sensitivity were detected. BLAST analysis against the M. avium subsp. paratuberculosis genome showed that the peptide is homologous to the MAP3985c gene sequence, encoding a hypothetical protein with an estimated molecular mass of 34.5 kDa. The MAP3985c ORF is part of a three-gene operon, and it is located downstream of the senX3/RegX3 transcriptional regulators in the M. avium subsp. paratuberculosis genome. MAP3985c shares homology with Rv0493c, a hypothetical protein of unknown function in M. tuberculosis. Interestingly, the Rv0493c operon is flanked by the senX3/RegX3 and the Gnrt transcriptional regulators.
FIG. 7.

MAP3985c binding to active Cdc42. MDBK cells were infected with wt M. avium subsp. paratuberculosis or with the ΔOx mutant for 30 min at 37°C. Cell lysates were then incubated with 20 μg of the PDB of Pak1 and a SwellGel-immobilized glutathione disc (GST-Pak1-PDB) at 4°C for 1 h. To remove unbound proteins, the resin was washed three times and the samples were eluted in 50 μl of 2× SDS sample buffer without β-mercaptoethanol. The eluted pull-down samples were analyzed on a 12% SDS-PAGE gel. The arrows point to the two bands that were excised from each lane of the gel and analyzed by mass spectrometry, because of prominence and because it correspond to the Cdc42 molecular weight. Lanes: MW, molecular weight marker; 1, uninfected cell lysates treated with 0.1 mM GTPγ; 2, lysates of uninfected cells treated with GDP; 3, lysates of MDBK cells infected with wt M. avium subsp. paratuberculosis; 4, lysates of MDBK cells infected with the ΔOx mutant. After sequencing of the 45-kDa band (approximately the size of the Cdc42 protein), we detected MAP3985c peptide in lane 3 but not in lanes 1, 2, and 4.
DISCUSSION
To cause infection through the gastrointestinal tract, bacterial pathogens, including M. avium subsp. paratuberculosis, are dependent on their ability to overcome the mucosal barrier. In this study, we have demonstrated that the MAP3464 gene encodes a NADH-flavin oxidoreductase associated with the invasion of epithelial cells. The involvement of oxidoreductases in pathogenicity, primarily through oxidative folding of secreted proteins or surface-presented factors such as toxins, adherence factors, pili, and components of the type III secretory system, has been demonstrated for a number of bacteria (28). For instance, Shigella flexneri disulfide oxidoreductase (DsbA) performs an essential role by facilitating the folding and transport to the outer membrane of the surface plasmid antigen (Spa32) protein so that it can participate in the secretion of invasion plasmid antigen (Ipa) proteins, which are essential for entering into epithelial cells (26). DsbA is known to be necessary for the functional maturation of the enterotoxin and for the biogenesis of the type IV pili in Vibrio cholera (17, 29). In enteropathogenic E. coli, DsbA is required for stabilizing the major-subunit structure of the type IV pili (30). In uropathogenic E. coli, DsbA catalyzes disulfide bond formation in a pilin-specific molecular chaperon, PapD, which in turn is required for the assembly of P-pili (7). In Yersinia pestis, a mutation in DsbA results in the unstable expression of an outer membrane protein, YscC, that constitutes the type III secretion system, which leads to the decreased translocation of Yop proteins (8). In each case, a mutation in DsbA usually causes the same phenotypic effect as a mutation in the gene encoding the virulence determinant itself.
Our study indicated that contact between wt M. avium subsp. paratuberculosis and bovine epithelial cells triggers the upregulation of the MAP3464 gene product. In fact, MAP3464 protein expression increased upon 30 min of exposure to MDBK cells. After being expressed, MAP3464 is likely transported to the cell surface, where it plays an essential role in the invasion of MDBK cells. The MAP3464 gene is located upstream of the MAP3465 and MAP3466 genes, which encode transmembrane proteins highly homologous to ABC transporters. ABC transporters, found in eukaryotes and prokaryotes, constitute a large superfamily of multisubunit permeases that act as mechanical pumps that couple ATP hydrolysis to either the uptake or the export of a wide variety of substrates across biological membranes (12). Considering the fact that MAP3464 and the two transporters are adjacent along the chromosome, it seems reasonable to hypothesize that the transporters might be implicated in the transport of MAP3464 from its place of expression in the cytoplasm to the periplasm.
An examination of surface-presented proteins revealed a 34-kDa protein on the surface of wt M. avium subsp. paratuberculosis. The ability of M. avium subsp. paratuberculosis to present the 34-kDa protein on the cell surface is associated with invasiveness, since the ΔOx mutant, incapable of presenting this surface-exposed protein, is less invasive. The disruption of the MAP3464 gene reduces the invasion capacity, perhaps as a consequence of the inefficient folding and translocation across the outer membrane of the 34-kDa protein. Similarly, a mutation in the Dsb protein of S. flexneri affected the folding structure of the Spa protein and its subsequent presentation in the outer membrane and reduced the bacterium's capacity to deliver Ipa proteins (26).
Intracellular pathogens have been shown to trigger host cell cytoskeleton rearrangement through activation of the Rho family of GTPases. For instance, the insertion of S. flexneri IpaC into the host plasma membrane results in changes in the actin cytoskeleton characteristic of the activation of the Rho family GTPases Cdc42 and Rac (24). Our results demonstrated that wt M. avium subsp. paratuberculosis induced the activation of Cdc42 and RhoA pathways of internalization 15 and 60 min after the infection of the host cell, respectively. In contrast, the ΔOx mutant failed to activate the Cdc42 pathway of internalization, but it was able to enter into the cells by use of the RhoA transduction pathway 60 min after infection. While wt M. avium subsp. paratuberculosis was able to enter into the cells 15 min after infection, the mutant needed approximately 45 min more to begin entering into the host cell. This delay in time caused a significant reduction in the infectiveness of the ΔOx mutant compared with that of wt M. avium subsp. paratuberculosis.
Associated to the Cdc42 of cells infected with wt M. avium subsp. paratuberculosis, we identified a peptide with homology to MAP3985c, a hypothetical protein with an estimated molecular mass of 34.5 kDa. We can speculate that the MAP3985c protein and the 34-kDa protein found on the surface of wt M. avium subsp. paratuberculosis might be the same protein, because both of them share the same molecular mass. Moreover, the 34-kDa protein was present on the surface of wt M. avium subsp. paratuberculosis but did not appear on the ΔOx mutant. Based on the fact that contact between the bacteria and epithelial cells resulted in the upregulation of MAP3464, we reasoned that downstream events, such as the presence of the 34-kDa protein on the cell surface and the binding of MAP3985c to Cdc42, are probably triggered by bacterial contact with the host cell as well. However, the secretion mechanism involved in the transport of MAP3985c inside of the host cell and the molecular mechanism that MAP3985c utilizes to activate Cdc42 remain unclear. A recent report suggests that direct GTPase mimicry and subcellular targeting of effectors may be a widely used mechanism in bacterium-host interactions (2). To fully demonstrate that MAP3985c functions as a Cdc42 effector, the overexpression of MAP3985c in the wt bacterium is being carried out.
Acknowledgments
This work was supported by grants from JDIP and USDA. The chromatography and mass spectrometric sequencing were supported in part by the National Institute of Environmental Health Science (NIH) under grant no. P30 ES00210.
We thank Denny Weber and Jill Bartlett for the editing and preparation of the manuscript and Brian Arbogast for assistance with mass spectrometry.
Editor: J. F. Urban, Jr.
Footnotes
Published ahead of print on 15 October 2007.
REFERENCES
- 1.Alonso, A., and G.-D. Portillo. 2004. Hijacking of eukaryotic functions by intracellular bacterial pathogens. Int. Microbiol. 7181-191. [PubMed] [Google Scholar]
- 2.Alto, N. M., F. Shao, C. S. Lazar, R. L. Brost, G. Chua, S. Mattoo, S. A. McMahon, P. Ghosh, T. R. Hughes, C. Boone, and J. E. Dixon. 2006. Identification of a bacterial type III effector family with G protein mimicry functions. Cell 124133-145. [DOI] [PubMed] [Google Scholar]
- 3.Bannantine, J. P., R. G. Barletta, J. R. Stabel, M. L. Paustian, and V. Kapur. 2004. Application of the genome sequence to address concerns that Mycobacterium avium subspecies paratuberculosis might be a foodborne pathogen. Foodborne Pathog. Dis. 13-15. [DOI] [PubMed] [Google Scholar]
- 4.Bannantine, J. P., J. F. Huntley, E. Miltner, J. R. Stabel, and L. E. Bermudez. 2003. The Mycobacterium avium subsp. paratuberculosis 35 kDa protein plays a role in invasion of bovine epithelial cells. Microbiology 1492061-2069. [DOI] [PubMed] [Google Scholar]
- 5.Chacon, O., L. E. Bermudez, and R. G. Barletta. 2004. Johne's disease, inflammatory bowel disease, and Mycobacterium paratuberculosis. Annu. Rev. Microbiol. 58329-363. [DOI] [PubMed] [Google Scholar]
- 6.Dam, T., L. Danelishvili, M. Wu, and L. E. Bermudez. 2006. The fadD2 gene is required for efficient Mycobacterium avium invasion of mucosal epithelial cells. J. Infect. Dis. 1931135-1142. [DOI] [PubMed] [Google Scholar]
- 7.Hultgren, S. J., F. Jacob-Dubuisson, C. H. Jones, and C. I. Branden. 1993. PapD and superfamily of periplasmic immunoglobulin-like pilus chaperones. Adv. Protein Chem. 4499-123. [DOI] [PubMed] [Google Scholar]
- 8.Jackson, M. W., and G. V. Plano. 1999. DsbA is required for stable expression of outer membrane protein YscC and for efficient Yop secretion in Yersinia pestis. J. Bacteriol. 1815126-5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linnabary, R. D., G. L. Meerdink, M. T. Collins, J. R. Stabel, R. W. Sweeney, M. K. Washington, S. J. Wells, R. J. Chiodini, D. Hansen, and R. H. Whitlock. 2001. Johne's disease in cattle, p. 1-10. Issue paper, vol. 17. Council for Agricultural Science and Technology, Ames, IA. [Google Scholar]
- 10.McGarvey, J. A., D. Wagner, and L. E. Bermudez. 2004. Differential gene expression in mononuclear phagocytes infected with pathogenic and non-pathogenic mycobacteria. Clin. Exp. Immunol. 136490-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miltner, E., K. Daroogheh, P. K. Mehta, S. L. Cirillo, J. D. Cirillo, and L. E. Bermudez. 2005. Identification of Mycobacterium avium genes that affect invasion of the intestinal epithelium. Infect. Immun. 734214-4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molle, V., D. Soulat, J. M. Jault, C. Grangeasse, A. J. Cozzone, and J. F. Prost. 2004. Two FHA domains on an ABC transporter, Rv1747, mediate its phosphorylation by PknF, a Ser/Thr protein kinase from Mycobacterium tuberculosis. FEMS Microbiol. Lett. 234215-223. [DOI] [PubMed] [Google Scholar]
- 13.Momotani, E., D. L. Whipple, A. B. Thiermann, and N. F. Cheville. 1988. Role of M cells and macrophages in the entrance of Mycobacterium paratuberculosis into domes of ileal Peyer's patches in calves. Vet. Pathol. 25131-137. [DOI] [PubMed] [Google Scholar]
- 14.Ott, S. L., S. J. Wells, and B. A. Wagner. 1999. Herd-level economic losses associated with Johne's disease on US dairy operations. Prev. Vet. Med. 40179-192. [DOI] [PubMed] [Google Scholar]
- 15.Parker, A. E., and L. E. Bermudez. 1997. Expression of the green fluorescent protein (GFP) in Mycobacterium avium as a tool to study the interaction between mycobacteria and host cells. Microb. Pathog. 22193-198. [DOI] [PubMed] [Google Scholar]
- 16.Patel, D., L. Danelishvili, Y. Yamazaki, M. Alonso, M. L. Paustian, J. P. Bannantine, L. Meunier-Goddik, and L. E. Bermudez. 2006. The ability of Mycobacterium avium subsp. paratuberculosis to enter bovine epithelial cells is influenced by preexposure to a hyperosmolar environment and intracellular passage in bovine mammary epithelial cells. Infect. Immun. 742849-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peek, J. A., and R. K. Taylor. 1992. Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 896210-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohatgi, R., L. Ma, H. Miki, M. Lopez, T. Kirchhausen, T. Takenawa, and M. W. Kirschner. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97221-231. [DOI] [PubMed] [Google Scholar]
- 19.Sangari, F. J., J. Goodman, and L. E. Bermudez. 2000. Mycobacterium avium enters intestinal epithelial cells through the apical membrane, but not by the basolateral surface, activates small GTPase Rho and, once within epithelial cells, expresses an invasive phenotype. Cell. Microbiol. 2561-568. [DOI] [PubMed] [Google Scholar]
- 20.Secott, T. E., T. L. Lin, and C. C. Wu. 2004. Mycobacterium avium subsp. paratuberculosis fibronectin attachment protein facilitates M-cell targeting and invasion through a fibronectin bridge with host integrins. Infect. Immun. 723724-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stender, S., A. Friebel, S. Linder, M. Rohde, S. Mirold, and W. D. Hardt. 2000. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 361206-1221. [DOI] [PubMed] [Google Scholar]
- 22.Stott, K., K. Saito, D. J. Thiele, and V. Massey. 1993. Old Yellow Enzyme. The discovery of multiple isozymes and a family of related proteins. J. Biol. Chem. 2686097-6106. [PubMed] [Google Scholar]
- 23.Tamme, R., E. Camp, R. D. Kortschak, and M. Lardelli. 2000. Nonspecific, nested suppression PCR method for isolation of unknown flanking DNA. BioTechniques 28895-899:902. [DOI] [PubMed] [Google Scholar]
- 24.Tran Van Nhieu, G., E. Caron, A. Hall, and P. J. Sansonetti. 1999. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 183249-3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verma, A., and G. M. Ihler. 2002. Activation of Rac, Cdc42 and other downstream signalling molecules by Bartonella bacilliformis during entry into human endothelial cells. Cell. Microbiol. 4557-569. [DOI] [PubMed] [Google Scholar]
- 26.Watarai, M., T. Tobe, M. Yoshikawa, and C. Sasakawa. 1995. Disulfide oxidoreductase activity of Shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc. Natl. Acad. Sci. USA 924927-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamazaki, Y., L. Danelishvili, M. Wu, M. Macnab, and L. E. Bermudez. 2006. Mycobacterium avium genes associated with the ability to form a biofilm. Appl. Environ. Microbiol. 72819-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu, J., and J. S. Kroll. 1999. DsbA: a protein-folding catalyst contributing to bacterial virulence. Microbes Infect. 11221-1228. [DOI] [PubMed] [Google Scholar]
- 29.Yu, J., S. McLaughlin, R. B. Freedman, and T. R. Hirst. 1993. Cloning and active site mutagenesis of Vibrio cholerae DsbA, a periplasmic enzyme that catalyzes disulfide bond formation. J. Biol. Chem. 2684326-4330. [PubMed] [Google Scholar]
- 30.Zhang, H. Z., and M. S. Donnenberg. 1996. DsbA is required for stability of the type IV pilin of enteropathogenic Escherichia coli. Mol. Microbiol. 21787-797. [DOI] [PubMed] [Google Scholar]