

Abstract
Excessive inflammation contributes to the pathogenesis of bacterial meningitis, which remains a serious disease despite treatment with antibiotics. Therefore, anti-inflammatory drugs have important therapeutic potential, and clinical trials have revealed that early treatment with dexamethasone significantly reduces mortality and morbidity from bacterial meningitis. Here we investigate the molecular mechanisms behind the inhibitory effect of dexamethasone upon the inflammatory responses evoked by Neisseria meningitidis and Streptococcus pneumoniae, two of the major causes of bacterial meningitis. The inflammatory cytokine response was dependent on Toll-like receptor signaling and was strongly inhibited by dexamethasone. Activation of the NF-κB pathway was targeted at several levels, including inhibition of IκB phosphorylation and NF-κB DNA-binding activity as well as upregulation of IκBα synthesis. Our data also revealed that the timing of steroid treatment relative to infection was important for achieving strong inhibition, particularly in response to S. pneumoniae. Altogether, we describe important targets of dexamethasone in the inflammatory responses evoked by N. meningitidis and S. pneumoniae, which may contribute to our understanding of the clinical effect and the importance of timing with respect to corticosteroid treatment during bacterial meningitis.
Bacterial meningitis is a severe acute infectious disease which remains a serious condition, with considerable mortality and morbidity worldwide. Among the most frequent causes of bacterial meningitis are Streptococcus pneumoniae and Neisseria meningitidis. It has been well established for decades that immunopathology, i.e., the exaggerated activation of the host's immune response induced by bacteria or their products, plays a major role in the pathogenesis of bacterial infection in the central nervous system (9). This view is underscored by the fact that mortality from bacterial meningitis, and from pneumococcal meningitis in particular, has not declined significantly for many years, even in the presence of appropriate antibiotic treatment. Large clinical trials have demonstrated that early treatment with glucocorticoids significantly reduces mortality and morbidity from bacterial meningitis in children and, as more recent reports have demonstrated, also in adults (11, 33, 40, 44). These results have led to the recommendation of introducing adjuvant glucocorticoids together with antibiotics for the early treatment of bacterial meningitis (44).
When pathogens enter the central nervous system, they replicate and, in this process, expose microbial material to host cells, which subsequently trigger an inflammatory response. This response is mediated by cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-6, IL-8, and prostaglandins produced by macrophage-equivalent brain cells, including astrocytes and microglia, as well as cerebral capillary endothelial cells (13, 40). The resultant leukocyte recruitment, meningeal inflammation, and increased permeability of the blood-brain barrier, if not orchestrated and regulated tightly, may cause cerebral edema, increased intracranial pressure, and neuronal injury (9).
The innate inflammatory immune response is of crucial importance for the early containment of infection but, at the same time, has the potential to result in immunopathology. The final outcome of infection therefore depends on an intricate balance between the pathogen and the host response. One of the central components of the innate immune system is the family of Toll-like receptors (TLRs). These pattern recognition receptors recognize evolutionarily conserved pathogen-associated molecular patterns present on most types of microorganisms (19). Once TLRs are activated, they signal to the host the presence of infection and trigger signaling cascades leading to antimicrobial and inflammatory responses involving both innate and adaptive immunity (19).
TLR ligand engagement results in intracellular signal transduction, including activation of nuclear factor κB (NF-κB) and mitogen-activated protein kinases (MAPKs). The TLR-activated signaling pathways proceed through adaptor proteins (most importantly MyD88) and lead to activation of the MAPKs and the inhibitory κB (IκB) protein kinase (IKK) complex. IKK in turn phosphorylates IκB and targets it for degradation, hence liberating NF-κB, which migrates to the nucleus and activates transcription of target genes (2, 12, 20, 35, 42).
Glucocorticoids are widely used due to their potent anti-inflammatory and immunosuppressive effects. However, the molecular mechanisms behind these effects are very complex and still not fully understood, although several targets have been identified. Since glucocorticoids are known to interfere with many signaling pathways and molecules involved in TLR signaling, it has been hypothesized that TLR signaling pathways may be important targets for glucocorticoid action and may explain many of their anti-inflammatory and immunosuppressive effects (32). A number of levels at which glucocorticoids can exert their multiple anti-inflammatory effects have been identified, including direct interaction with the transcriptional machinery, interference with upstream signal transduction, and modulation of RNA stability (32). Briefly, glucocorticoids bind specifically to the intracellular glucocorticoid receptor α, thereby promoting dissociation from heat shock protein 90 and subsequent translocation to the nucleus, where this ligand-activated transcription factor can activate the expression of genes with anti-inflammatory effects, including lipocortin, IL-1 receptor antagonist, IL-10, and IκBα genes (1, 4), through binding to glucocorticoid response elements (6). Another major anti-inflammatory mechanism is glucocorticoid-mediated repression of a whole array of genes encoding proinflammatory mediators, such as cytokines, chemokines, and leukocyte adhesion molecules (32). This effect is mainly achieved through direct protein-protein interactions between the glucocorticoid-glucocorticoid receptor complex and the transcription factors NF-κB and AP-1, thereby reducing or preventing interaction with the essential coactivator CBP/p300, resulting in inhibition of their transactivating potential. At the level of signal transduction, glucocorticoids can inhibit upstream proinflammatory signaling through both the NF-κB and MAPK pathways (32). Dexamethasone has been demonstrated to inhibit activation of the MAPKs extracellular signal-regulated kinase 1/2 (ERK1/2), Jun N-terminal kinase (JNK), and p38 by a mechanism involving upregulation and decreased degradation of MAPK phosphatase 1, thereby preventing phosphorylation and activation of these MAPKs (8, 15, 25, 43), and the NF-κB pathway seems also to be targeted at a level upstream of IκB degradation (45).
Here we investigate the effect and mechanism of action of dexamethasone on the inflammatory responses evoked by N. meningitidis and S. pneumoniae, two of the main causes of bacterial meningitis.
MATERIALS AND METHODS
Cell culture.
Peripheral blood mononuclear cells (PBMCs) were isolated from blood obtained from healthy adult donors by Isopaque-Ficoll separation. The blood was diluted, laid on top of Ficoll-Paque (Amersham Biosciences), and centrifuged at 600 × g for 30 min at room temperature. The PBMC-containing interphase was isolated, and the cells were washed in phosphate-buffered saline containing 100 μg of heparin per ml. Subsequently, the cells were centrifuged at 200 × g for 15 min at room temperature and resuspended in RPMI 1640 medium containing 5% heat-inactivated fetal calf serum (FCS). The cells were seeded in 96-well tissue culture plates at a density of 2.0 × 105 cells per well and left overnight to settle before further treatment. The human monocytic cell line THP-1 was grown in RPMI 1640 medium supplemented with 10% FCS and antibiotics. For experiments, cells were seeded in 96- and 6-well tissue culture plates at densities of 2.0 × 105 and 4.0 × 106 cells per well, respectively, and left for 2 h to settle before further treatment. The RAW 264.7-derived cell lines RAW TNF-α 3′ untranslated region (UTR) AU+ and RAW TNF-α 3′ UTR AU÷ (16, 36, 46) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FCS, antibiotics, and 500 μg/ml G418 (Roche, Basel, Switzerland). For experiments, the cells were seeded in six-well tissue culture plates at a density of 2 × 105 cells/well and left for 4 h before further treatment. The cells stably contain a reporter system where chloramphenicol acetyltransferase (CAT) mRNA is expressed constitutively. In one of the cell lines, the 3′ UTR of the CAT-encoding mRNA was taken from wild-type TNF-α (RAW TNF-α 3′ UTR AU+), whereas in the other cell line, the 3′ UTR was again taken from TNF-α, but the AU-rich region (AUR) was mutated (RAW TNF-α 3′ UTR AU÷). The idea is that any observed differences in CAT protein levels can be ascribed to the AUR in the 3′ UTR, which is a major regulator of mRNA stability (22).
Bacteria and reagents.
The bacteria used were the N. meningitidis strain NGO93 and the S. pneumoniae strain SK1013. The bacteria were grown overnight in brain heart infusion broth with 10% Levinthal broth (Statens Serum Institute, Copenhagen, Denmark), reaching a concentration of 18.0 × 108 ± 2.2 × 108 bacteria per ml, as determined in a Thoma counting chamber. Pam3CSK4, lipopolysaccharide (LPS; ultrapure from Escherichia coli O111:B4), and oligodeoxynucleotide (ODN) M362 were all obtained from Invivogen (San Diego, CA). TNF-α was purchased from R&D Systems. The MyD88 homodimerization inhibitory peptide was obtained from Imgenex (San Diego, CA). Dexamethasone was obtained from Pharmacia (Uppsala, Sweden), and cycloheximide was obtained from Sigma-Aldrich (St. Louis, MO).
Purification of RNA and RT-PCR.
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA) according to the recommendations of the manufacturer. Briefly, cells were lysed in TRIzol, and chloroform was added, followed by phase separation by centrifugation. RNA was precipitated with isopropanol and pelleted by centrifugation. Pellets were washed with 80% ethanol and redissolved in RNase-free water. For cDNA generation, 1 μg of RNA was subjected to reverse transcription (RT) with oligo(dT) as a primer and with Expand reverse transcriptase (both from Roche). Prior to RT-PCR, RNA was treated with DNase I (Ambion, Austin, TX) to remove any contaminating DNA, the absence of which was confirmed in control experiments in which the reverse transcriptase enzyme was omitted (data not shown). The cDNA was amplified by PCR using the following primers: for IL-8, 5′-TTG TGA GGA CAT GTG GAA GC-3′ (forward) and 5′-ACA CAG CTG GCA ATG ACA AG-3′ (reverse); for IκBα, 5′-CGG AAT TCC AGG CGG CCG AGC GCC CC-3′ (forward) and 5′-GGG GTA CCT CAT AAC GTC AGA CGC TG-3′ (reverse); and for β-actin, 5′-CCA ACC GTG AAA AGA TGA CC-3′ (forward) and 5′-GCA GTA ATC TCC TTC TGC ATC C-3′ (reverse). The primers were obtained from DNA Technology (Aarhus, Denmark).
Preparation of whole-cell extracts.
To assay for phosphorylation of IκBα, p38, and JNK, cells were seeded in six-well plates as described above and treated with bacteria as specified in the text. At different time points poststimulation, cells were lysed using a Bio-Plex cell lysis kit (Bio-Rad, Hercules, CA) according to the recommendations of the manufacturer. Briefly, the cells were washed with 3 ml cell wash buffer per well and treated with 1 ml lysing solution supplemented with phenylmethylsulfonyl fluoride, followed by incubation for 20 min at 4°C. The suspension was centrifuged at 4,500 × g for 20 min at 4°C, and supernatants were harvested as whole-cell extracts.
Preparation of nuclear extracts.
To isolate nuclear proteins, we used a nuclear extraction kit (Active Motif, Carlsbad, CA). Briefly, cells were washed twice with ice-cold phosphate-buffered saline supplemented with phosphatase inhibitors, scraped off the plate, and spun down (2,000 × g for 1 min) before resuspension in 250 μl 1× hypotonic buffer for 15 min on ice. Twenty-five microliters of the supplied detergent was added, and the mixture was vortexed for 10 s and centrifuged at 14,000 × g for 30 s. The supernatants were removed, and 25 μl of complete lysis buffer was added to the nuclei, which were incubated for 30 min at 4°C with rocking. The samples were vortexed and centrifuged at 14,000 × g for 10 min at 4°C. Supernatants containing nuclear proteins were harvested and transferred to new prechilled tubes.
NF-κB DNA-binding activity.
Enzyme-linked immunosorbent assay (ELISA)-based measurement of the DNA-binding activity of the nuclear NF-κB subunit p65 was performed according to the manufacturer's protocol (Active Motif, Carlsbad, CA). Briefly, 5 μg of nuclear extract was used per sample in duplicate in a 96-well plate precoated with a consensus oligonucleotide for NF-κB (5′-GGGACTTTCC-3′). After washing of wells to remove nonspecific binding, a specific antibody to the NF-κB subunit p65 was added. After antibody binding, the plate was washed again before adding a horseradish peroxidase-conjugated secondary antibody. The peroxidase substrate was added, and the colorimetric change was measured as the optical density at 450 nm.
Luminex technology for detection of cytokines and phosphoproteins.
Cytokine expression and phosphoprotein levels were measured using Luminex technology and Luminex kits purchased from Bio-Rad (total IκBα, phospho-IκBα, p38, JNK, and STAT2) and Biosource (Camarillo, CA) (human IL-8, IL-6, and TNF-α). Briefly, the filter plates were washed with assay buffer, and freshly vortexed antibody-conjugated beads were added to each well. The plate was washed with assay buffer, and samples and standards were added. For detection of cytokines, samples were diluted between 4 and 10 times in growth medium, and for detection of phosphoproteins, 45 μg of protein in 50 μl lysis buffer was used per sample. After a brief shake (30 s at 1,100 rpm), the plate was incubated with shaking (300 rpm) either at room temperature in the dark for 45 min to 2 h (cytokine assay) or overnight at 4°C (phosphoprotein assay). After a wash step, a detection antibody was added to each well, and the plate was shaken (300 rpm) and incubated at room temperature in the dark for 45 min to 2 h (cytokine and phosphoprotein assays). Subsequently, the plate was washed and incubated for 10 min with 50 μl of a streptavidin-phycoerythrin solution, with shaking (30 s at 1,100 rpm and 10 min at 300 rpm). Finally, the plate was washed, 125 μl of assay buffer was added to each well, and the plate was shaken for 10 s at 1,100 rpm and read immediately on a Bio-Plex reader.
Measurement of CAT levels.
Cells were seeded and treated as described above for the appropriate amount of time. For cell lysis and measurement of CAT levels, a CAT ELISA kit from Roche (Basel, Switzerland) was used.
Statistical analysis.
The data are presented as means ± standard errors of the means (SEM). The statistical significance was estimated with the Wilcoxon rank sum test. P values of <0.05 were considered statistically significant.
RESULTS
Dexamethasone inhibits IL-8 production induced by N. meningitidis and S. pneumoniae.
In human PBMCs, N. meningitidis and S. pneumoniae trigger a potent inflammatory response involving several cytokines and chemokines (30). In an initial set of experiments, we examined the ability of the two bacterial species to induce the expression of cytokines. PBMCs were treated with increasing doses of N. meningitidis and S. pneumoniae, and accumulation of IL-8 in the culture supernatants was measured. Both bacteria strongly induced expression of the cytokine, with N. meningitidis being slightly more potent (Fig. 1A). In order to investigate the effect of dexamethasone on proinflammatory cytokine production induced in the presence of these bacteria, PBMCs were seeded in culture and treated as indicated in Fig. 1B to D. Both bacteria and the purified TLR ligands (LPS for TLR4, Pam3Csk4 for TLR2, and ODN M362 for TLR9) induced IL-8, IL-6, and TNF-α, all of which were strongly inhibited by dexamethasone (P < 0.05 for all stimuli). The inhibitory activity was not due to cytotoxic effects of the steroid, as assessed by trypan blue staining (data not shown), and was also specific, since the ability of alpha interferon to phosphorylate STAT2 was not modified by pretreatment with dexamethasone (data not shown).
FIG. 1.

Dexamethasone inhibits induction of proinflammatory cytokines by N. meningitidis and S. pneumoniae. (A) PBMCs were seeded and treated for 20 h with N. meningitidis or S. pneumoniae at increasing doses, from 9 × 104 bacteria/ml to 9 × 107 bacteria/ml. Supernatants were harvested, and IL-8 levels were measured by Luminex technology. (B to D) PBMCs were treated with 1 μM dexamethasone 1 h before stimulation of the cells with LPS (100 ng/ml), Pam3Csk4 (200 ng/ml), ODN M362 (1 μM), or 9 × 107 bacteria/ml of N. meningitidis (N.m.) or S. pneumoniae (S.p.). Twenty hours later, supernatants were harvested and cytokine levels were measured by Luminex technology. (E and F) PBMCs and THP-1 cells were treated with dexamethasone at doses from 1 nM to 1.0 μM 1 h prior to stimulation with 9 × 107 bacteria/ml of N. meningitidis or S. pneumoniae or with LPS (100 ng/ml). Twenty hours later, supernatants were harvested and cytokine levels were measured by Luminex technology. The data are shown as means for triplicate cultures ± SEM. (G) Total RNA was harvested from THP-1 cells pretreated with dexamethasone (1 μM) for 1 h and treated with bacteria (9 × 107 bacteria/ml) for 5 h. IL-8 and β-actin mRNAs were detected by RT-PCR. Similar results were obtained in two or three independent experiments. UT, untreated cells.
Next, we preincubated PBMCs and the human monocytic cell line THP-1 with different doses of dexamethasone 30 min before the addition of N. meningitidis and S. pneumoniae. After 20 h of incubation, the supernatants were analyzed for IL-8. As shown in Fig. 1E and F, both N. meningitidis and S. pneumoniae induced large amounts of IL-8, which was inhibited in a dose-dependent manner by dexamethasone. For N. meningitidis and LPS, the cytokine response was inhibited significantly by all concentrations of dexamethasone used (1 nM to 1 μM), whereas for S. pneumoniae significant inhibition was seen when dexamethasone was present at concentrations from 10 nM to 1 μM. Finally, we examined IL-8 mRNA expression in THP-1 cells and found that dexamethasone inhibited expression of the cytokine, although it exerted a less pronounced effect on S. pneumoniae-induced IL-8 than on that induced by N. meningitidis (Fig. 1G).
N. meningitidis and S. pneumoniae induce cytokine expression through a TLR-dependent mechanism.
We previously investigated the pattern of TLR activation by bacteria causing meningitis and found that N. meningitidis activates TLR2, TLR4, and TLR9, whereas S. pneumoniae activates TLR2 and TLR9 (30). Therefore, we hypothesized that bacterially induced IL-8 production was TLR dependent. To test this, we inhibited TLR signaling, using a cell-permeative MyD88 inhibitory peptide or a control peptide. MyD88 exists as a homodimer when recruited to activated TLRs. The inhibitor peptide contains a sequence from the MyD88 TIR homodimerization domain. MyD88 monomers bind to this inhibitor peptide, thereby blocking MyD88 homodimerization. The cells were treated with the peptide 24 h prior to stimulation, and supernatants were harvested at 20 h poststimulation for measurement of IL-8. As shown in Fig. 2, the ability of both N. meningitidis and S. pneumoniae to induce IL-8 production was strongly inhibited in the presence of the MyD88 inhibitory peptide in both PBMCs (Fig. 2A) and THP-1 cells (Fig. 2B). The specificity of the MyD88 inhibitory peptide was shown by its ability to inhibit IL-8 induction by LPS but not by TNF-α, the latter of which signals independently of TLRs and MyD88 (26). Thus, the two bacteria induced cytokine expression largely through a TLR-dependent mechanism.
FIG. 2.

N. meningitidis and S. pneumoniae induce inflammatory cytokine expression through TLRs. PBMCs (A) or THP-1 cells (B) were seeded and treated with control peptide (Con pep.) or MyD88 inhibitory peptide (100 μM; MyD88 pep.) for 24 h before stimulation for 20 h with 9 × 107 bacteria/ml of N. meningitidis (N.m.) or S. pneumoniae (S.p.), 100 ng/ml of LPS, or 25 ng/ml of TNF-α. Levels of IL-8 in the supernatants were measured by Luminex technology. The data are shown as means for triplicate cultures ± SEM. Similar results were obtained in two independent experiments.
Kinetics of dexamethasone-mediated inhibition of TLR signaling.
In clinical trials of dexamethasone treatment of patients with bacterial meningitis, timing has proved to be important for achievement of significant clinical effects (11). In order to examine if the action of dexamethasone was also dependent on the time of addition relative to the addition of bacteria in our cell culture system and to gain further insight into the mechanism of action of glucocorticoids, we examined the consequences of dexamethasone added prior to, concomitant with, or following stimulation of PBMCs or THP-1 cells with relevant bacteria (Fig. 3). N. meningitidis potently induced IL-8 in both cellular systems, and this response was completely abrogated when dexamethasone was added 2 h prior to or concomitant with bacteria and partially inhibited when the steroid was added at later time points. The inhibition was highly significant even when dexamethasone was added at 7 h postinfection. S. pneumoniae-induced IL-8 production was strongly inhibited when dexamethasone was added between 2 h before and 2 h after the bacteria were added. However, in contrast to the pattern observed with N. meningitidis, dexamethasone was no longer able to mediate significant inhibition of IL-8 production if it was added at later time points. These results indicate that in the case of N. meningitidis, dexamethasone seems to have a maximal effect when it can interfere with signaling at early time points but also has some potency when included several hours after bacterial infection. With respect to S. pneumoniae, dexamethasone similarly works at early time points, fully inhibiting the IL-8 response, but does not have any effect when added later than 2 h after treatment with bacteria.
FIG. 3.
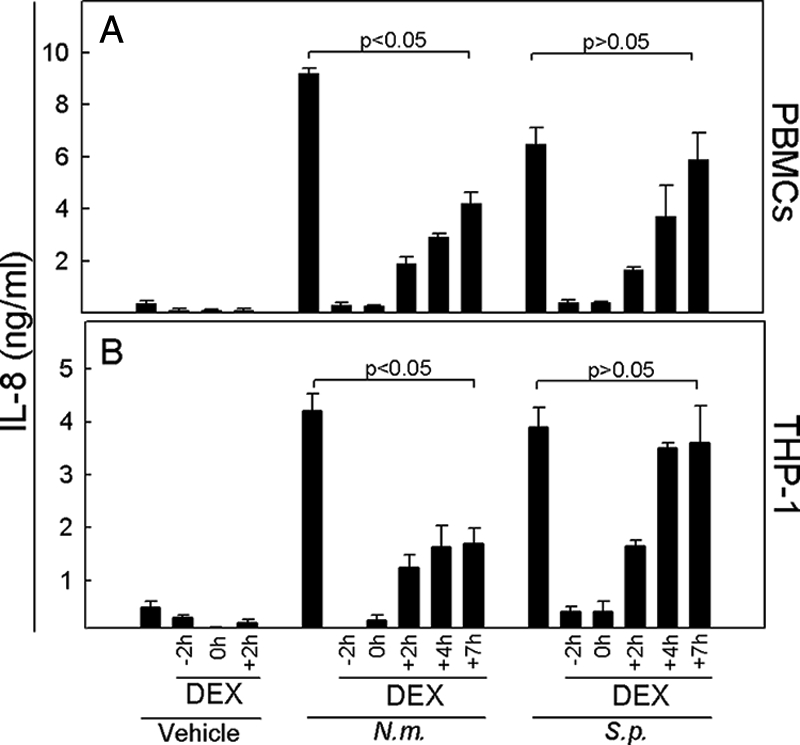
N. meningitidis and S. pneumoniae display differential sensitivity to timing of dexamethasone treatment with respect to induction of cytokine expression. PBMCs (A) or THP-1 cells (B) were treated with 1.0 μM of dexamethasone at the indicated time points before stimulation with 9 × 107 bacteria/ml of N. meningitidis (N.m.) or S. pneumoniae (S.p.). At 20 h poststimulation, supernatants were harvested and levels of IL-8 were determined. The data are shown as means for triplicate cultures ± SEM. Similar results were obtained in two to four independent experiments.
Dexamethasone inhibits N. meningitidis- and S. pneumoniae-induced TLR signaling by interfering with upstream signal transduction.
The data described above strongly suggested that dexamethasone interferes with the bacterium-induced proinflammatory response at an upstream level relatively soon after bacterial challenge. Glucocorticoids are known to interfere with several signaling pathways at different levels (1, 5, 7, 17, 37, 43). In subsequent experiments, we focused on the transcription factor NF-κB, which we reasoned may be activated by these bacteria and also could represent relevant targets for dexamethasone action. THP-1 cells pretreated with dexamethasone for 2 h were incubated for 2 h in the presence of N. meningitidis or S. pneumoniae, and whole-cell extracts were isolated and analyzed by Luminex technology to determine the levels of total and phosphorylated IκBα. As shown in Fig. 4A, N. meningitidis and S. pneumoniae induced three- to fourfold increases in IκBα phosphorylation. Although dexamethasone significantly inhibited the responses induced by both bacteria, it exerted the strongest effect on N. meningitidis-induced IκBα phosphorylation. Following IκBα phosphorylation, the inhibitory protein is ubiquitinated and degraded by the proteasome pathway. Accordingly, total levels of IκBα in cells decreased in response to treatment with bacteria or LPS (Fig. 4B), and this was largely prevented if cells had been pretreated with dexamethasone. Interestingly, we also observed that dexamethasone treatment alone led to a significant and reproducible induction of IκBα mRNA and protein (Fig. 4B and C). The final steps in NF-κB activation are translocation to the nucleus, binding to specific sites in gene promoters, and activation of NF-κB-driven gene transcription. To investigate the effect of dexamethasone on the DNA-binding activity of NF-κB, we also harvested nuclear extracts from cells pretreated with dexamethasone and stimulated with bacteria for 2 h and subsequently measured DNA binding of the NF-κB subunit p65. As shown in Fig. 4D, both bacterial species induced the DNA-binding activity of p65, and as seen at the level of P-IκBα, dexamethasone inhibited S. pneumoniae p65 DNA binding less efficiently than that of N. meningitidis.
FIG. 4.
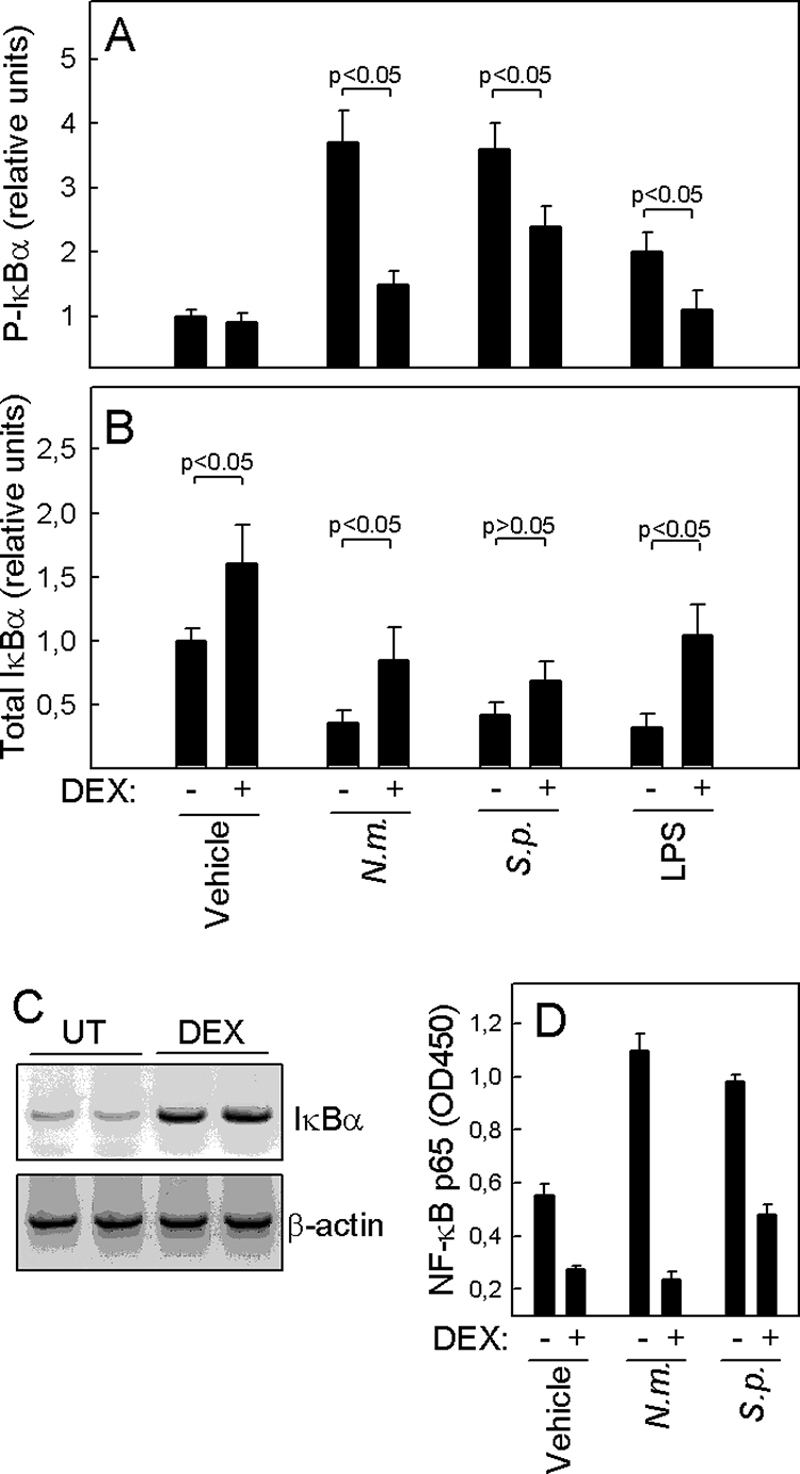
Dexamethasone inhibits cellular signaling induced by bacteria at the upstream level. THP-1 cells were treated with 1.0 μM of dexamethasone 1 h before stimulation with 9 × 107 bacteria/ml of N. meningitidis (N.m.) or S. pneumoniae (S.p.). At 2 h poststimulation, the cells were lysed and whole-cell and nuclear extracts were harvested. (A and B) Levels of total and phosphorylated IκBα in whole-cell extracts were measured by Luminex technology. (C) Total RNA was harvested from THP-1 cells either left untreated (UT) or incubated for 4 h in the presence of 1.0 μM dexamethasone. IκBα and β-actin mRNAs were amplified by RT-PCR and visualized by ethidium bromide staining of agarose gels. (D) Nuclear extracts were analyzed for NF-κB p65 DNA-binding activity. The data are shown as means for duplicate cultures ± SEM. Similar results were obtained in two or three independent experiments.
Taken together, the two different bacteria induced IκB phosphorylation followed by IκB degradation and NF-κB activation, and dexamethasone inhibited N. meningitidis-induced signaling more potently than that induced by S. pneumoniae, as already observed at the level of IL-8 mRNA synthesis (Fig. 1G).
Effect of dexamethasone on bacterially induced cytokine production is independent of de novo protein synthesis.
The findings above suggested that dexamethasone inhibits signaling to NF-κB both by inducing synthesis of IκBα and by preventing phosphorylation of this inhibitory protein. To examine if inhibition of IκBα phosphorylation was dependent on de novo protein synthesis or if dexamethasone was mediating this function independently of its activity as a transcription factor, we treated cells with the protein synthesis inhibitor cycloheximide and subsequently added dexamethasone, followed by bacteria or LPS. Total cell lysates were isolated after 2 h of incubation with bacteria, and the levels of phosphorylated IκBα were determined. As also shown in Fig. 4A, we found that N. meningitidis, S. pneumoniae, and LPS all induced IκBα phosphorylation, which was significantly inhibited by dexamethasone (Fig. 5). Importantly, the ability of dexamethasone to prevent phosphorylation of IκBα was also observed in cells treated with cycloheximide, thus suggesting that dexamethasone exerted this function independently of the induction of de novo protein synthesis.
FIG. 5.
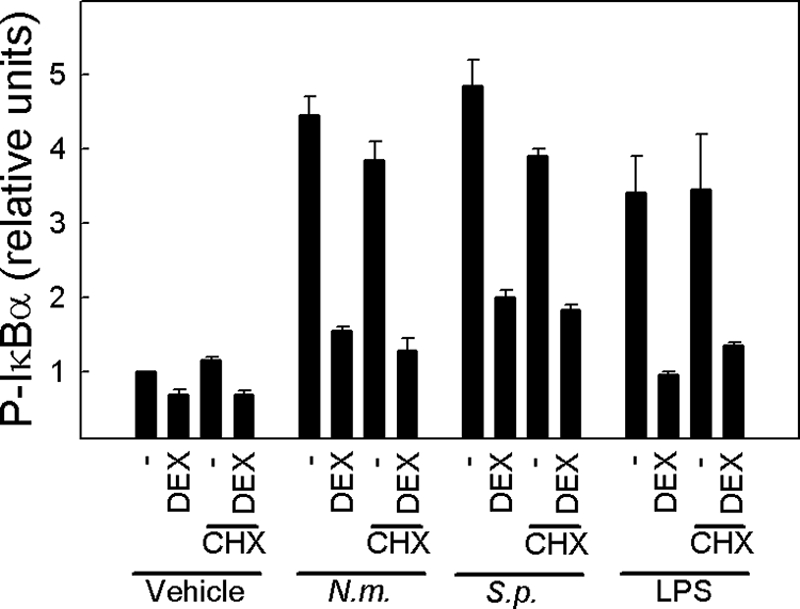
Dexamethasone inhibits cellular signaling induced by bacteria, independent of de novo protein synthesis. THP-1 cells were treated with cycloheximide (20 μg/ml; CHX) 30 min before receiving 1.0 μM of dexamethasone. One hour later, the cells were stimulated with 9 × 107 bacteria/ml of N. meningitidis (N.m.) or S. pneumoniae (S.p.) for 2 h, and whole-cell extracts were isolated. Phosphorylation of IκBα was measured by Luminex technology. The data are shown as means for duplicate cultures ± SEM. Similar results were obtained in two independent experiments.
Bacterial infection enhances mRNA stability, which is not affected by dexamethasone.
In addition to being regulated at the transcriptional level, many proinflammatory mediators are regulated at the level of mRNA stability through AURs in the 3′ UTR of the mRNA (22). We wanted to examine if the ability of S. pneumoniae and N. meningitidis to stimulate expression of proinflammatory cytokines involves stabilization of mRNA and if this is counteracted by glucocorticoids. For this purpose, we used a previously reported system with two cell lines derived from the macrophage-like cell line RAW264.7 (16, 36, 46). These cells were treated with dexamethasone 2 h prior to incubation with bacteria, and cellular lysates were prepared at 20 h postinfection. As shown in Fig. 6, the addition of either bacterium led to an increase in the levels of CAT, with S. pneumoniae being far more potent than N. meningitidis. The ability of bacteria to stabilize TNF-α mRNA was largely AUR dependent, although a minor stabilizing activity of S. pneumoniae remained in the RAW TNF-α 3′ UTR AU÷ cells. Importantly, dexamethasone did not affect CAT expression, regardless of which other stimuli were given. In separate experiments, we found that dexamethasone did work in the cellular system used, since LPS-induced expression of IL-6 and TNF-α was strongly inhibited (data not shown). Thus, N. meningitidis and, in particular, S. pneumoniae trigger AUR-dependent stabilization of mRNAs, and this activity is not counteracted by dexamethasone.
FIG. 6.

TNF-α mRNA stability is increased by N. meningitidis and S. pneumoniae through an AUR-dependent mechanism, which is not affected by dexamethasone. RAW TNF-α 3′ UTR AU+ and RAW TNF-α 3′ UTR AU÷ cells were seeded and left for 4 h before the addition of 1 μM dexamethasone. Two hours later, 9 × 107 bacteria/ml of N. meningitidis (N.m.) or S. pneumoniae (S.p.) were added to the cells as indicated. Twenty hours later, the cells were lysed and CAT levels were measured by ELISA. The data are shown as means ± SEM. Similar results were obtained in three independent experiments.
DISCUSSION
The molecular mechanisms of dexamethasone action have been studied extensively, and dexamethasone is known to interfere with proinflammatory signal transduction, gene expression, and protein synthesis at various levels (32). However, the precise targets of dexamethasone with respect to TLR-mediated signaling and cytokine production are less well defined. Since dexamethasone is widely used clinically to inhibit or dampen inflammation, whether it is triggered by infectious microorganisms or autoimmunity, and since TLRs play important roles in inducing such inflammation, we were interested in studying the molecular targets of dexamethasone-mediated inhibition of inflammation triggered by TLRs. In this study, we therefore investigated how dexamethasone inhibits proinflammatory signaling induced by live N. meningitidis and S. pneumoniae, two leading causes of bacterial meningitis, as large clinical trials have demonstrated the beneficial effect of early treatment with dexamethasone (11).
It has been proposed that TLR signaling pathways are key targets for the anti-inflammatory and immunosuppressive effects of glucocorticoids (32, 34, 35). Furthermore, LPS-induced inflammation has been demonstrated to be inhibited by glucocorticoids (27). We and others have previously demonstrated that S. pneumoniae activates TLRs 2 and 9 (3, 30, 47), whereas N. meningitidis signals through TLRs 2, 4, and 9 (18, 28, 30, 48). In our experimental system, we found that the vast majority of IL-8 produced in the presence of bacteria could be inhibited when TLR signaling through MyD88 was blocked. Therefore, the bacterially induced cytokine production observed was largely mediated through TLRs. However, we cannot formally exclude that the residual IL-8 measured in the presence of the MyD88 inhibitor may be due to TLR-independent pathways activated by the bacteria, which has indeed been described for S. pneumoniae (23). The nature of potential TLR-independent (or at least MyD88-independent) signaling mechanisms triggered by bacteria remains unknown.
Aiming at understanding the molecular targets of dexamethasone in bacterially induced, TLR-mediated proinflammatory signaling, we studied the diverse levels upon which dexamethasone might potentially act. First, we found that both IκBα phosphorylation and degradation as well as NF-κB DNA-binding activity were potently inhibited by dexamethasone. The finding that IκBα phosphorylation was affected strongly suggests a target upstream of this molecule. This might be IKK as well as other kinases or adaptor molecules involved in upstream TLR-mediated signaling (21). A previously described mechanism of direct interaction of glucocorticoids with p65 may also be involved (1), but our experimental setup does not allow us to determine the relative contributions of direct p65 inhibition and inhibition of IκB phosphorylation. Furthermore, we identified a separate mechanism by which dexamethasone itself induced increases in the levels of total cellular IκBα mRNA and protein, which ultimately may result in binding and inhibition of free transcriptionally active nuclear or cytoplasmic p65. For different experimental systems, similar mechanisms have been reported, including glucocorticoid-induced IκBα synthesis and upregulation of IκBα mRNA expression in the brain (5, 37). Finally, we observed that regardless of whether IκBα phosphorylation, NF-κB DNA binding, or IL-8 production was measured, dexamethasone seemed to exert a more powerful effect on N. meningitidis-induced activities than on those triggered by S. pneumoniae.
JNK and p38, both belonging to the family of MAPKs, are prominent targets of dexamethasone as well (7, 17, 41, 43), but since the THP-1 cell line did not activate the MAPKs in response to either N. meningitidis or S. pneumoniae, the question of a possible effect of dexamethasone on these MAPKs could not be addressed.
Our data on the kinetics of dexamethasone-mediated inhibition of cytokine production provide some additional insights into the mechanisms of action of dexamethasone. First, there may be several different mechanisms operating for dexamethasone-mediated inhibition of N. meningitidis-induced IL-8 production, since the response was fully inhibited when dexamethasone was added prior to or concomitant with bacteria and only partially inhibited at later time points. These results may be explained by the existence of an early mechanism, involving inhibition of upstream signaling as demonstrated, including inhibition of IκBα phosphorylation and NF-κB DNA-binding activity, where the presence of dexamethasone is required before or at least concomitant with bacterial challenge, and a late mechanism, possibly involving induction or upregulation of other cellular factors. The latter mechanism may be dependent upon de novo protein synthesis. For instance, Imasato et al. demonstrated that Haemophilus influenzae-induced upregulation of TLR2 was enhanced by dexamethasone by a mechanism involving upregulation of MKP-1, which in turn results in dephosphorylation and inactivation of p38 (17). However, in our experimental system, the inhibitory action of dexamethasone upon IκBα phosphorylation did not seem to be dependent upon protein synthesis.
As suggested by our data, different inhibitory mechanisms may exist, depending on whether inflammatory signaling is induced by N. meningitidis or S. pneumoniae. Both bacteria have in common that dexamethasone must be present before or concomitant with bacterial challenge in order to have the maximum effect, but we observed a clear difference with respect to the effect at later time points, where dexamethasone could partially inhibit IL-8 induced by meningococci, in contrast to the absence of any inhibitory effect exerted upon IL-8 induced by pneumococci. This idea is further supported by our previous finding that N. meningitidis and S. pneumoniae use distinct yet overlapping sets of TLRs, and possibly TLR-independent signaling as well (30). Taken together, looking at IκBα phosphorylation, NF-κB DNA-binding activity, and IL-8 production, there is a tendency towards dexamethasone inhibiting S. pneumoniae-induced inflammation to a lesser degree than that for inflammation induced by N. meningitidis.
In addition to regulation at the level of transcription, control of mRNA stability also represents an important level of regulation of inflammatory gene expression (22). AURs in the 3′ UTRs of mRNAs are targeted by specific constitutive and stimulus-regulated RNA-binding proteins capable of either stabilizing or destabilizing the mRNA (10, 22). We found that both N. meningitidis and S. pneumoniae stabilized TNF-α mRNA in an AUR-dependent manner, with S. pneumoniae being a particularly potent activator of this response. While it has long been known that purified and synthetic TLR ligands can stabilize mRNA through AUR elements (16, 38, 39), to our knowledge this is the first report showing how live N. meningitidis and S. pneumoniae interact with this important step in the inflammatory response. Although glucocorticoids have been reported to destabilize mRNAs for cyclooxygenase 2, monocyte chemoattractant protein 1, and inducible NO synthase (24, 25, 39), we did not observe any effect of steroid treatment on TNF-α mRNA stability, despite all these mRNAs being regulated by AURs in the 3′ UTR. This could indicate that dexamethasone utilizes a mechanism that is not yet well characterized to destabilize only a subset of transcripts bearing the AUR signature.
In a large randomized clinical trial of dexamethasone treatment of adults with bacterial meningitis, timing has proved to be an important issue, since mortality and morbidity were reduced only when dexamethasone was given prior to or concomitant with the initiation of antibiotic treatment (11). Furthermore, subgroup analysis indicated that dexamethasone may be particularly effective for patients with pneumococcal meningitis (11, 29). In this study, we have identified some of the cellular targets of dexamethasone in inflammatory signaling induced by N. meningitidis and S. pneumoniae and, more specifically, addressed the questions regarding the importance of timing and the possible more important effect upon pneumococcal meningitis.
Pneumococcal meningitis has the poorer prognosis with regards to mortality and also carries a relatively great risk of acquiring permanent sequelae. This may be due to the more overwhelming nature of the S. pneumoniae-induced inflammatory response, which even increases early after antibiotic treatment, since penicillin enhances bacterial lysis mediated by pneumococcal autolysin, hence liberating bacterial cell wall components and DNA (14, 31), which activate TLRs and elicit strong signals for enhanced inflammation. Similar mechanisms may also be operating during meningococcal meningitis, although to a lesser extent. Therefore, the early presence of dexamethasone is required in order to prevent excessive inflammation, and this may be particularly important during pneumococcal meningitis, for which our data indicate that dexamethasone is indeed effective only at early time points. Taken together, our results suggest that dexamethasone must be given early to patients with bacterial meningitis in order to inhibit TLR-dependent proinflammatory signaling and gene expression, including inhibition at various levels of the transcription factor NF-κB, as demonstrated in this study.
Acknowledgments
This work was supported by grants from the LEO Pharma Research Foundation, Kong Christian den Tiendes Fond, Beckett Fonden, and The Danish Medical Research Council (grant 271-06-0438).
We thank Mogens Kilian for a critical reading of the manuscript and invaluable discussions, as well as technicians Jonna Guldberg, Tove Findahl, and Kirsten Stadel Pedersen for excellent assistance.
Editor: A. Camilli
Footnotes
Published ahead of print on 15 October 2007.
REFERENCES
- 1.Adcock, I. M., and G. Caramori. 2001. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol. Cell Biol. 79376-384. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4499-511. [DOI] [PubMed] [Google Scholar]
- 3.Albiger, B., S. Dahlberg, A. Sandgren, F. Wartha, K. Beiter, H. Katsuragi, S. Akira, S. Normark, and B. Henriques-Normark. 2007. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell. Microbiol. 9633-644. [DOI] [PubMed] [Google Scholar]
- 4.Almawi, W. Y., and O. K. Melemedjian. 2002. Negative regulation of nuclear factor-κB activation and function by glucocorticoids. J. Mol. Endocrinol. 2869-78. [DOI] [PubMed] [Google Scholar]
- 5.Auphan, N., J. A. DiDonato, C. Rosette, A. Helmberg, and M. Karin. 1995. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science 270286-290. [DOI] [PubMed] [Google Scholar]
- 6.Beato, M., P. Herrlich, and G. Schutz. 1995. Steroid hormone receptors: many actors in search of a plot. Cell 83851-857. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharyya, S., D. E. Brown, J. A. Brewer, S. K. Vogt, and L. J. Muglia. 2007. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 1094313-4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caelles, C., J. M. Gonzalez-Sancho, and A. Munoz. 1997. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 113351-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhuri, A., and P. O. Behan. 2004. Fatigue in neurological disorders. Lancet 363978-988. [DOI] [PubMed] [Google Scholar]
- 10.Dean, J. L., G. Sully, A. R. Clark, and J. Saklatvala. 2004. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 161113-1121. [DOI] [PubMed] [Google Scholar]
- 11.de Gans, J., and D. van de Beek. 2002. Dexamethasone in adults with bacterial meningitis. N. Engl. J. Med. 3471549-1556. [DOI] [PubMed] [Google Scholar]
- 12.Dunne, A., and L. A. O'Neill. 2003. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE 2003re3. [DOI] [PubMed] [Google Scholar]
- 13.Fowler, M. I., R. O. Weller, J. E. Heckels, and M. Christodoulides. 2004. Different meningitis-causing bacteria induce distinct inflammatory responses on interaction with cells of the human meninges. Cell. Microbiol. 6555-567. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Bustos, J., and A. Tomasz. 1989. Mechanism of pneumococcal cell wall degradation in vitro and in vivo. J. Bacteriol. 171114-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez, M. V., J. M. Gonzalez-Sancho, C. Caelles, A. Munoz, and B. Jimenez. 1999. Hormone-activated nuclear receptors inhibit the stimulation of the JNK and ERK signalling pathways in endothelial cells. FEBS Lett. 459272-276. [DOI] [PubMed] [Google Scholar]
- 16.Han, J., T. Brown, and B. Beutler. 1990. Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level. J. Exp. Med. 171465-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imasato, A., C. Desbois-Mouthon, J. Han, H. Kai, A. C. Cato, S. Akira, and J. D. Li. 2002. Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of Toll-like receptor 2. J. Biol. Chem. 27747444-47450. [DOI] [PubMed] [Google Scholar]
- 18.Ingalls, R. R., E. Lien, and D. T. Golenbock. 2000. Differential roles of TLR2 and TLR4 in the host response to gram-negative bacteria: lessons from a lipopolysaccharide-deficient mutant of Neisseria meningitidis. J. Endotoxin Res. 6411-415. [PubMed] [Google Scholar]
- 19.Iwasaki, A., and R. Medzhitov. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5987-995. [DOI] [PubMed] [Google Scholar]
- 20.Jiang, Z., M. Zamanian-Daryoush, H. Nie, A. M. Silva, B. R. Williams, and X. Li. 2003. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem. 27816713-16719. [DOI] [PubMed] [Google Scholar]
- 21.Katsuyama, K., M. Shichiri, H. Kato, T. Imai, F. Marumo, and Y. Hirata. 1999. Differential inhibitory actions by glucocorticoid and aspirin on cytokine-induced nitric oxide production in vascular smooth muscle cells. Endocrinology 1402183-2190. [DOI] [PubMed] [Google Scholar]
- 22.Khabar, K. S. 2005. The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J. Interferon Cytokine Res. 251-10. [DOI] [PubMed] [Google Scholar]
- 23.Koedel, U., T. Rupprecht, B. Angele, J. Heesemann, H. Wagner, H. W. Pfister, and C. J. Kirschning. 2004. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain 1271437-1445. [DOI] [PubMed] [Google Scholar]
- 24.Korhonen, R., A. Lahti, M. Hamalainen, H. Kankaanranta, and E. Moilanen. 2002. Dexamethasone inhibits inducible nitric-oxide synthase expression and nitric oxide production by destabilizing mRNA in lipopolysaccharide-treated macrophages. Mol. Pharmacol. 62698-704. [DOI] [PubMed] [Google Scholar]
- 25.Lasa, M., M. Brook, J. Saklatvala, and A. R. Clark. 2001. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol. Cell. Biol. 21771-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, Z. G. 2005. Molecular mechanism of TNF signaling and beyond. Cell Res. 1524-27. [DOI] [PubMed] [Google Scholar]
- 27.Ma, W., K. Gee, W. Lim, K. Chambers, J. B. Angel, M. Kozlowski, and A. Kumar. 2004. Dexamethasone inhibits IL-12p40 production in lipopolysaccharide-stimulated human monocytic cells by down-regulating the activity of c-Jun N-terminal kinase, the activation protein-1, and NF-κB transcription factors. J. Immunol. 172318-330. [DOI] [PubMed] [Google Scholar]
- 28.Massari, P., P. Henneke, Y. Ho, E. Latz, D. T. Golenbock, and L. M. Wetzler. 2002. Cutting edge: immune stimulation by neisserial porins is Toll-like receptor 2 and MyD88 dependent. J. Immunol. 1681533-1537. [DOI] [PubMed] [Google Scholar]
- 29.McIntyre, P. B., C. S. Berkey, S. M. King, U. B. Schaad, T. Kilpi, G. Y. Kanra, and C. M. Perez. 1997. Dexamethasone as adjunctive therapy in bacterial meningitis. A meta-analysis of randomized clinical trials since 1988. JAMA 278925-931. [DOI] [PubMed] [Google Scholar]
- 30.Mogensen, T. H., S. R. Paludan, M. Kilian, and L. Ostergaard. 2006. Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2, 4, and 9 in species-specific patterns. J. Leukoc. Biol. 80267-277. [DOI] [PubMed] [Google Scholar]
- 31.Moore, L. J., A. C. Pridmore, M. E. Lee, and R. C. Read. 2005. Induction of pro-inflammatory cytokine release by human macrophages during exposure of Streptococcus pneumoniae to penicillin is influenced by minimum inhibitory concentration ratio. Int. J. Antimicrob. Agents 26188-196. [DOI] [PubMed] [Google Scholar]
- 32.Moynagh, P. N. 2003. Toll-like receptor signalling pathways as key targets for mediating the anti-inflammatory and immunosuppressive effects of glucocorticoids. J. Endocrinol. 179139-144. [DOI] [PubMed] [Google Scholar]
- 33.Odio, C. M., I. Faingezicht, M. Paris, M. Nassar, A. Baltodano, J. Rogers, X. Saez-Llorens, K. D. Olsen, and G. H. McCracken, Jr. 1991. The beneficial effects of early dexamethasone administration in infants and children with bacterial meningitis. N. Engl. J. Med. 3241525-1531. [DOI] [PubMed] [Google Scholar]
- 34.Ogawa, S., J. Lozach, C. Benner, G. Pascual, R. K. Tangirala, S. Westin, A. Hoffmann, S. Subramaniam, M. David, M. G. Rosenfeld, and C. K. Glass. 2005. Molecular determinants of crosstalk between nuclear receptors and Toll-like receptors. Cell 122707-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Neill, L. A. 2006. How Toll-like receptors signal: what we know and what we don't know. Curr. Opin. Immunol. 183-9. [DOI] [PubMed] [Google Scholar]
- 36.Paludan, S. R., S. Ellermann-Eriksen, V. Kruys, and S. C. Mogensen. 2001. Expression of TNF-α by herpes simplex virus-infected macrophages is regulated by a dual mechanism: transcriptional regulation by NF-κB and activating transcription factor 2/Jun and translational regulation through the AU-rich region of the 3′ untranslated region. J. Immunol. 1672202-2208. [DOI] [PubMed] [Google Scholar]
- 37.Quan, N., L. He, W. Lai, T. Shen, and M. Herkenham. 2000. Induction of IκBα mRNA expression in the brain by glucocorticoids: a negative feedback mechanism for immune-to-brain signaling. J. Neurosci. 206473-6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raj, N. B., and P. M. Pitha. 1983. Two levels of regulation of beta-interferon gene expression in human cells. Proc. Natl. Acad. Sci. USA 803923-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reddy, K. V., G. Bhattacharjee, G. Schabbauer, A. Hollis, K. Kempf, M. Tencati, M. O'Connell, M. Guha, and N. Mackman. 2004. Dexamethasone enhances LPS induction of tissue factor expression in human monocytic cells by increasing tissue factor mRNA stability. J. Leukoc. Biol. 76145-151. [DOI] [PubMed] [Google Scholar]
- 40.Saez-Llorens, X., and G. H. McCracken, Jr. 2003. Bacterial meningitis in children. Lancet 3612139-2148. [DOI] [PubMed] [Google Scholar]
- 41.Sakai, A., J. Han, A. C. Cato, S. Akira, and J. D. Li. 2004. Glucocorticoids synergize with IL-1β to induce TLR2 expression via MAP kinase phosphatase-1-dependent dual inhibition of MAPK JNK and p38 in epithelial cells. BMC Mol. Biol. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato, S., M. Sugiyama, M. Yamamoto, Y. Watanabe, T. Kawai, K. Takeda, and S. Akira. 2003. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 1714304-4310. [DOI] [PubMed] [Google Scholar]
- 43.Swantek, J. L., M. H. Cobb, and T. D. Geppert. 1997. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor α (TNF-α) translation: glucocorticoids inhibit TNF-α translation by blocking JNK/SAPK. Mol. Cell. Biol. 176274-6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van de Beek, D., J. de Gans, P. McIntyre, and K. Prasad. 2004. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect. Dis. 4139-143. [DOI] [PubMed] [Google Scholar]
- 45.Vital, A. L., M. Goncalo, M. T. Cruz, A. Figueiredo, C. B. Duarte, and M. C. Lopes. 2003. Dexamethasone prevents granulocyte-macrophage colony-stimulating factor-induced nuclear factor-κB activation, inducible nitric oxide synthase expression and nitric oxide production in a skin dendritic cell line. Mediators Inflamm. 1271-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willeaume, V., V. Kruys, T. Mijatovic, and G. Huez. 1995. Tumor necrosis factor-α production induced by viruses and by lipopolysaccharides in macrophages: similarities and differences. J. Inflamm. 461-12. [PubMed] [Google Scholar]
- 47.Yoshimura, A., E. Lien, R. R. Ingalls, E. Tuomanen, R. Dziarski, and D. Golenbock. 1999. Cutting edge: recognition of gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 1631-5. [PubMed] [Google Scholar]
- 48.Zughaier, S. M., Y. L. Tzeng, S. M. Zimmer, A. Datta, R. W. Carlson, and D. S. Stephens. 2004. Neisseria meningitidis lipooligosaccharide structure-dependent activation of the macrophage CD14/Toll-like receptor 4 pathway. Infect. Immun. 72371-380. [DOI] [PMC free article] [PubMed] [Google Scholar]