

Abstract
The collectins surfactant-associated protein A (SP-A) and SP-D are components of innate immunity that are present before birth. Both proteins bind pathogens and assist in clearing infection. The significance of SP-A and SP-D as components of the neonatal immune system has not been investigated. To determine the role of SP-A and SP-D in neonatal immunity, wild-type, SP-A null, and SP-D null mice were bred in a bacterium-laden environment (corn dust bedding) or in a semisterile environment (cellulose fiber bedding). When reared in the corn dust bedding, SP-A null pups had significant mortality (P < 0.001) compared to both wild-type and SP-D null pups exposed to the same environment. The mortality of the SP-A null pups was associated with significant gastrointestinal tract pathology but little lung pathology. Moribund SP-A null newborn mice exhibited Bacillus sp. and Enterococcus sp. peritonitis. When the mother or newborn produced SP-A, newborn survival was significantly improved (P < 0.05) compared to the results when there was a complete absence of SP-A in both the mother and the pup. Significant sources of SP-A likely to protect a newborn include the neonatal lung and gastrointestinal tract but not the lactating mammary tissue of the mother. Furthermore, exogenous SP-A delivered by mouth to newborn SP-A null pups with SP-A null mothers improved newborn survival in the corn dust environment. Therefore, a lack of SP-D did not affect newborn survival, while SP-A produced by either the mother or the pup or oral exogenous SP-A significantly reduced newborn mortality associated with environmentally induced infection in SP-A null newborns.
Newborns have an increased risk for developing infection compared to older children and adults (67). This may be a result of the relatively immunocompromised status of a newborn making the transition from the protected and sterile life in utero to the outside world. For example, in humans and mice, antigen-presenting cells (dendritic cells) and immunoglobulin levels are reduced during the first weeks to years of life (2a, 65). In contrast, certain protein components of the innate immune system, such as the collectins, are present and functional prior to birth (7, 18, 46, 50, 61).
The collectins are a family of proteins characterized by collagenous and carbohydrate-binding domains (9). They include surfactant-associated protein A (SP-A), SP-D, and mannan-binding lectin (MBL). MBL is a serum protein produced by hepatocytes, while SP-A and SP-D are proteins secreted locally by epithelial cells (20). Collectins have highly conserved domains that bind microbial components and particles, including lipopolysaccharide, viruses, fungal cell walls, and pollen and dust mite glycoproteins (9, 20). MBL participates in innate immune system function by complement-mediated pathogen killing (24). SP-A and SP-D promote pathogen agglutination and opsonization in addition to neutrophil chemotaxis (35, 52, 58, 64). More recently, SP-A and SP-D have also been found to have direct microbiocidal properties (31, 72).
SP-A is detected in the human fetal lung as early as at 20 weeks during gestation (25) and on day 17 during gestation in the mouse fetal lung (7). In humans, SP-D is also detected in the lung in the late second trimester of gestation and just prior to parturition in rodents (8, 61, 69). SP-A null adult mice do not exhibit altered lung physiology. Conversely, over time SP-D null mice develop a complex pulmonary phenotype that consists of an accumulation of lipid-laden alveolar macrophages and emphysematous structural changes (4, 68). Specifically, at 2 weeks of age increased numbers of macrophages are present in the lung, which is followed by the development of abnormal lung structure at 3 weeks of age. While the lung cellular and inflammatory response to pathogens is impaired in both SP-A and SP-D null mice, increased mortality as a result of spontaneous bacterial infection has not been observed in these animals (4, 33-35). Thus, SP-A and SP-D are present prior to birth, but the absence of either collectin does not seem to place animals at increased risk for death when they are reared in a standard laboratory environment. The effect of the absence of SP-A or SP-D on the health of newborn mice has yet to be determined.
The most abundant surfactant protein in lungs is SP-A (70). SP-A is encoded by two similar genes (SP-A1 and SP-A2) in humans (23), while rodents, such as mice, have only one SP-A gene (45). SP-D is encoded by one gene in humans and by one gene in rodents (28, 47). SP-A and SP-D mRNA are present at nonalveolar sites in humans that may be critical to the observations reported in the current study, specifically in the intestines and mammary tissues (26, 37, 40). SP-A1 and SP-A2 mRNA have been detected in the human small intestine and colon (37, 41). In both of these studies the amount of SP-A gene expression in the intestines is significantly less than that in the lung. SP-A mRNA and protein have also been detected in human breast tissue and ductal epithelial cells, respectively (5). SP-D mRNA has been localized to human intestine and mammary tissues, while SP-D protein has been detected in human small intestine epithelial cells (40). A thorough evaluation of extrapulmonary sites of SP-A production in the C57BL/6 strain of mice was performed using reverse transcription PCR (RT-PCR) by Akiyama et al. (2). In this study, SP-A and SP-D mRNA were present primarily in the lung but also exhibited various patterns of extrapulmonary expression. Both surfactant genes were expressed in the esophagus and stomach, while only SP-A mRNA was detected in the jejunum. Neither SP-A nor SP-D mRNA was detected in murine mammary tissues. In another rodent study, SP-A protein was found to be constitutively expressed in the epithelial cells of the large and small intestines, as shown by immunohistochemistry, immunoblot analysis, and two-dimensional electrophoresis (56). Together, these human and rodent data demonstrate that both SP-A and SP-D are produced at extrapulmonary sites that may be pertinent to newborn survival, specifically in the mammary gland and gastrointestinal (GI) tract.
Given that SP-A and SP-D are components of the innate immune system present at birth, when other immune components are not present, we hypothesized that these proteins may prevent infection in the neonate. We used a murine model of environmental exposure to bacteria and endotoxin-laden organic dust (corn dust) in order to mimic a nonhygienic environment (15). Using this model, we demonstrated that wild-type mice bred in a nonhygienic corn dust environment had significantly smaller litter sizes than wild-type mice bred in a control (semisterile) environment. Adult SP-A null or SP-D null mice did not have increased mortality when they were maintained in the corn dust environment. We observed significant mortality in the SP-A null newborn pups but not in the SP-D null newborn pups reared in the corn dust bedding. The SP-A null newborn pups also developed gastrointestinal pathology and Bacillus sp. or Enterococcus sp. peritoneal infection. Both maternal SP-A production and neonatal SP-A production independently had positive effects on newborn survival in the nonhygienic environment. Finally, exogenous purified human SP-A delivered by mouth to the newborn SP-A null pups improved survival in the corn dust bedding environment.
MATERIALS AND METHODS
Animals.
Male and female C3HeB/FeJ mice from Jackson Laboratories (Bar Harbor, ME) and Swiss Black mice from Taconic (Hudson, NY) were purchased at 6 weeks of age to create breeding colonies at our animal care facility. SP-A null mice with a C3H/Hen background were generously provided for these studies by F. McCormack (University of Cincinnati). These mice were derived from SP-A null mice originally produced using a Swiss Black genetic background (30). Both the C3HeB/FeJ and C3H/Hen mice are substrains of C3H mice, and they have been shown to differ from each other by only 1 of 1,638 nucleotide polymorphisms examined (51). Mice belonging to the C3HeB/FeJ substrain were used as the primary wild-type control animals and have been used previously in our laboratory to study the environmental effects of bacteria and endotoxin (14-16). The SP-D null mice with the Swiss Black background were generously provided by J. Whitsett (University of Cincinnati) and were compared to wild-type Swiss Black mice (Taconic). All mice were housed in isolation cubicles with microisolator lids on individual cages. In all studies, mice that served as controls were maintained within the same isolation cubicle in individual cages adjacent to the experimental mice during the same time period. All pups were born and allowed to mature in the presence of both parents in either the control or corn dust bedding environment. Pups were weaned at 21 days of age. Mice were provided food and water ad libitum. All procedures were performed according to protocols approved by the Animal Care and Use Committee at the University of Iowa.
Environmental exposure model.
In order to create a nonhygienic environment rich in organic microbes, breeding pairs and their litters were exposed to an organic dust used as bedding material, as previously described (15). Corn dust collected from the drying system at a local grain elevator during October was used to create the nonhygienic environment. Two batches of corn dust were used in the experiments. Batch A corn dust was used for experimental exposures from 2003 to 2005 and has previously been characterized (15). Batch B corn dust was used for experimental exposures from 2005 to 2007. The control environment for all experiments consisted of a cellulose fiber bedding, Cellu-Dri (Shepherd Specialty Papers, Kalamazoo, MI), the bedding material routinely used in our animal care facility.
Endotoxin measurements.
The endotoxin contents of the bedding and purified human SP-A were determined by the kinetic chromogenic Limulus amebocyte lysate assay (Whittaker Bioproducts, Walkersville, MD) as previously reported (63). The endotoxin content of the purified SP-A was 0.012 ng/μg protein.
Necropsy and histological examination.
Necropsies for animals that were euthanized with an overdose of isoflurane when they appeared to be “critically ill” (dehydrated with reduced spontaneous movement and a distended abdomen) were photographed. Histological examination of the lower respiratory tract and GI tissues was performed using mice that were ∼24 h old. The GI system (i.e., the stomach to the distal large bowel) was dissected en block and then placed into fixative (zinc-formalin or 10% formalin) for 7 days. The right heart was then flushed with ice-cold phosphate-buffered saline (PBS), and the lungs were inflated with fixative via the trachea at a pressure of 25 cm H2O, as previously described (15). All tissues were embedded in paraffin, and 5-μm-thick sections were mounted on glass slides and then stained with hematoxylin and eosin.
Bacterial cultures for the bedding and animals.
Bacterial identification and quantification of bacteria in the bedding materials were performed using standard microbiological techniques. Using sterile technique, peritoneal fluid, blood, and minced lung were collected on a sterile culture swab (BBL CultureSwab; Becton, Dickinson and Co., Sparks, MD) and submitted for bacterial identification. Bacteria were identified by either the Clinical Microbiology Laboratory or the University Hygienic Laboratory at the University of Iowa.
Heterozygous breeding and newborn survival.
Crossing C3HeB/FeJ (SP-A+/+) mice with C3H/Hen (SP-A−/−) mice allowed us to generate SP-A heterozygous breeder mice. The heterozygous breeders were then paired with SP-A null breeders using the strategy shown in Fig. 1. Using the heterozygous breeding strategies ensured that the SP-A gene mutation was controlled for while the C3H substrain backgrounds were randomly mixed. Two different crosses were performed. In the first cross (cross A), the female was heterozygous for SP-A and the male was SP-A null. In the second cross (cross B), the female was SP-A null and the male was heterozygous for SP-A. Since the progeny of these crosses were either SP-A null (50%) or SP-A heterozygous (50%), there were four mother/neonate outcomes, which are indicated in Fig. 1. Throughout this portion of the study, all breeding pairs and their offspring were observed two or three times per day. The total number of pregnancies was defined as the number of pregnancies that resulted in the birth of live pups. Only pups that were alive at the time of birth were used for data collection.
FIG. 1.

Heterozygous breeding strategy. The offspring of C3HeB/FeJ SP-A+/+ and C3H/Hen SP-A−/− breeding pairs were used to generate SP-A+/− breeders. In breeding pair cross A, the female was heterozygous for SP-A, and in cross B, the male was heterozygous for SP-A. These SP-A+/− breeders were crossed with C3H/Hen SP-A null mice, thus separating the effects of maternal SP-A production from the effects of neonatal SP-A production on the offspring. The SP-A genotypes of mothers and offspring are indicated. The mother and pup SP-A phenotypes are indicated by a plus sign (able to produce SP-A) or a minus sign (not able to produce SP-A). For example, the phenotype of an SP-A+/− mother with an SP-A+/− pup resembled the wild-type phenotype, while an SP-A+/− mother with an SP-A−/− pup allowed us to study the influence of maternal SP-A on an SP-A null pup. An SP-A−/− mother with an SP-A+/− pup allow us to study the influence of endogenous SP-A from the pup in the absence of maternal SP-A, while an SP-A−/− mother with an SP-A−/− pup allow us to study the SP-A null phenotype. All animals used for this breeding strategy were exposed to batch B corn dust bedding. WT, wild type.
SP-A genotyping.
Genotyping for all breeders and their offspring was performed with DNA isolated from tail clips or ear punches, using tail lysis buffer (Viagen Biotech, Los Angeles, CA). PCR primers amplified the SP-A DNA sequence that had been deleted to create the mutated SP-A null mice (30). The SP-A primers amplified a region that included portions of SP-A exon 3-4 and intron 3 (forward primer GCAGAGATGGGAGAGATGGTATCAA and reverse primer ATGGACCTCCATTAGCATGTGGGA). PCR primers were also used to amplify the neomycin insert placed into the SP-A gene (forward primer TGAATGAACTGCAGGACGAG and reverse primer ATACTTTCTCGGCAGGAGCA) in the SP-A null mice. The two PCRs amplified the wild-type and mutated SP-A genes, respectively. The DNA template was denatured at 94°C and annealed at 60°C and extension was performed at 74°C for 25 cycles. PCR products were electrophoresed on a 1.0% agarose gel and photographed.
SP-A mRNA determination.
Real-time RT-PCR analysis of SP-A mRNA was performed using lactating and nonlactating mammary tissues and newborn intestinal tissues. RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA). A one-step RT-PCR was performed using the SuperScript III Platinum One-Step quantitative RT-PCR system (Invitrogen). The samples were analyzed using 6-carboxyfluorescein-labeled SP-A and 18S rRNA (housekeeping gene) primers (TaqMan gene expression assays; Applied Biosciences, Foster City, CA) and a Stratagene Mx3000P instrument. The cycle thresholds (CT) for both SP-A mRNA and 18S mRNA were used to determine relative SP-A gene expression, using the 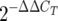 calculation, and the GI tract value was used as the reference value (38). The lower limit of detection for the CT was 40 cycles. The following
calculation, and the GI tract value was used as the reference value (38). The lower limit of detection for the CT was 40 cycles. The following 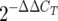 calculation was used: ΔCT = SP-A CT − 18S mRNA CT, with the GI tract value used as the reference value (ΔΔCT = test tissue ΔCT − GI tract ΔCT).
calculation was used: ΔCT = SP-A CT − 18S mRNA CT, with the GI tract value used as the reference value (ΔΔCT = test tissue ΔCT − GI tract ΔCT).
SP-A purification and administration.
The method used to isolate and purify SP-A from lavage fluid obtained from alveolar proteinosis patients has previously been described (48). Briefly, the lavage material was pelleted and delipidated with isopropyl ether and 1-butanol. The aqueous phase was collected and precipitated with 100% ethanol. The precipitated pellet was resuspended in 20 mM KH2PO4 and further purified using an Affi-gel Blue column (Bio-Rad, Hercules, CA). The column flowthrough was dialyzed against distilled H2O, and the protein concentration was determined by a Bradford assay (Bio-Rad). The purity of the SP-A was assessed by electrophoresis on a polyacrylamide gel, followed by staining with Coomassie blue (GelCode Blue stain; Pierce, Rockford, IL).
Purified human SP-A was stored at −80°C until it was used. The protein was diluted in sterile PBS just prior to administration. Newborn mice received purified human SP-A (5 μg/5 μl) delivered per os twice in the first 24 h of life via a flexible gel loading tip and pipette. This dose is within the range used in murine studies of pulmonary infection (35, 39). Sham-fed littermates received 5 μl sterile PBS via the same delivery technique.
Statistical evaluation.
Log rank analysis was used to determine the relationships between the different outcomes presented in Kaplan-Meier survival curves (SigmaStat, version 3.0). A chi-square test was used to evaluate the genetic segregation of the SP-A gene mutation. For all other statistical comparisons, Student's t test was performed. A P value of <0.05 was considered significant. All data were expressed as means ± standard errors of the means.
RESULTS
Environmental exposure.
Two batches of corn dust were used in the following experiments. Corn dust batch A has been characterized previously (15). Batch A corn dust contained 57 times more endotoxin than the control bedding. Consistent with these findings, the average endotoxin content in the second corn dust batch (batch B) was 259 to ± 82 endotoxin units (EU)/mg (n = 4), while the control bedding material contained significantly lower levels of endotoxin (average, 11.1 ± 3.3 EU/mg) (P < 0.001). We have previously determined that the presence of mice living in cage bedding does not contribute to environmental endotoxin levels. In our exposure model, the only significant contribution to environmental endotoxin levels was the contribution from the bedding materials (15). The following bacteria and fungi were identified and quantified in the corn dust bedding: in batch A, Klebsiella oxytoca, Klebsiella pneumoniae, Enterobacter sp., Pseudomonas putida, Staphylococcus sp., Proteus sp., Enterococcus sp., Bacillus sp. (not Bacillus anthracis), and Rhizopus; and in batch B, K. pneumoniae, Microbacterium sp., Oerskovia sp., Bacillus sp. (including Bacillus cereus), Chryseobacterium sp., Enterobacter sp., other gram-negative rods, Acremonium sp., Mucor sp., Cladosporium sp., and Aspergillus sp. The analyses of corn dust batches A and B were performed by the Clinical Microbiology Laboratory at the University of Iowa and the University Hygienic Laboratory. The concentration of bacteria in batch A has been reported previously (15). Batch B contained 7,100 CFU/mg bacteria and 9,600 CFU/mg fungi. In comparison, only two morphologies of Bacillus sp. were grown from the control cellulose bedding material.
Corn dust environment reduces the litter size of wild-type mice.
In our previous studies, C3HeB/FeJ mice were bred in a corn dust bedding environment in order to create a perinatal immune stimulus (15). We performed a retrospective analysis of the litter sizes produced by breeding pairs exposed to corn dust bedding (batch A [see above]) and by breeding pairs exposed to control bedding. There was a significant reduction in the number of pups per litter at the time of weaning (on day 21 of life) for animals exposed to corn dust bedding compared to animals exposed to control bedding (2.93 ± 0.37 and 5.34 ± 0.43 pups/litter, respectively; P < 0.001). This translated to a ∼45% reduction in litter size at the time of weaning as a result of exposure to the corn dust environment.
SP-A, but not SP-D, is critical for newborn survival in the corn dust environment.
SP-A null mice, SP-D null mice, and the corresponding control wild-type mice were bred in corn dust (batch A [see above]) or control bedding. Each of the SP-A and SP-D null breeding pairs produced at least two litters of pups. The litter size at weaning, the number of pregnancies, and the number of adult breeder deaths were recorded during a 5-month period (Table 1). As we have previously observed, the C3HeB/FeJ wild-type mice had smaller litters in the corn dust bedding than the animals exposed to the control bedding (P < 0.05). The four SP-A null breeding pairs maintained in the corn dust environment had 10 pregnancies that resulted in the birth of live pups after 19.5 to 20 days of gestation. While all 10 pregnancies produced live pups, none of the pups survived longer than 48 h (Table 1). In contrast, the SP-A null breeding pairs maintained in control bedding produced ∼5 pups/litter in 24 pregnancies. Similar to the C3HeB/FeJ wild-type mice, the Swiss Black wild-type mice produced smaller litters in the corn dust bedding than in the control bedding; however, the difference did not reach statistical significance (Table 1). Further, there was no significant effect of the corn dust bedding on the survival of SP-D null pups. During the 5-month time period only 1 of 28 adult mice exposed to corn dust bedding died. This animal was a C3HeB/FeJ wild-type mouse. Finally, to ensure that the SP-A null breeders could successfully rear pups, one pair maintained in corn dust was placed into control bedding. This breeding pair had previously delivered two litters in the corn dust bedding environment, but all of the pups had died. Placing this breeding pair into the control bedding allowed them to have three subsequent litters of pups with an average of 3.5 ± 2.1 pups/litter at weaning.
TABLE 1.
Effect of corn dust on adult and newborn survivala
| Mice | Genotype | Bedding | No. of pregnancies | No. of adult deaths/total no. | No. of pups/litter (mean ± SEM) |
|---|---|---|---|---|---|
| C3H | SP-A+/+ | Control | 17 | 0/20 | 5.8 ± 0.7 |
| Corn dust | 14 | 1/8 | 2.8 ± 0.6b | ||
| SP-A−/− | Control | 24 | 0/10 | 5.0 ± 0.4 | |
| Corn dust | 10 | 0/8 | 0c | ||
| Swiss Black | SP-D+/+ | Control | 12 | 0/6 | 8.5 ± 1.0 |
| Corn dust | 9 | 0/6 | 6.1 ± 1.2 | ||
| SP-D−/− | Control | 14 | 0/6 | 8.7 ± 0.5 | |
| Corn dust | 7 | 0/6 | 7.9 ± 0.7 |
Adult deaths and litter size at the time of weaning for SP-A+/+ and SP-A−/− mice and SP-D+/+ and SP-D−/− mice exposed to either control or corn dust bedding (batch A) were recorded over the period of time that the breeding pairs were together. The survival data for adult breeders housed in both environmental conditions are expressed as the number of deaths/total number of adult animals in each experimental condition. Litter size was defined as the number of pups at the time of weaning (day 21 of life).
P = 0.012 for a comparison of SP-A+/+ mice in corn dust and SP-A+/+ mice in control bedding.
P < 0.001 for a comparison of SP-A−/− mice in corn dust and SP-A−/− mice in control bedding.
SP-A null pups survive in a sterile but high-endotoxin environment.
Autoclaving the corn dust (batch A [see above] Table 1) for 30 min at 120°C rendered it sterile. The endotoxin content of the autoclaved corn dust, however, remained high (170 EU/mg) compared to the content of the control bedding. Three SP-A null breeding pairs were placed in the autoclaved corn dust bedding, six litters were born, and the average number of pups/litter at the time of weaning was 2.67 ± 1.63. Thus, sterilizing the corn dust allowed the SP-A null offspring to survive until weaning. The litter size for the SP-A null breeding pairs maintained in autoclaved corn dust did not return to the baseline value (5.0 ± 0.4 pups/litter) observed when SP-A null mice were born in control, semisterile bedding (P < 0.05).
GI pathology in newborn SP-A null mice exposed to corn dust bedding.
Necropsy and histological examination of the newborn SP-A null mice born in the corn dust bedding were performed, and the results were compared to the results for SP-A null mice born in control bedding and also to the results for wild-type mice born in both environments. Figure 2 shows a photograph of a representative of many SP-A null mice born in the corn dust bedding. The pups appeared to be critically ill (i.e., they had gasping respiration, bloated abdomens, signs of dehydration) at the time that they were euthanized. Gross abnormalities were observed in the intestines, while other abdominal and thoracic organs appeared to be normal. These characteristics were present in critically ill SP-A null mice exposed to corn dust but were not observed in age-matched wild-type mice exposed to the same environment (data not shown). The gross morphology of the GI tract, from the stomach to the distal large bowel, from a healthy 24-h-old wild-type mouse was compared to that of the GI tracts from two SP-A null mice reared in the corn dust bedding (Fig. 3). The stomach and proximal small bowel of the wild-type mouse were white and full of milk, whereas the SP-A null mice had empty bile-colored stomachs and proximal small bowels. Histological examination of lung tissues from 24-h-old wild-type and SP-A null mice reared in both environments revealed no difference in lung structure between the genetic strains or between the environmental conditions. Blood congestion was however consistently observed only in the lungs of SP-A null mice reared in corn dust and critically ill at 24 h of age. In the GI tract, the most consistent structural difference observed between the small bowel of SP-A null mice exposed to corn dust and the small bowel of SP-A null mice reared in control bedding or the small bowel of wild-type mice reared in either bedding was a marked dilation of the intestinal lumen (Fig. 4). Histological differences were also observed in the GI tracts of 24-h-old SP-A null mice exposed to corn dust compared to the GI tracts of SP-A null mice reared in control bedding (Fig. 5). Specifically, in the stomachs of SP-A null mice exposed to corn dust, we observed neutrophil accumulation and sloughing epithelial cells with condensed nuclei (Fig. 5B and E). The proximal small bowel of critically ill SP-A null pups reared in corn dust bedding had more readily detectable neutrophils within and marginated along the walls of moderately congested gastric blood vessels (Fig. 5D and F).
FIG. 2.

Representative photograph of thoracic and abdominal organs in an SP-A null mouse born and reared in corn dust bedding. The animal appeared to be critically ill at approximately 24 h of age. The abdominal and thoracic organs are labeled. Abnormal intestinal gas was observed throughout the intestines (asterisks). Bar = 5 mm.
FIG. 3.

Representative GI tracts from SP-A null and wild type (WT) mice born and reared in the corn dust environment. The mouse on the left died after 8.5 h of life, while the other animals were euthanized after 24 h of life. An asterisk indicates the stomach, and an arrow indicates the site of the proximal intestine leaving the stomach. The stomach of the wild-type mouse is full of white milk, while the stomachs of the SP-A null mice contain little milk and their proximal intestines are bile colored.
FIG. 4.

Structure of the proximal small bowel of 24-h-old wild-type and SP-A null mice born and reared in control and corn dust bedding environments. The proximal small bowel structures were similar in the two strains of mice born in control bedding. The only consistent structural alteration observed in the SP-A null mice that appeared to be critically ill in the corn dust bedding was dilation of the intestinal lumen (L) and a paucity of fecal matter. The images are representative of four to six mice per condition. Bar =100 μm.
FIG. 5.

Inflammatory changes in the GI tract of critically ill SP-A null mice exposed to corn dust bedding. Twenty-four-hour-old SP-A null mice born and reared in control or corn dust bedding were examined. Representative histological sections from stomachs (A, B, and E) and small intestines (C, D, and F) are shown. SP-A null mice born in corn dust bedding (B) frequently exhibited sloughing gastric epithelial cells with condensed nuclei (asterisk) compared to SP-A null mice born in control bedding (A). On average, we observed 0.80 ± 0.26 sloughing gastric cell/high-power field in the SP-A null mice, which is significantly more (P < 0.05) than we observed in the SP-A null mice exposed to control bedding (0.25 ± 0.13 cell/high-power field). An example of this is shown in panel E. Neutrophils were also observed within and marginating along the gastric blood vessels of critically ill SP-A null pups in corn dust bedding (arrows in panel B). Also, SP-A null mice reared in corn dust bedding frequently exhibited inflammatory changes in the proximal small bowel (D) compared to SP-A null mice reared in control bedding (C). Specifically, neutrophils were frequently observed within the blood vessels at the base of the villi and also outside blood vessel walls (black arrows in panel D). Additionally, collections of mononuclear cells were also observed only in the ill SP-A null newborns exposed to corn dust bedding (open arrow in panel D). These changes in the proximal small bowel are shown at a higher magnification in panel F. These inflammatory findings were not observed in any of the age-matched SP-A null mice born in control bedding. The images are representative of four to six mice per condition. Bar = 50 μm.
Bacteria isolated from critically ill newborn SP-A null mice.
Gross examination of critically ill SP-A null mice born and reared in corn dust bedding frequently revealed abdominal distention. During necropsy, fluid was often noted in the peritoneal space, which was never observed in healthy newborn mice. Bacterial cultures of peritoneal fluid obtained from critically ill SP-A null pups reared in corn dust were obtained using a sterile culture swab and sterile technique (Table 2). Bacillus sp. (not B. anthracis) was isolated from the peritoneum of three of five ill SP-A null pups, while Entercoccus sp. was isolated once. In contrast, three blood cultures and one lung tissue culture from these animals were negative for bacteria.
TABLE 2.
Bacterial culture data for SP-A null newborn mice reared in corn dust beddinga
| Ill appearance | Source | Culture results |
|---|---|---|
| Yes | Peritoneum | Bacillus sp., Enterococcus sp. |
| Blood | Culture negative | |
| Yes | Peritoneum | Culture negative |
| No | Peritoneum | Culture negative |
| Yes | Peritoneum | Bacillus sp. |
| Blood | Culture negative | |
| Yes | Peritoneum | Bacillus sp. |
| Lung | Culture negative | |
| Yes | Peritoneum | Culture negative |
| Blood | Culture negative |
Twenty-four-hour-old SP-A null mice born in corn dust bedding were described as critically ill if they had at least two of the following characteristics: distended abdomen, dehydration, decreased spontaneous movements, and abnormal respiration pattern. The source of the bacterial culture is indicated, as are the culture results.
Effects of maternal and neonatal SP-A on newborn survival.
Data for survival from birth to day 21 were collected for offspring born to heterozygous breeding pairs (Fig. 1) or wild-type breeding pairs maintained on corn dust bedding (batch B [see above]) or control bedding. Tissue samples for genotyping were available for 93% of all live born offspring and correlated with survival. The anticipated SP-A genotypes of the pups generated according to the heterozygous breeding shown in Fig. 1 were 50% SP-A−/− and 50% SP-A+/−. The offspring genotype data for the heterozygous breeding closely followed Mendel's law of segregation (P = 0.789, as determined by the chi-square test with 1 degree of freedom), with 47% of the offspring having an SP-A+/− genotype and 53% of the offspring having an SP-A−/− genotype. Of note, corn dust batch B used for this and the remainder of our studies was not as lethal to the SP-A null pups as batch A was. The rates of survival of wild-type and SP-A null pups reared in the control bedding did not differ (Fig. 6A). Exposure to corn dust significantly reduced the survival of wild-type pups (P = 0.022) compared to the survival of wild-type pups reared in control bedding (71 and 89%, respectively) (Fig. 6A and B). There was no difference in pup survival when SP-A heterozygous pups born to heterozygous mothers were compared to wild-type pups born to wild-type mothers in the corn dust bedding (Fig. 6B). However, a significant reduction in pup survival (P = 0.048) was found when the survival of SP-A null pups born to SP-A null mothers was compared to the survival of SP-A heterozygous pups born to SP-A heterozygous mothers and reared in the same corn dust environment (52 and 73%, respectively). Finally, if either the mother or the pup produced SP-A, the survival of the pup in the corn dust environment was significantly improved (P = 0.048) compared with the survival when there was a lack of SP-A in both the mother and the pup (Fig. 6C).
FIG. 6.

Kaplan-Meier survival curves for newborn mice born and reared in control bedding compared with curves for mice born and reared in corn dust bedding. The mothers' and pups' SP-A genotypes, the bedding, the number of mice per group, and the percent survival are indicated. (A) Survival of wild-type and SP-A null pups in control bedding. The survival data were not different for the wild-type and SP-A null pups. (B) Survival of wild-type and SP-A heterozygous pups (offspring of SP-A heterozygous dames) born and raised in corn dust bedding (batch B). The data demonstrate there was no difference in survival between heterozygous pups of heterozygous mothers and wild-type pups of wild-type mothers. (C) Survival of SP-A heterozygous and SP-A null offspring generated from the SP-A heterozygous breeding pairs (Fig. 1) born and reared in corn dust bedding (batch B) and survival curves from panels A and B. In panel C, three major outcome groups, indicated by brackets 1 to 3, were identified. These distinct groups are distinguished by the absence of exposure to corn dust (bracket 1), the presence of SP-A in the mother, the pup, or both plus exposure to corn dust bedding (bracket 2), and the complete absence of SP-A in the mother and the pup plus exposure to corn dust bedding (bracket 3). Wild-type pup survival was significantly reduced (P = 0.022) (one asterisk) when the pups were exposed to corn dust bedding compared to pups exposed to control bedding (71 and 89%, respectively). The survival rates of heterozygous pups or SP-A null pups with heterozygous mothers (bracket 2) were not statistically different from each other (P = 0.676). A pup's survival in corn dust bedding was significantly improved (P = 0.048) (two asterisks) if either the mother or the pup was heterozygous for SP-A compared to a complete absence of SP-A in both the pup and the mother.
Sources of extrapulmonary maternal and newborn SP-A.
The SP-A mRNA contents of lactating mammary tissues and newborn intestinal tract tissues were analyzed using real-time RT-PCR. Murine lung tissue served as the positive control for SP-A mRNA; the average CT for this control was 20.23 ± 0.24, while the average CT for the intestine was 30.22 ± 0.73. In comparison, the CT for SP-A mRNA in lactating mammary tissues collected 12 h to 12 days postpartum was uniformly greater than 40 (n = 5), suggesting that the SP-A gene is not expressed in the mouse lactating mammary gland. Using the 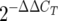 calculation to determine relative SP-A gene expression, the newborn intestine contained 5,000-fold less SP-A mRNA than the lung tissue (1.0 ± 0.65 versus 5,256 ± 1.2; three samples/group).
calculation to determine relative SP-A gene expression, the newborn intestine contained 5,000-fold less SP-A mRNA than the lung tissue (1.0 ± 0.65 versus 5,256 ± 1.2; three samples/group).
Orally administered purified human SP-A improves survival of SP-A null newborn pups.
SP-A null pairs were allowed to breed in the corn dust bedding (batch B [see above]). Newborn pups were fed purified human SP-A (5 μg) twice during the first 24 h of life. Littermates fed sterile PBS (diluent) in place of the SP-A served as the control group. At 5 days of life, the survival rate of the SP-A null newborn pups which received enteral SP-A was significantly improved compared to that of the SP-A null pups that received PBS (81.3 and 45.0%, respectively; P = 0.027, as determined by log rank analysis with 16 and 20 pups/group, respectively) (Fig. 7). At day 21 of life, the positive effect of SP-A treatment on SP-A null pup survival in corn dust bedding compared to the survival of SP-A null pups that did not receive SP-A persisted (P = 0.035).
FIG. 7.
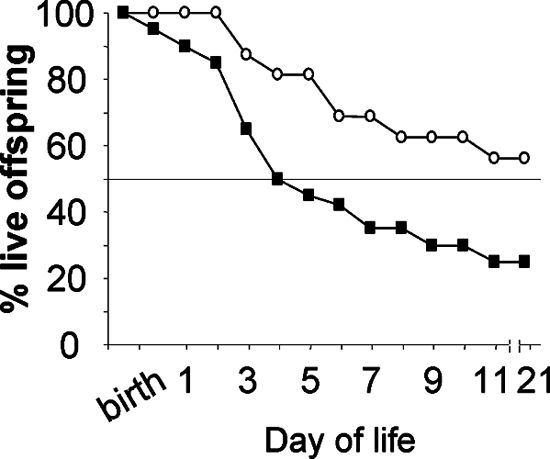
Kaplan-Meier survival curves for SP-A null newborn mice with and without oral SP-A treatment. Newborn SP-A null mice born and reared in corn dust bedding were either given two doses of SP-A protein (5 μg/5 μl) (○) or the same volume of sterile PBS (▪) twice in the first 24 h of life. At 5 days of life, the mice treated with SP-A had a significantly better chance of survival (P = 0.027) than their littermates treated with diluent (PBS). At day 21 of life, this trend for improved survival in the mice that received SP-A compared to the SP-A null mice that did not receive SP-A continued (P = 0.034).
DISCUSSION
We have developed a murine model that permits study of the effects of exposure early in life to a nonhygienic environment on the developing newborn (15). Large amounts of organic dust are created during harvest time in rural communities, and environmental exposure to corn dust is common. In the present study, we used our experimental model to evaluate the roles of SP-A and SP-D in neonatal immunity. Because newborn humans and mice rely on maternal production of immune modulating factors (such as immunoglobulins and cellular immunity) in utero and postpartum, we chose to use environmental conditions in which the mother, developing fetus, and newborn were all exposed to the same environment. We also chose a complex environment (corn dust) as a screening tool, in order to evaluate the effects of common environmental pathogens and immune modulating bacterial components, such as endotoxin. Using this model, we determined that SP-A, but not SP-D, is critically important for survival and prevention of infection in newborns. We also observed that Bacillus sp. may be an important pathogen in the SP-A null immunocompromised state.
The use of corn dust bedding resulted in a decrease in litter size in both C3HeB/FeJ (inbred) and Swiss Black (outbred) wild-type mice. Despite the variable lethality associated with the two different batches of bedding used in our studies, exposure to bacteria in the corn dust played a significant role in newborn morbidity and mortality. While our experimental design did not determine the inciting event that adversely affected the health of animals exposed to corn dust, infection was a consistent component.
The observed significant newborn mortality was unexpected in light of what is known about SP-A and SP-D null mice. Specifically, both strains of mice, while immmunocompromised, do not require strict pathogen-free conditions (Francis McCormack, University of Cincinnati, personnel communication). Both colletins are components of the innate immune system with the ability to bind many bacteria and viruses (71). Numerous studies have demonstrated that SP-A null mice have a reduced ability to clear pathogens from the lung (32, 34). Conversely, despite developing a profound inflammatory response in the lung following bacterial exposure, SP-D null mice can clear bacteria similar to wild-type animals (36). Therefore, while both collectins can bind bacteria, they are functionally distinct with regard to bacterial clearance. Our data support the hypothesis that there are functional differences between SP-A and SP-D that result in a negative impact of corn dust bedding on SP-A null newborn mice but not on SP-D null newborn mice.
SP-A is most abundant in the lung, where it is produced by type II epithelial cells and Clara cells (70). For this reason, we expected to find the newborn lung most adversely affected in the corn dust environment by a lack of SP-A protein. Inflammatory infiltrates were not observed in the lungs of SP-A null newborns reared in either environment. We did, however, observe blood congestion in lung tissue of critically ill SP-A null mice. We hypothesized that this finding is consistent with lung injury associated with a systemic inflammatory process. Specifically, in murine models systemically delivered endotoxin is associated with pulmonary vascular endothelial injury (43, 49). Thus, a primary lung insult is not required for a pulmonary injury to occur. Our data indicate that another organ system, such as the GI tract, was primarily infected and that the associated systemic events contributed to increased pup mortality.
In our mouse model, the intestinal tract of the SP-A null newborns exposed to corn dust had striking abnormalities consistent with inflammation and poor nutritional intake. We also detected bacteria within the peritoneal space of newborn SP-A null mice exposed to the nonhygienic environment. The initiating event(s) leading to infection and ultimately death in the SP-A null pups was not identified in the present study. Ingestion of bacteria while the pups were nursing along with an inability to clear these bacteria from the GI tract is one plausible mechanism. The pathology and infection observed within the intestines and surrounding peritoneal space may have been the result of a disease pathway involving bacterial growth and translocation. Alternatively, disease could also have been the result of combined pathogen and endotoxin exposure leading to a proinflammatory state. Studies supporting this hypothesis demonstrated that in the absence of SP-A, alveolar macrophages have increased activation of NF-κB, a proinflammatory nuclear transcription factor, due to reduced levels of the inhibitory protein I-κB (73). In our mouse model, a cytokine surge in the animals exposed to corn dust might result in poor feeding and shock consistent with a previously described in vivo animal model of neonatal sepsis (53).
The bacterium most commonly detected in the peritoneal space of critically ill SP-A null newborn mice was Bacillus sp. The genus Bacillus is comprised of many closely related species of aerobic, spore-forming, gram-positive, rod-shaped bacteria with flagella (60). These organisms are hardy environmental bacteria commonly found in soil and on plants. B. anthracis is a member of this genus, but it is nonmotile and only rarely causes gastrointestinal infections. In contrast, many B. cereus strains are associated with GI illness (59). Toxins with cytotoxic and emetic properties are produced by numerous Bacillus spp., including non-B. cereus strains (12). Bacillus sp. infection typically does not cause severe illness beyond food poisoning, unless the host falls into one of two relatively immunocompromised categories, premature newborns or cancer patients (13). Bacillus sp. can be a cause of sepsis and bacteremia in premature infants (1, 19, 55). It has been isolated from the peritoneal space and associated with intestinal perforations in a 28-week gestational infant (17), and it was the cause of refractory infection in a 24-week gestational infant (66). Thus, Bacillus sp. is a serious pathogen in high-risk populations, such as newborns, and it may have been the inciting pathogen in our mouse model. To date, interactions between SP-A and Bacillus spp. or their toxins have not been evaluated.
The evidence that SP-A is functional in extrapulmonary sites is relatively new. One example is the function of SP-A in the initiation of parturition (7). Other examples are based on association, specifically genetic association studies. For example, SP-A is found in the human nasal epithelium (27). A polymorphism of the SP-A2 gene is associated with increased risk of meningococcemia, an infectious process that requires colonization of the nasopharynx by Neisseria meningitides (21). GI tract infection or pathology associated with a lack of SP-A production has not previously been characterized. Since a lack of SP-A is associated with reduced bacterial clearance during a lung infection, unlike a lack of SP-D (36), a role for SP-A in the clearance of pathogenic intestinal bacteria in the newborn could be hypothesized.
Two very similar genes, SP-A1 and SP-A2, encode the human SP-A protein, while the mouse has only one SP-A gene (23, 29, 44, 45). In the human population, there are four SP-A1 alleles and five SP-A2 alleles that occur with a frequency of >1% in the general population (10). One of these alleles has been linked to an increased risk for neonatal respiratory distress syndrome and is associated with a reduction in SP-A mRNA levels (22, 54). Therefore, we speculated that GI diseases of the newborn associated with infection, such as necrotizing enterocolitis, may be linked to altered SP-A gene expression early in life.
Our data demonstrate that SP-A produced by either the mother or the newborn is effective in improving pup survival in a nonhygienic environment. The mechanisms by which indirect SP-A (maternal source) and endogenous SP-A (neonatal source) confer newborn immunoprotection appear to be different. We hypothesized that neonatal immunoprotection was conferred by SP-A protein secreted into the newborn digestive tract or by ingestion via maternal milk. SP-A protein secretion by epithelial cells of the lung has been characterized previously (11, 42). Therefore, we speculated that endogenous SP-A secreted by intestinal epithelial cells may have allowed the SP-A heterozygous pups to survive in the corn dust bedding despite having a SP-A null mother. The improved survival in the corn dust bedding of SP-A null newborn pups fed purified human SP-A strongly supports this hypothesis. In contrast, the ability of a heterozygous female to protect her SP-A null pup from early mortality in a nonhygienic environment does not appear to be a direct effect of SP-A production in the mammary tissue. Instead, we hypothesized that SP-A expressed in the mother may have influenced the maternal immune system function and allowed her to pass on, via either the placenta or the mammary gland, cellular or humoral immune components that are vital to the pup for survival in a nonhygienic environment. The ability of SP-A to modulate both the innate and adaptive immune systems in adults has been well established (3, 6), thus making this a plausible mechanism for the indirect effects of maternal SP-A on offspring.
SP-A protein is highly conserved among mammalian, avian, reptilian, and amphibian species and lungfish (62). Phylogenetic evidence suggests that the emergence of SP-A-like proteins occurred very early in the course of evolution. The earliest detection of an SP-A-like protein was in structures such as the swim bladder that developed from the embryonal gut in fish and preceded the evolution of the lung. Therefore, as our data indicate, SP-A may have significant immunological importance in the newborn intestinal tract, conferring immunoprotection against common environmental pathogens. Further, delivery of exogenous SP-A to the GI tract is a novel and effective use of a protein with an innate immune system function.
Acknowledgments
This work was supported by a Parker B. Francis Pulmonary Fellowship, by NIEHS/NIH grant P30 ES05605, and by The Children's Miracle Network.
We thank Rebecca Oberly (National Jewish Center, Denver, CO) for purification of the human SP-A. We also thank Nervana Metwali (University of Iowa) for endotoxin analysis of the bedding materials and purified SP-A.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 29 October 2007.
REFERENCES
- 1.Adler, A., G. Gottesman, T. Dolfin, S. Arnon, R. Regev, S. Bauer, and I. Litmanovitz. 2005. Bacillus species sepsis in the neonatal intensive care unit. J. Infect. 51390-395. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama, J., A. Hoffman, C. Brown, L. Allen, J. Edmondson, F. Poulain, and S. Hawgood. 2002. Tissue distribution of surfactant proteins A and D in the mouse. J. Histochem. Cytochem. 50993-996. [DOI] [PubMed] [Google Scholar]
- 2a.Braun, J., and E. R. Stiehm. 1996. The B-lymphocyte system, p. 35-74. In E. R. Stiehm (ed.), Immunological disorders in infants and children, 4th ed. W. B. Saunders Co., Philadelphia, PA.
- 3.Borron, P. J., E. A. Mostaghel, C. Doyle, E. S. Walsh, M. G. McHeyzer-Williams, and J. R. Wright. 2002. Pulmonary surfactant proteins A and D directly suppress CD3+/CD4+ cell function: evidence for two shared mechanisms. J. Immunol. 1695844-5850. [DOI] [PubMed] [Google Scholar]
- 4.Botas, C., F. Poulain, J. Akiyama, C. Brown, L. Allen, J. Goerke, J. Clements, E. Carlson, A. M. Gillespie, C. Epstein, and S. Hawgood. 1998. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc. Natl. Acad. Sci. USA 9511869-11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braidotti, P., C. Cigala, D. Graziani, B. Del Curto, E. Dessy, G. Coggi, S. Bosari, and G. G. Pietra. 2001. Surfactant protein A expression in human normal and neoplastic breast epithelium. Am. J. Clin. Pathol. 116721-728. [DOI] [PubMed] [Google Scholar]
- 6.Brinker, K. G., H. Garner, and J. R. Wright. 2003. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am. J. Physiol. Lung Cell Mol. Physiol. 284L232-L241. [DOI] [PubMed] [Google Scholar]
- 7.Condon, J. C., P. Jeyasuria, J. M. Faust, and C. R. Mendelson. 2004. Surfactant protein secreted by the maturing mouse fetal lung acts as a hormone that signals the initiation of parturition. Proc. Natl. Acad. Sci. USA 1014978-4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crouch, E., K. Rust, W. Marienchek, D. Parghi, D. Chang, and A. Persson. 1991. Developmental expression of pulmonary surfactant protein D (SP-D). Am. J. Respir. Cell Mol. Biol. 513-18. [DOI] [PubMed] [Google Scholar]
- 9.Crouch, E. C. 1998. Collectins and pulmonary host defense. Am. J. Respir. Cell Mol. Biol. 19177-201. [DOI] [PubMed] [Google Scholar]
- 10.DiAngelo, S., Z. Lin, G. Wang, S. Phillips, M. Ramet, J. Luo, and J. Floros. 1999. Novel, non-radioactive, simple and multiplex PCR-cRFLP methods for genotyping human SP-A and SP-D marker alleles. Dis. Markers 15269-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle, I. R., H. A. Barr, and T. E. Nicholas. 1994. Distribution of surfactant protein A in rat lung. Am. J. Respir. Cell Mol. Biol. 11405-415. [DOI] [PubMed] [Google Scholar]
- 12.From, C., R. Pukall, P. Schumann, V. Hormazabal, and P. E. Granum. 2005. Toxin-producing ability among Bacillus spp. outside the Bacillus cereus group. Appl. Environ. Microbiol. 711178-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaur, A. H., and J. L. Shenep. 2001. The expanding spectrum of disease caused by Bacillus cereus. Pediatr. Infect. Dis. J. 20533-534. [DOI] [PubMed] [Google Scholar]
- 14.George, C. L., H. Jin, C. L. Wohlford-Lenane, M. E. O'Neill, J. C. Phipps, P. O'Shaughnessy, J. N. Kline, P. S. Thorne, and D. A. Schwartz. 2001. Endotoxin responsiveness and subchronic grain dust-induced airway disease. Am. J. Physiol. Lung Cell Mol. Physiol. 280L203-L213. [DOI] [PubMed] [Google Scholar]
- 15.George, C. L., M. L. White, K. Kulhankova, A. Mahajan, P. S. Thorne, J. M. Snyder, and J. N. Kline. 2006. Early exposure to a nonhygienic environment alters pulmonary immunity and allergic responses. Am. J. Physiol. Lung Cell Mol. Physiol. 291L512-L522. [DOI] [PubMed] [Google Scholar]
- 16.George, C. L., M. L. White, M. E. O'Neill, P. S. Thorne, D. A. Schwartz, and J. M. Snyder. 2003. Altered surfactant protein A gene expression and protein metabolism associated with repeat exposure to inhaled endotoxin. Am. J. Physiol. Lung Cell Mol. Physiol. 285L1337-L1344. [DOI] [PubMed] [Google Scholar]
- 17.Girisch, M., M. Ries, M. Zenker, R. Carbon, R. Rauch, and M. Hofbeck. 2003. Intestinal perforations in a premature infant caused by Bacillus cereus. Infection 31192-193. [DOI] [PubMed] [Google Scholar]
- 18.Goss, K. L., A. R. Kumar, and J. M. Snyder. 1998. SP-A2 gene expression in human fetal lung airways. Am. J. Respir. Cell Mol. Biol. 19613-621. [DOI] [PubMed] [Google Scholar]
- 19.Hilliard, N. J., R. L. Schelonka, and K. B. Waites. 2003. Bacillus cereus bacteremia in a preterm neonate. J. Clin. Microbiol. 413441-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmskov, U., S. Thiel, and J. C. Jensenius. 2003. Collections and ficolins: humoral lectins of the innate immune defense. Annu. Rev. Immunol. 21547-578. [DOI] [PubMed] [Google Scholar]
- 21.Jack, D. L., J. Cole, S. C. Naylor, R. Borrow, E. B. Kaczmarski, N. J. Klein, and R. C. Read. 2006. Genetic polymorphism of the binding domain of surfactant protein-A2 increases susceptibility to meningococcal disease. Clin. Infect. Dis. 431426-1433. [DOI] [PubMed] [Google Scholar]
- 22.Karinch, A. M., D. E. deMello, and J. Floros. 1997. Effect of genotype on the levels of surfactant protein A mRNA and on the SP-A2 splice variants in adult humans. Biochem. J. 32139-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katyal, S. L., G. Singh, and J. Locker. 1992. Characterization of a second human pulmonary surfactant-associated protein SP-A gene. Am. J. Respir. Cell Mol. Biol. 6446-452. [DOI] [PubMed] [Google Scholar]
- 24.Kawasaki, N., T. Kawasaki, and I. Yamashina. 1989. A serum lectin (mannan-binding protein) has complement-dependent bactericidal activity. J. Biochem. (Tokyo) 106483-489. [DOI] [PubMed] [Google Scholar]
- 25.Khoor, A., M. E. Gray, W. M. Hull, J. A. Whitsett, and M. T. Stahlman. 1993. Developmental expression of SP-A and SP-A mRNA in the proximal and distal respiratory epithelium in the human fetus and newborn. J. Histochem. Cytochem. 411311-1319. [DOI] [PubMed] [Google Scholar]
- 26.Khubchandani, K. R., and J. M. Snyder. 2001. Surfactant protein A (SP-A): the alveolus and beyond. FASEB J. 1559-69. [DOI] [PubMed] [Google Scholar]
- 27.Kim, J. K., S. S. Kim, K. W. Rha, C. H. Kim, J. H. Cho, C. H. Lee, J. G. Lee, and J. H. Yoon. 2007. Expression and localization of surfactant proteins in human nasal epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 292L879-L884. [DOI] [PubMed] [Google Scholar]
- 28.Kolble, K., J. Lu, S. E. Mole, S. Kaluz, and K. B. Reid. 1993. Assignment of the human pulmonary surfactant protein D gene (SFTP4) to 10q22-q23 close to the surfactant protein A gene cluster. Genomics 17294-298. [DOI] [PubMed] [Google Scholar]
- 29.Korfhagen, T. R., M. D. Bruno, S. W. Glasser, P. J. Ciraolo, J. A. Whitsett, D. L. Lattier, K. A. Wikenheiser, and J. C. Clark. 1992. Murine pulmonary surfactant SP-A gene: cloning, sequence, and transcriptional activity. Am. J. Physiol. 263L546-L554. [DOI] [PubMed] [Google Scholar]
- 30.Korfhagen, T. R., M. D. Bruno, G. F. Ross, K. M. Huelsman, M. Ikegami, A. H. Jobe, S. E. Wert, B. R. Stripp, R. E. Morris, S. W. Glasser, C. J. Bachurski, H. S. Iwamoto, and J. A. Whitsett. 1996. Altered surfactant function and structure in SP-A gene targeted mice. Proc. Natl. Acad. Sci. USA 939594-9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuzmenko, A. I., H. Wu, S. Wan, and F. X. McCormack. 2005. Surfactant protein A is a principal and oxidation-sensitive microbial permeabilizing factor in the alveolar lining fluid. J. Biol. Chem. 28025913-25919. [DOI] [PubMed] [Google Scholar]
- 32.LeVine, A. M., M. D. Bruno, K. M. Huelsman, G. F. Ross, J. A. Whitsett, and T. R. Korfhagen. 1997. Surfactant protein A-deficient mice are susceptible to group B streptococcal infection. J. Immunol. 1584336-4340. [PubMed] [Google Scholar]
- 33.LeVine, A. M., J. Elliott, J. A. Whitsett, A. Srikiatkhachorn, E. Crouch, N. DeSilva, and T. Korfhagen. 2004. Surfactant protein-D enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 31193-199. [DOI] [PubMed] [Google Scholar]
- 34.LeVine, A. M., J. Gwozdz, J. Stark, M. Bruno, J. Whitsett, and T. Korfhagen. 1999. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J. Clin. Investig. 1031015-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeVine, A. M., K. E. Kurak, J. R. Wright, W. T. Watford, M. D. Bruno, G. F. Ross, J. A. Whitsett, and T. R. Korfhagen. 1999. Surfactant protein-A binds group B streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-A-deficient mice. Am. J. Respir. Cell Mol. Biol. 20279-286. [DOI] [PubMed] [Google Scholar]
- 36.LeVine, A. M., J. A. Whitsett, J. A. Gwozdz, T. R. Richardson, J. H. Fisher, M. S. Burhans, and T. R. Korfhagen. 2000. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J. Immunol. 1653934-3940. [DOI] [PubMed] [Google Scholar]
- 37.Lin, Z., D. deMello, D. S. Phelps, W. A. Koltun, M. Page, and J. Floros. 2001. Both human SP-A1 and Sp-A2 genes are expressed in small and large intestine. Pediatr. Pathol Mol. Med. 20367-386. [PubMed] [Google Scholar]
- 38.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 39.Madan, T., U. Kishore, M. Singh, P. Strong, H. Clark, E. M. Hussain, K. B. Reid, and P. U. Sarma. 2001. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J. Clin. Investig. 107467-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madsen, J., A. Kliem, I. Tornoe, K. Skjodt, C. Koch, and U. Holmskov. 2000. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J. Immunol. 1645866-5870. [DOI] [PubMed] [Google Scholar]
- 41.Madsen, J., I. Tornoe, O. Nielsen, C. Koch, W. Steinhilber, and U. Holmskov. 2003. Expression and localization of lung surfactant protein A in human tissues. Am. J. Respir. Cell Mol. Biol. 29591-597. [DOI] [PubMed] [Google Scholar]
- 42.Mason, R. J., M. C. Lewis, K. E. Edeen, K. McCormick-Shannon, L. D. Nielsen, and J. M. Shannon. 2002. Maintenance of surfactant protein A and D secretion by rat alveolar type II cells in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 282L249-L258. [DOI] [PubMed] [Google Scholar]
- 43.Matsuda, N., Y. Hattori, Y. Takahashi, J. Nishihira, S. Jesmin, M. Kobayashi, and S. Gando. 2004. Therapeutic effect of in vivo transfection of transcription factor decoy to NF-κB on septic lung in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 287L1248-L1255. [DOI] [PubMed] [Google Scholar]
- 44.McCormick, S. M., and C. R. Mendelson. 1994. Human SP-A1 and SP-A2 genes are differentially regulated during development and by cAMP and glucocorticoids. Am. J. Physiol. 266L367-L374. [DOI] [PubMed] [Google Scholar]
- 45.Moore, K. J., M. A. D'Amore-Bruno, T. R. Korfhagen, S. W. Glasser, J. A. Whitsett, N. A. Jenkins, and N. G. Copeland. 1992. Chromosomal localization of three pulmonary surfactant protein genes in the mouse. Genomics 12388-393. [DOI] [PubMed] [Google Scholar]
- 46.Mori, K., N. Kurihara, S. Hayashida, M. Tanaka, and K. Ikeda. 2002. The intrauterine expression of surfactant protein D in the terminal airways of human fetuses compared with surfactant protein A. Eur. J. Pediatr. 161431-434. [DOI] [PubMed] [Google Scholar]
- 47.Motwani, M., R. A. White, N. Guo, L. L. Dowler, A. I. Tauber, and K. N. Sastry. 1995. Mouse surfactant protein-D. cDNA cloning, characterization, and gene localization to chromosome 14. J. Immunol. 1555671-5677. [PubMed] [Google Scholar]
- 48.Oberley, R. E., K. A. Ault, T. L. Neff, K. R. Khubchandani, E. C. Crouch, and J. M. Snyder. 2004. Surfactant proteins A and D enhance the phagocytosis of Chlamydia into THP-1 cells. Am. J. Physiol. Lung Cell Mol. Physiol. 287L296-L306. [DOI] [PubMed] [Google Scholar]
- 49.O'Dea, K. P., A. J. Young, H. Yamamoto, J. L. Robotham, F. M. Brennan, and M. Takata. 2005. Lung-marginated monocytes modulate pulmonary microvascular injury during early endotoxemia. Am. J. Respir. Crit. Care Med. 1721119-1127. [DOI] [PubMed] [Google Scholar]
- 50.Ogasawara, Y., Y. Kuroki, M. Shiratori, H. Shimizu, K. Miyamura, and T. Akino. 1991. Ontogeny of surfactant apoprotein D, SP-D, in the rat lung. Biochim. Biophys. Acta 1083252-256. [DOI] [PubMed] [Google Scholar]
- 51.Petkov, P. M., Y. Ding, M. A. Cassell, W. Zhang, G. Wagner, E. E. Sargent, S. Asquith, V. Crew, K. A. Johnson, P. Robinson, V. E. Scott, and M. V. Wiles. 2004. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 141806-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pikaar, J. C., W. F. Voorhout, L. M. van Golde, J. Verhoef, J. A. Van Strijp, and J. F. van Iwaarden. 1995. Opsonic activities of surfactant proteins A and D in phagocytosis of gram-negative bacteria by alveolar macrophages. J. Infect. Dis. 172481-489. [DOI] [PubMed] [Google Scholar]
- 53.Premer, D. M., R. Goertz, M. K. Georgieff, M. C. Mammel, and S. J. Schwarzenberg. 2002. Muscle proteolysis and weight loss in a neonatal rat model of sepsis syndrome. Inflammation 2697-101. [DOI] [PubMed] [Google Scholar]
- 54.Ramet, M., R. Haataja, R. Marttila, J. Floros, and M. Hallman. 2000. Association between the surfactant protein A (SP-A) gene locus and respiratory-distress syndrome in the Finnish population. Am. J. Hum Genet. 661569-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ronnestad, A., T. G. Abrahamsen, S. Medbo, H. Reigstad, K. Lossius, P. I. Kaaresen, I. E. Engelund, L. M. Irgens, and T. Markestad. 2005. Septicemia in the first week of life in a Norwegian national cohort of extremely premature infants. Pediatrics 115e262-268. [DOI] [PubMed] [Google Scholar]
- 56.Rubio, S., T. Lacaze-Masmonteil, B. Chailley-Heu, A. Kahn, J. R. Bourbon, and R. Ducroc. 1995. Pulmonary surfactant protein A (SP-A) is expressed by epithelial cells of small and large intestine. J. Biol. Chem. 27012162-12169. [DOI] [PubMed] [Google Scholar]
- 57.Reference deleted.
- 58.Schagat, T. L., J. A. Wofford, K. E. Greene, and J. R. Wright. 2003. Surfactant protein A differentially regulates peripheral and inflammatory neutrophil chemotaxis. Am. J. Physiol. Lung Cell Mol. Physiol. 284L140-L147. [DOI] [PubMed] [Google Scholar]
- 59.Schoeni, J. L., and A. C. Wong. 2005. Bacillus cereus food poisoning and its toxins. J. Food Prot. 68636-648. [DOI] [PubMed] [Google Scholar]
- 60.Sinchaikul, S., B. Sookkheo, S. Topanuruk, H. F. Juan, S. Phutrakul, and S. T. Chen. 2002. Bioinformatics, functional genomics, and proteomics study of Bacillus sp. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 771261-287. [DOI] [PubMed] [Google Scholar]
- 61.Stahlman, M. T., M. E. Gray, W. M. Hull, and J. A. Whitsett. 2002. Immunolocalization of surfactant protein-D (SP-D) in human fetal, newborn, and adult tissues. J. Histochem. Cytochem. 50651-660. [DOI] [PubMed] [Google Scholar]
- 62.Sullivan, L. C., C. B. Daniels, I. D. Phillips, S. Orgeig, and J. A. Whitsett. 1998. Conservation of surfactant protein A: evidence for a single origin for vertebrate pulmonary surfactant. J. Mol. Evol. 46131-138. [DOI] [PubMed] [Google Scholar]
- 63.Thorne, P. S. 2000. Inhalation toxicology models of endotoxin- and bioaerosol-induced inflammation. Toxicology 15213-23. [DOI] [PubMed] [Google Scholar]
- 64.Tino, M. J., and J. R. Wright. 1999. Surfactant proteins A and D specifically stimulate directed actin-based responses in alveolar macrophages. Am. J. Physiol. 276L164-L174. [DOI] [PubMed] [Google Scholar]
- 65.Tschernig, T., A. S. Debertin, F. Paulsen, W. J. Kleemann, and R. Pabst. 2001. Dendritic cells in the mucosa of the human trachea are not regularly found in the first year of life. Thorax 56427-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tuladhar, R., S. K. Patole, T. H. Koh, R. Norton, and J. S. Whitehall. 2000. Refractory Bacillus cereus infection in a neonate. Int. J. Clin. Pract. 54345-347. [PubMed] [Google Scholar]
- 67.Watson, R. S., J. A. Carcillo, W. T. Linde-Zwirble, G. Clermont, J. Lidicker, and D. C. Angus. 2003. The epidemiology of severe sepsis in children in the United States. Am. J. Respir. Crit. Care Med. 167695-701. [DOI] [PubMed] [Google Scholar]
- 68.Wert, S. E., M. Yoshida, A. M. LeVine, M. Ikegami, T. Jones, G. F. Ross, J. H. Fisher, T. R. Korfhagen, and J. A. Whitsett. 2000. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc. Natl. Acad. Sci. USA 975972-5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong, C. J., J. Akiyama, L. Allen, and S. Hawgood. 1996. Localization and developmental expression of surfactant proteins D and A in the respiratory tract of the mouse. Pediatr. Res. 39930-937. [DOI] [PubMed] [Google Scholar]
- 70.Wright, J. R. 1997. Immunomodulatory functions of surfactant. Physiol. Rev. 77931-962. [DOI] [PubMed] [Google Scholar]
- 71.Wright, J. R. 2005. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 558-68. [DOI] [PubMed] [Google Scholar]
- 72.Wu, H., A. Kuzmenko, S. Wan, L. Schaffer, A. Weiss, J. H. Fisher, K. S. Kim, and F. X. McCormack. 2003. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Investig. 1111589-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu, Y., S. Adam, L. Hamann, H. Heine, A. J. Ulmer, U. Buwitt-Beckmann, and C. Stamme. 2004. Accumulation of inhibitory κB-alpha as a mechanism contributing to the anti-inflammatory effects of surfactant protein-A. Am. J. Respir. Cell Mol. Biol. 31587-594. [DOI] [PubMed] [Google Scholar]