

Abstract
Both morphogenesis and antibiotic production in the streptomycetes are initiated in response to starvation, and these events are coupled. We previously described a transposon-generated mutant in Streptomyces coelicolor, SE293, that resulted in a bld strain that overproduced the antibiotic actinorhodin. The SCO1135 open reading frame identified by the insertion encodes a member of the TetR family of transcriptional regulators. Here we show that a constructed deletion of the SCO1135 open reading frame resulted in the same morphological and antibiotic production phenotype as the insertion mutant. The constructed deletion also resulted in constitutive expression of SCO1135 transcript, as well as that of the gene cluster immediately adjacent to it, SCO1134-1132, which encodes a putative molybdopterin binding complex. A His6-tagged version of the SCO1135 protein product was shown to bind the intergenic region between SCO1135 and SCO1134, which contains the apparent transcription start sites for each gene mapped by primer extension analysis. Increased expression of the SCO1134-1132 transcript in the SCO1135 deletion mutant also resulted in increased expression of xanthine dehydrogenase activity, confirming the predictions about these open reading framed based on protein similarity. We have designated the SCO1134-1142 gene cluster xdhABC and the regulator encoded by SCO1135 xdhR. We speculate that the inappropriate expression of xanthine dehydrogenase affects purine salvaging pathways at the onset of development, creating artificially high concentrations of both GTP and ppGpp and perturbing the pathways these molecules participate in for the initiation of morphogenesis and antibiotic production.
The interpretation of environmental signals plays a key role in the ability of an organism to survive starvation, adapt to environmental changes, and initiate new programs of development. Bacteria must respond quickly and effectively to the constantly changing environments in which they live, and regulatory proteins that respond to small molecule signals or morphogens serve to activate or repress the transcription of genes that allow the organism to adapt. The regulator proteins control interconnected and often complex networks that involve the interaction of multiple signaling pathways. Signal transduction typically involves the binding of a small molecule to a transcriptional regulator to modulate its activity on gene expression. The TetR family is a common class of transcriptional regulator with more than 2,000 members found in a wide variety of bacteria, but only about 100 have been fully characterized (21). The first member of the group, TetR, was identified in Escherichia coli and controls expression of the genes encoding a tetracycline efflux pump responsible for drug resistance conferred by Tn10. In the absence of tetracycline, TetR binds to the tet promoter and represses transcription of the efflux pump genes. When tetracycline enters the cell it binds TetR and causes conformational changes within the protein that abolish protein binding, thus relieving repression (23). RsrA, another member of this class, has been shown to activate transcription of rpoS, a sigma factor responsible for transcription of stationary-phase genes in Pseudomonas putida, in response to cell density (3).
TetR-like regulators contain a conserved helix-turn-helix DNA-binding domain, form homodimers, and generally act as repressors of transcription. They function to regulate a wide range of cellular activities including drug efflux, antibiotic production, amino acid metabolism, and development (21). There are 151 predicted TetR-like transcriptional regulators in the Streptomyces coelicolor genome (2, 21). Two (ActII and CprB) are repressors of actinorhodin biosynthesis, two (Pip and PqrA) are repressors of drug resistance genes, and one (ScbR) is a repressor of γ-butarylactone synthesis (1, 6-8, 27).
Both morphogenesis and antibiotic production in the streptomycetes are initiated in response to starvation. Upon sensing starvation, the substrate mycelia release small molecules that act as signals for the initiation of aerial hyphal growth, as well as for the production of antibiotics (29, 30). As the aerial hyphae grow, they coil and septate into uninucleoid cells that give rise to spores. Most of what is known about this process comes from the study of mutants that fail to produce aerial hyphae, called bld mutants, or those that initiate aerial hyphal growth but fail to produce mature spores, called whi mutants (13).
We previously described a transposon-generated mutant, SE293, that resulted in a bld strain that overproduced the antibiotic actinorhodin (25). The SE293 mutant also required arginine for growth on minimal medium (25). The SCO1135 open reading frame identified by the insertion encodes a member of the TetR family of transcriptional regulators. Here we show that a constructed deletion of the SCO1135 open reading frame resulted in the same phenotype as the insertion mutant but was prototrophic, suggesting that the auxotrophy of the original mutant resulted from a second, unrelated mutation. Deletion of the SCO1135 open reading frame resulted in constitutive expression of the SCO1135 transcript, as well as that of a molybdopterin binding complex encoded by the gene cluster immediately adjacent to it, and increased the levels of xanthine dehydrogenase (XDH) activity, confirming the predictions about these open reading frames based on protein similarity. The SCO1135 gene product is a DNA-binding protein that interacts directly with the intergenic region between SCO1135 and SCO1134, which contains the apparent transcription start sites for both genes, to repress transcription.
MATERIALS AND METHODS
Strains and growth conditions.
General techniques for bacterial growth were performed as described previously for S. coelicolor (16) and E. coli (24), respectively. S. coelicolor strains were grown on mannitol-soya flour (MS) agar medium with the addition of 10 mmol MgCl2 for mating experiments. Antibiotic selections were applied by overlay with soft nutrient agar (NSA). Streptomyces RNA was isolated from cells grown on cellophane discs placed on top of maltose-yeast extract-malt extract (MYM) agar. E. coli strains and growth conditions for the preparation of cosmids for marker replacement in Streptomyces were as described previously (9). The S. coelicolor strains used in the present study were M145 (SCP1− SCP2−), SE293/xdhR (M145 SCO1135::Tn5::apr), and ΔxdhR::aac(3)IV [M145 SCO1135::acc(3)IV]. E. coli strains ET12576 [Δ(dam dcm)] containing the nontransmissible helper plasmid pUZ8002 and BL21(DE3) [F− dcm ompT hsdS (rB− mB−) galλ(DE3)] were used for mutant construction and protein expression, respectively. Primers used to amplify fragments for construction of the His tag fusion, construction of mutants, primer extension reactions, and reverse transcriptase PCRs are listed in Table 1.
TABLE 1.
Primers used in this study
| Primer and purpose | Sequence |
|---|---|
| For construction of His6-tagged XdhR protein | |
| His6-XdhR Forward | AACCCGAAAGGAGGACATATGCCGCAGCCGAAGAAG |
| His6-XdhR Reverse | GTGGAGACCGCCGAATTCGTCGAGCGCGCG |
| For RT-PCR | |
| XdhA RT Forward | CACCCTCGCCGACGTCCAGCGCC |
| XdhA RT Reverse | TGCCTTCGATGGTGGTGATCTCG |
| XdhR RT Forward | GCTCGGACGCCCAGCGCAACCGC |
| XdhR RT Reverse | CCATCCACTGGCGCAGGGCCAGG |
| WhiB Forward | GTCGACGACGCGGACGAGGAA |
| WhiB Reverse | AGATGCCGAAGCGCTCGTCGT |
| WhiG Forward | TGTGGCGGTCGTACAAGACGA |
| WhiG Reverse | ATCGCGTACGTCTCGAACTTG |
| WhiH Forward | AGCTGGGCCAGATGATCGTCT |
| WhiH Reverse | AAGGCACGCCATTCGATGATG |
| HrdB Forward | CGGCCGCAAGGTACGAGTTGATGA |
| HrdB Reverse | CCATGACAGAGACGGACTCGGCG |
| For construction of xdhR mutant | |
| XdhA Upstream | CGGGCAGGGCACGTTCTACCGCAACTTCCCGAACCGCGAATTCCGGGGATCCGTCGACC |
| XdhA Downstream | GACGCCCCGAACGCCGCTACCGCTTCCGGGGCGCGGTCATGTAGGCTGGAGCTGCTTC |
| P1 | ATTCCGGGGATCCGTCGACCTGCA |
| P2 | TGTAGGCTGGAGCTGCTTCGAAGT |
| For primer extension | |
| XdhA PE | AGACACCAAAGAAGGCTGATCAT |
| XdhR PE | AACCCGAAAGGAGGACGAGTGCC |
| PE seq Forward | GCGCTCGCGGTTGCGCTGGGCGT |
| PE seq Reverse | GTCAGCTGGTGCTTCTCGCCGTT |
Construction and confirmation of deletion mutants.
A deletion of the SCO1135 open reading frame was constructed by using the PCR targeting method described by Gust et al. The ΔxdhR::aac(3)IV mutant was made by replacing the SCO1135 open reading frame with the apramycin resistance cassette [acc(3)IV]. The deletion extended from position +200 with respect to the translation start site to the end of the open reading frame, eliminating 130 of the 196 amino acids, allowing analysis of the 5′ end of the transcript of this gene in the deletion mutant itself. The presence and location of the apramycin resistance cassette was detected using the specific primers P1 and P2. The extent and location of the deletion was also confirmed by PCR. Marker replacement of the deletion with a wild-type copy of xdhR was accomplished by the same method using cosmid 2STG38.
Scanning electron microscopy.
The procedure used in this analysis was as previously described (22). Samples from colonies grown for 5 days on MYM agar plates were mounted on a an aluminum stub with O.C.T. compound, submerged in liquid nitrogen slush at approximately −210°C and transferred to a Gatan Alto 2500 cryostage and cryoprep chamber (Gatan UK, Oxford, United Kingdom) attached to a LEO 982 field emission scanning electron microscope (LEO Electron Microscopy, Inc., Thornwood, NY). Samples were sublimated to remove surface frost at −95°C for 3 min, coated with platinum, placed on the cryostage in the main chamber of the microscope, at approximately −140°C, and viewed at 5.0 kV.
RT-PCR.
RNA was isolated from S. coelicolor M145 and the ΔxdhR::aac(3)IV mutant after 24, 48, and 72 h of growth on MYM agar medium overlaid with cellophane discs as for primer extension analysis. The One-Step PCR kit (Qiagen) was used with primers specific for each gene. The forward and reverse primers for xdhA, xdhR, whiG, whiB, whiH, and hrdB are listed in Table 1. Reaction mixtures contained 10 pmol of each primer and 100 ng of RNA in a total volume of 20 μl. Each primer was first tested using chromosomal DNA as a template and without a reverse transcription (RT) cycle to test for DNA contamination in the RNA. Reactions were run for up to 35 cycles with wild-type RNA, sampling every 5 cycles between 20 and 35 in order to determine the linear range of product formation. In all cases the 25th cycle was in the linear range and was chosen as the assay point. The experiments were done in triplicate, and the experiment shown in the figure is representative. HrdB was used as a control for RNA concentration. Products were displayed on a 1% agarose gel and visualized by staining with ethidium bromide.
Gel retardation assays.
To construct a His-tagged version of the XdhR protein, the coding region was amplified by PCR from S. coelicolor genomic DNA using the primers His6-XdhR Forward and His6-XdhR Reverse (Table 1). The fragment was digested with NdeI (NEB) and EcoRI (NEB) and cloned into NdeI/EcoRI-digested pET28a. XbaI and HindIII digestion confirmed the correct orientation and fusion to the His tag. Expression and purification of the His6-XdhR protein from E. coli was as previously described (12). Purified His6-XdhR protein (5, 10, or 20 μg) or crude cell extract (20 μg of total protein) was mixed with the 305-bp PCR-generated DNA fragment containing the intergenic region between SCO1134 and SCO1135 used for primer extension experiments. The fragment was 5′ end labeled with [γ-32P]ATP (MP Biomedicals) using T4 polynucleotide kinase (Promega) and purified on a 1% agarose gel. The labeled DNA fragment (1 ng; 6,000 cpm) was incubated with cell extracts or purified His-tagged XdhR protein for 20 min at 30°C in 20 μl (total volume) of binding buffer (20 mM Tris, 10 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [pH 8.0]) containing 1 μg of sonicated salmon sperm DNA and 3 μg of bovine serum albumin. For competitive inhibition of the binding reaction, 100 ng of unlabeled fragment or 100 μg of sonicated salmon sperm was added to 5 μg of the purified His6-XdhR protein. Reactions were displayed on a nondenaturing 6% acrylamide Tris-borate-EDTA gel and visualized by autoradiography.
RNA isolation and primer extension analysis.
Primer extension reactions were carried out as described previously (24). RNA was isolated from cells grown for 48 h on MYM agar plates overlaid with cellophane discs. Primers were labeled by using [γ-32P]ATP 6,000 Ci/ml and OptiKinase (USB) according to the manufacturer's instructions. Then, 40 μg of RNA was hybridized to 1 pmol of either XdhA primer extension (PE) primer or XdhR PE-labeled primer by heating reactions to 65°C for 20 min and then allowing them to cool to room temperature for 10 min. The annealed primer and RNA mixture was added to a reaction mixture containing 40 mM sodium pyrophosphate and 1 U of avian myeloblastosis virus reverse transcriptase (Promega), followed by incubation at 42°C for 30 min. A 305-bp fragment, generated by PCR using the primers PE seq Forward and PE seq Reverse from chromosomal DNA, was used as a template. This fragment was also used to generate the DNA sequence ladder with the Fmol Cycle DNA Sequencing System (Promega) with 1 pmol of 5′ primer labeled with 6,000 Ci of γ-32P/ml. Products from the DNA sequencing reactions were separated on a 6% denaturing polyacrylamide gel, and bands were visualized by autoradiography.
XDH and AOR assays.
Cells were grown for 36 h at 30°C in YEME medium, harvested by centrifugation at 3,500 × g for 10 min, and resuspended in extraction buffer (0.1 M Tris-HCl [pH 7.6], 10 mM MgCl2, 6 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride). Extracts were obtained by first homogenization by sonication, followed by centrifugation at 12,000 × g for 15 min at 4° (16). XDH activity was measured spectroscopically as conversion of NAD+ to NADH at 340 nm in 50 mM sodium pyrophosphate buffer (pH 8.5) containing 0.2 mM EDTA, 0.15 mM xanthine, and 0.5 mM NAD+ (14). Then, 20 μg of cell extract was added, and measurements were taken at 5-, 10-, 15-, and 20-min intervals. The aldehyde oxidoreductase (AOR) activity was determined by reduction of the electron acceptor 2,6-dichlorophenol-indophenol (DCPIP) at 600 nm in 50 mM Tris-HCl buffer (pH 7.6) containing 35 μM DCPIP and 50 μM acetylaldehyde (28). Next, 20 μg of cell extract was added, and measurements were taken at 5-, 10-, 15-, and 20-min intervals. Xanthene dehydrogenase (XDH) and AOR activities were normalized to glutamate dehydrogenase activity. The glutamate dehydrogenase activity was assayed by measuring NADH oxidation at 340 nm in 50 mmol of triethanolamine buffer containing 50 mmol of ammonium sulfate, 200 nmol of NADH, and 10 mmol of 2-oxoglutarate. Reactions were measured every 6 min (20). Protein concentrations were determined by the Bradford assay (4).
RESULTS
A deletion of the xdhR open reading frame results in a bld mutant phenotype.
A mutation in xdhR was first identified by a transposon insertion into the SCO1135 open reading frame (25). The location of SCO1135 and the organization of the genes on the chromosome near it are shown in Fig. 1. The xdhR open reading frame encodes a TetR family transcriptional regulator and is located directly adjacent to but in the opposite orientation of a gene cluster, xdhA (SCO1134), xdhB (SCO1133), and xdhC (SCO1132), predicted to encode a molybdopterin binding protein complex. The transposon mutant was completely defective in morphogenesis, failing to make the aerial hyphae associated with the initiation of development but overproduced the blue pigment associated with the polyketide antibiotic actinorhodin. While the original mutant had a single copy of the transposon, it also required arginine for growth on minimal medium. The arginine auxotrophy was presumed to be unrelated to the morphological phenotype (25). A deletion of the xdhR open reading frame was constructed by using the targeted marker replacement method developed by Gust et al. (9). The deletion mutant, ΔxdhR::aac(3)IV, had the same morphological phenotype as the insertion mutant but was able to grow on minimal medium, suggesting that the arginine auxotrophy was most likely a second and unrelated mutation.
FIG. 1.

Organization of the S. coelicolor genome showing the xdhR open reading frame identified by transposon insertion and adjacent genes. Gene notations are based on the Sanger Centre Sequencing Projects (http://www.sanger.ac.uk/Projects/S_coelicolor).
Scanning electron microscopy of the ΔxdhR::aac(3)IV mutant (Fig. 2) grown on MYM sporulation medium revealed normal substrate mycelia but no evidence of aerial hyphae or spores. Unlike many bld mutants that are substantially delayed in morphogenesis but will eventually produce some spores, the ΔxdhR::aac(3)IV mutant never produces either aerial hyphae or spores even with prolonged (more than 10 days) growth on sporulation agar.
FIG. 2.

Scanning electron micrographs of wild-type S. coelicolor (M145) and the xdhR mutant [ΔxdhR::aac(3)IV] at ×2,000 magnification. Both strains were grown for 5 days at 30°C on MYM agar medium.
The morphogenic and antibiotic production phenotypes of the ΔxdhR::aac(3)IV mutant were not complemented with the wild-type allele of xdhR introduced on a pSET plasmid integrated at the φC31 attachment site. Similar experiments with the original SE293 transposon insertion mutant also failed to show complementation by the wild-type allele (25). To test whether the observed phenotypes were due to the xdhR mutation, cosmid 2STG38 was introduced into the ΔxdhR::aac(3)IV mutant, and exconjugants containing a marker replacement of the mutation with the wild-type allele were obtained. The resulting strain was wild type. This suggests that the xdhR mutation, in fact, caused the defects in morphogenic and antibiotic production and while it is not clear why the mutation is not complemented by a wild-type copy of the gene, it raises the possibility that the xdhR gene product does not work in trans.
The xdhR gene product acts to repress its own transcription, as well as the transcription of an adjacent gene cluster that encodes a putative molybdoterin binding complex.
To test whether a mutation in xdhR affected regulation of itself or the adjacent gene cluster, RT-PCRs were performed with RNA isolated from wild-type S. coelicolor and the ΔxdhR::aac(3)IV mutant that had been grown for 24, 36, 48, or 60 h on MYM sporulation medium. This solid medium supports morphological development of the wild type, and the stages of development are clearly detectable. Cells harvested after 16 h were growing vegetatively (no aerial mycelia present). Aerial mycelia began to appear between 24 and 36 h and were abundant between 36 and 48 h, as was the blue pigment associated with actinorhodin production. The gray pigment associated with mature spores was evident at 60 h.
As shown in Fig. 3, transcript from xdhA, the first of a series of the genes adjacent to xdhR, was detected at low levels during growth phase and at higher levels as development proceeded with a peak in wild-type cells coincident with the aerial hyphae production. Transcription of xdhA was constitutive in the ΔxdhR::aac(3)IV mutant. RT-PCRs were also performed with RNA isolated from wild-type S. coelicolor and the ΔxdhR::aac(3)IV mutant to examine the effect of the mutant on its own expression. As shown in Fig. 3, in the wild-type strain, transcript from xdhR was similar to that of the adjacent cluster, present at low levels during growth phase and at higher levels as development proceeded with a peak in wild-type cells coincident with the aerial hyphae production. The transcription of xdhR was constitutive in the ΔxdhR::aac(3)IV mutant. The same RNA preparations used for this analysis were used for detection of the hrdB transcript, which served as a control for the level of RNA (Fig. 3). The PCR products shown are samples taken during the exponential phase of the PCR so that the amount of product is representative of quantitative differences in RNA level. These data suggest that the xdhR gene product acts to repress, either directly or indirectly, transcription of itself as well as the genes in the adjacent gene cluster.
FIG. 3.
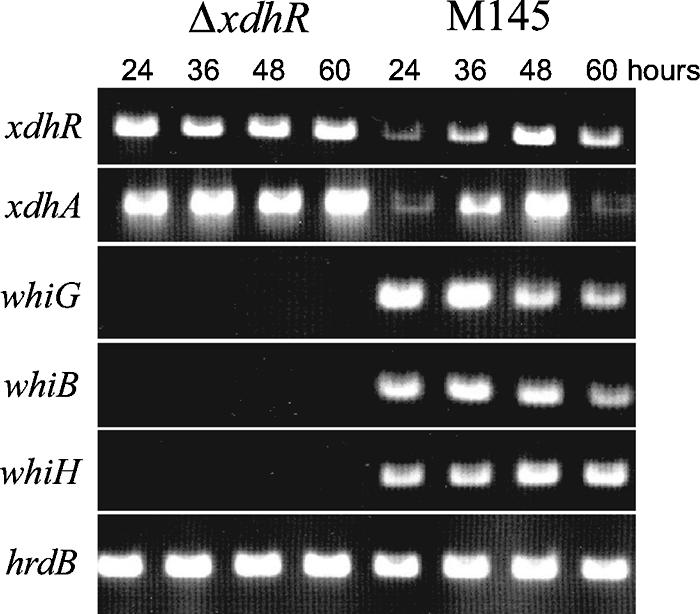
RT-PCR analysis of transcripts from various morphological mutants in wild-type S. coelicolor (M145) and the xdhR deletion mutant [ΔxdhR::aac(3)IV]. hrdB was used as a control for RNA.
The xdhR gene product binds the intergenic region between xdhR and xdhA in vitro.
To test whether the XdhR protein made direct contact with the xdhR and/or xdhA promoter regions, an His6-XdhR protein was constructed and expressed in E. coli and used in a gel mobility shift assay with a DNA fragment containing the intergenic region between xdhR and xdhA. As shown in Fig. 4, crude extracts from E. coli cells expressing the His6-XdhR protein retarded the promoter-containing fragment, whereas extracts from uninduced cells did not. Furthermore, the amount of probe shifted in the retardation assay was reduced in the presence of unlabeled probe but not nonspecific DNA, suggesting that binding of XdhR to the promoter-containing fragment was sequence or motif specific.
FIG. 4.

Gel mobility shift assays using a DNA fragment containing the intergenic region between xdhR and xdhA and the His6-XdhR protein. Lane 1, labeled fragment; lane 2, crude extract from uninduced cells containing the His6-XdhR construction; lane 3, crude extract from induced cells containing the His6-XdhR construction; lanes 4 to 6, labeled fragment with 5, 10, or 20 μg of purified His6-XdhR protein; lane 7, labeled fragment with 100 ng of unlabeled promoter-containing fragment and 5 μg of purified His6-XdhR protein; lane 9, labeled fragment with 100 μg of sonicated salmon sperm DNA and 5 μg of purified His6-XdhR protein.
Primer extension analysis (Fig. 5A) was used to identify apparent transcription start sites for xdhR and xdhA. The start site of xdhR maps to an adenine or guanine nucleotide located 19 or 20 nucleotides upstream of the annotated translational start codon. The sequences centered at −10 and −35 with respect to the apparent start site show little homology to known consensus sequences for RNA polymerase binding. The start site for xdhA maps to guanine or adenine located 18 or 19 nucleotides upstream of the annotated translational start site and like xdhR the −10 and −35 regions show little homology to known RNA polymerase binding sites. The distance between the apparent transcription start sites of xdhA and xdhR is 150 bp. While the leader region of these predicted transcripts is relatively short, each contains a potential ribosome-binding site, in fact, the putative ribosome-binding site for xdhR was previously annotated in the genome sequence.
FIG. 5.

(A) Primer extension analysis of transcripts originating upstream of xdhR and xdhA. (B) DNA sequence upstream of the apparent transcription start sites (indicated in boldface italics) of xdhR and xdhA. Potential RNA polymerase recognition sequences are underlined. The annotated translation starts are indicated by boldface with no italics.
The xdhR mutant is defective in the expression of other developmental genes.
The ΔxdhR::aac(3)IV mutation results in a severe bld phenotype. Many of the bld mutants affect the expression, either directly or indirectly, of whi genes. To test the effect of the ΔxdhR::aac(3)IV mutation on the expression of other developmental genes, RT-PCR was performed on RNA isolated from wild-type S. coelicolor M145 and the ΔxdhR::aac(3)IV mutant. Primers specific for whiG, whiB, whiH, xdhA, xdhR, and hrdB, a constitutively expressed gene as a control for RNA, were used. Although transcripts from whiG, whiB, and whiH were readily detected from the wild-type strain, no expression of these genes was detected in the ΔxdhR::aac(3)IV mutant. The whiG, whiB, and whiH genes play early roles in the cascade of whi gene expression, and their expression is dependent on several bld genes. xdhR is clearly one of them.
The SCO1132-34 gene cluster likely encodes a XDH/AOR enzyme complex.
The predicted protein products of the xdhA (SCO1134), xdhB (SCO1133), xdhC (SCO1132) gene cluster show significant similarity to a molybdopterin binding protein in the AOR/XDH family (19). XdhC shows 63% similarity (50% identity) to the molybdodenum binding subunit of S. erythraea XDH; xdhB shows 73% similarity (61% identity) to the flavin adenine dinucleotide (FAD)-binding subunit and xdhA shows 73% similarity (60% identity) to the [2Fe-2S] binding subunit (http://www.ncbi.nlm.nih.gov/BLAST/). Molybdenum is often bound to a pterin cofactor, and the synthesis of the molybdopterin cofactor has been shown to require more than 12 proteins that are highly conserved in all organisms (11). Molybdenum-containing enzymes perform a variety of functions, but all involve oxidation or reduction reactions. These enzymes take advantage of the ability of molybdenum to exist in a variety of oxidation states under physiological conditions. This allows the enzyme to catalyze redox reactions that require the movement of one or two electrons (11). Molybdenum enzymes are grouped in three families: the AOR/XDH family, the sulfite oxidase family, and the dimethyl sulfoxide reductase family. XDH enzymes are sometimes made up of multienzyme complexes or a single protein with multiple enzymatic functions or domains. Like many XDH and AOR proteins, the putative heterotrimeric protein encoded by the xdhABC gene cluster (Fig. 6) is composed a [2Fe-2S] iron-sulfur binding subunit, a FAD binding subunit, and a molybdopterin binding/dimerization subunit (17). The enzyme catalyzes the reactions that convert hypoxanthine into xanthine and then xanthine to uric acid.
FIG. 6.

(A) Comparison of the predicted products of xdhABC with XDH and AOR. (B) Enzyme assays using cell extracts from either the wild type (M145) or the xdhR deletion mutant [ΔxdhR::aac(3)IV]. The AOR activity was determined by the reduction of DCPIP. The XDH activity was measured spectroscopically as the conversion of NAD+ to NADH at 340 nm.
To test whether this gene cluster in fact encoded an XDH complex, enzyme assays were preformed on extracts from wild type and ΔxdhR::aac(3)IV mutant cells. XDH activity was determined spectrophotometrically by assaying conversion of NAD+ to NADH at 340 nm in the presence of xanthine. In the presence of xanthine, the rate of NAD+ to NADH increased 2.7-fold in the ΔxdhR::aac(3)IV mutant strain. AOR activity was assayed by reduction of DCPIP as measured by increased absorbance at 600 nm, and there was no significant difference between the wild type and the ΔxdhR::aac(3)IV mutant for this activity. Bradford assays were used to determine the protein concentration of cell extracts. All enzyme activities were normalized to glutamate dehydrogenase activity.
DISCUSSION
Scanning electron microscopy of the ΔxdhR::aac(3)IV mutant showed that the mutation resulted in the complete loss of morphological development (no aerial mycelium even after prolonged incubation on sporulation medium) while apparently increasing actinorhodin antibiotic production. RT-PCR analysis of transcripts from xdhR and xdhABC, the first gene in an adjacent gene cluster, suggested that xdhR acts to repress its own transcription, as well as the transcription of xdhABC. Gel mobility shift experiments using His6-XdhR protein and the xdhR-xdhA intergenic region showed that the XdhR protein binds this region directly in vitro and primer extension analysis identified apparent transcription start sites for xdhR and xdhABC within this region. XDH assays showed that the level of enzyme activity was significantly increased in the xdhR mutant supporting the prediction from BLAST analysis that the xdhABC gene cluster encodes a XDH complex. RT-PCR analysis of the transcription of other genes required for morphological development in S. coelicolor showed that xdhR is required for the transcription of whiG, whiB, and whiH.
XdhR is a member of the TetR family of transcriptional regulators and, like other members of this group, is a DNA-binding protein that acts to repress transcription. XDH is a molybdo-flavoenzyme that participates in purine catabolism and catalyzes the conversion of hypoxanthine to xanthine and of xanthine to uric acid. XDH is a heterodimeric protein composed of two [2Fe-2S] cluster-binding domains, an FAD binding domain, and a domain for dimerization and binding of the Moco molybdenum cofactor (5). Electrons from the substrate are passed from the Moco center to FAD by the two [2Fe-2S] clusters. Once electrons reach the FAD site, they are transferred either to molecular oxygen or to NAD+ to form NADH. In E. coli the conversion of hypoxanthine to xanthine by XDH plays a role in the purine salvage pathway. Deletion mutants of xdhA were sensitive to exogenous adenine, a phenotype previously shown to be due to inefficient conversion of adenine to guanine due to decreased xanthine availability (18, 31).
Why should the overexpression of XDH result in loss of morphogenesis while increasing antibiotic production? The purine tetraphosphate, ppGpp, is produced under conditions of amino acid limitation by the activity of RelA (10), which phosphorylates GTP to ppGpp and pppGpp. These molecules have been implicated in the sensing of nutritional shifts in both E coli and S. coelicolor. Interestingly, S. coelicolor relA mutants are defective in antibiotic production and delayed in morphogenesis, but the delay in morphogenesis occurs only under conditions of nitrogen limitation (26). Antibiotic production is restored by overexpression of relA, suggesting that increased levels of ppGpp restore antibiotic production. Streptomyces clavuligerus relA mutants are defective in antibiotic production and are bld under all conditions (15). In S. clavuligerus, ppGpp synthesis is accompanied by depletion of the cellular GTP pool (15). If, as in E. coli, the XDH encoded by the xdhABC cluster is involved in purine salvage, under starvation conditions such as those that signal the initiation of morphogenesis and antibiotic production in Streptomyces species, induction of purine salvage might be a signal for the initiation of antibiotic production and morphological development. Inappropriate expression of this XDH might interfere with this signaling pathway and lead to a defect in the ability of the organism to appropriately sense or interpret starvation signals. Overexpression of a cluster could result in an abnormally high level of xanthine in the cells and an elevated level of GTP during the time when ppGpp synthesis is strongest. High levels of ppGpp synthesis that result in depletion of the intracellular GTP pool is normally enough to trigger morphological differentiation in Streptomyces species (15). A large GTP pool could cause an arrest in morphological development, while at the same time increased ppGpp production could cause overproduction of actinorhodin.
Acknowledgments
We thank Karen Stirrett for many thoughtful discussions during the course of the study and assistance in the construction of mutants, Michael W. W. Adams for suggesting the XDH assays and for providing the tools and equipment to do them, and David Brown for help with preparation of the manuscript.
This study was supported by a grant from Microbia, Inc., Cambridge, MA, to J.W. B.H. was also supported by predoctoral training grant GM07103 from the National Institute for General Medical Sciences to the Genetics Department of the University of Georgia.
Footnotes
Published ahead of print on 26 October 2007.
REFERENCES
- 1.Aigle, B., A. Wietzorrek, E. Takano, and M. J. Bibb. 2000. A single amino acid substitution in region 1.2 of the principal sigma factor of Streptomyces coelicolor A3(2) results in pleiotropic loss of antibiotic production. Mol. Microbiol. 37995-1004. [DOI] [PubMed] [Google Scholar]
- 2.Bentley, S. D., K. F. Chater, A. M. Cerdeno-Tarraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C. H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M. A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill, and D. A. Hopwood. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417141-147. [DOI] [PubMed] [Google Scholar]
- 3.Bertani, I., and V. Venturi. 2004. Regulation of the N-acyl homoserine lactone-dependent quorum-sensing system in rhizosphere Pseudomonas putida WCS358 and cross-talk with the stationary-phase RpoS sigma factor and the global regulator GacA. Appl. Environ. Microbiol. 705493-5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 5.Bray, R. C., M. J. Barber, H. Dalton, D. J. Lowe, and M. P. Coughlan. 1975. Iron-sulphur systems in some isolated multi-component oxidative enzymes. Biochem. Soc. Trans. 3479-482. [DOI] [PubMed] [Google Scholar]
- 6.Caballero, J. L., F. Malpartida, and D. A. Hopwood. 1991. Transcriptional organization and regulation of an antibiotic export complex in the producing Streptomyces culture. Mol. Gen. Genet. 228372-380. [DOI] [PubMed] [Google Scholar]
- 7.Cho, Y. H., E. J. Kim, H. J. Chung, J. H. Choi, K. F. Chater, B. E. Ahn, J. H. Shin, and J. H. Roe. 2003. The pqrAB operon is responsible for paraquat resistance in Streptomyces coelicolor. J. Bacteriol. 1856756-6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folcher, M., R. P. Morris, G. Dale, K. Salah-Bey-Hocini, P. H. Viollier, and C. J. Thompson. 2001. A transcriptional regulator of a pristinamycin resistance gene in Streptomyces coelicolor. J. Biol. Chem. 2761479-1485. [DOI] [PubMed] [Google Scholar]
- 9.Gust, B., G. L. Challis, K. Fowler, T. Kieser, and K. F. Chater. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. USA 1001541-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haseltine, W. A., and R. Block. 1973. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc. Natl. Acad. Sci. USA 701564-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hille, R. 2002. Molybdenum and tungsten in biology. Trends Biochem. Sci. 27360-367. [DOI] [PubMed] [Google Scholar]
- 12.Hillerich, B., and J. Westpheling. 2006. A new GntR family transcriptional regulator in Streptomyces coelicolor is required for morphogenesis and antibiotic production and controls transcription of an ABC transporter in response to carbon utilization. J. Bacteriol. 1887477-7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopwood, D. A., H. Wildermuth, and H. M. Palmer. 1970. Mutants of Streptomyces coelicolor defective in sporulation. J. Gen. Microbiol. 61397-408. [DOI] [PubMed] [Google Scholar]
- 14.Ichimori, K., M. Fukahori, H. Nakazawa, K. Okamoto, and T. Nishino. 1999. Inhibition of xanthine oxidase and xanthine dehydrogenase by nitric oxide: nitric oxide converts reduced xanthine-oxidizing enzymes into the desulfo-type inactive form. J. Biol. Chem. 2747763-7768. [DOI] [PubMed] [Google Scholar]
- 15.Jin, W., Y. G. Ryu, S. G. Kang, S. K. Kim, N. Saito, K. Ochi, S. H. Lee, and K. J. Lee. 2004. Two relA/spoT homologous genes are involved in the morphological and physiological differentiation of Streptomyces clavuligerus. Microbiology 1501485-1493. [DOI] [PubMed] [Google Scholar]
- 16.Kieser, T., M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, United Kingdom.
- 17.Kisker, C., H. Schindelin, D. Baas, J. Retey, R. U. Meckenstock, and P. M. Kroneck. 1998. A structural comparison of molybdenum cofactor-containing enzymes. FEMS Microbiol. Rev. 22503-521. [DOI] [PubMed] [Google Scholar]
- 18.Levine, R. A., and M. W. Taylor. 1982. Mechanism of adenine toxicity in Escherichia coli. J. Bacteriol. 149923-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendel, R. R., and F. Bittner. 2006. Cell biology of molybdenum. Biochem. Biophys. Acta 1763621-625. [DOI] [PubMed] [Google Scholar]
- 20.Perez-de la Mora, M., J. Mendez-Franco, R. Salceda, and J. R. Riesgo-Escovar. 1989. A glutamate dehydrogenase-based method for the assay of l-glutamic acid: formation of pyridine nucleotide fluorescent derivatives. Anal. Biochem. 180248-252. [DOI] [PubMed] [Google Scholar]
- 21.Ramos, J. L., M. Martinez-Bueno, A. J. Molina-Henares, W. Teran, K. Watanabe, X. Zhang, M. T. Gallegos, R. Brennan, and R. Tobes. 2005. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69326-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryding, N. J., M. J. Bibb, V. Molle, K. C. Findlay, K. F. Chater, and M. J. Buttner. 1999. New sporulation loci in Streptomyces coelicolor A3(2). J. Bacteriol. 1815419-5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saenger, W., P. Orth, C. Kisker, W. Hillen, and W. Hinrichs. 2000. The tetracycline repressor: a paradigm for a biological switch. Angew. Chem. Int. Ed. Engl. 392042-2052. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook, J., and D. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Sprusansky, O., L. Zhou, S. Jordan, J. White, and J. Westpheling. 2003. Identification of three new genes involved in morphogenesis and antibiotic production in Streptomyces coelicolor. J. Bacteriol. 1856147-6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun, J., A. Hesketh, and M. Bibb. 2001. Functional analysis of relA and rshA, two relA/spoT homologues of Streptomyces coelicolor A3(2). J. Bacteriol. 1833488-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takano, E., R. Chakraburtty, T. Nihira, Y. Yamada, and M. J. Bibb. 2001. A complex role for the γ-butyrolactone SCB1 in regulating antibiotic production in Streptomyces coelicolor A3(2). Mol. Microbiol. 411015-1028. [DOI] [PubMed] [Google Scholar]
- 28.Thapper, A., M. G. Rivas, C. D. Brondino, B. Ollivier, G. Fauque, I. Moura, and J. J. Moura. 2006. Biochemical and spectroscopic characterization of an aldehyde oxidoreductase isolated from Desulfovibrio aminophilus. J. Inorg. Biochem. 10044-50. [DOI] [PubMed] [Google Scholar]
- 29.Willey, J., J. Schwedock, and R. Losick. 1993. Multiple extracellular signals govern the production of a morphogenetic protein involved in aerial mycelium formation by Streptomyces coelicolor. Genes Dev. 75895-903. [DOI] [PubMed] [Google Scholar]
- 30.Willey, J. M., A. Willems, S. Kodani, and J. R. Nodwell. 2006. Morphogenetic surfactants and their role in the formation of aerial hyphae in Streptomyces coelicolor. Mol. Microbiol. 59731-742. [DOI] [PubMed] [Google Scholar]
- 31.Xi, H., B. L. Schneider, and L. Reitzer. 2000. Purine catabolism in Escherichia coli and function of xanthine dehydrogenase in purine salvage. J. Bacteriol. 1825332-5341. [DOI] [PMC free article] [PubMed] [Google Scholar]