

Abstract
To assess the contributions of single-strand DNases (ssDNases) to recombination in a recBCD+ background, we studied 31 strains with all combinations of null alleles of exonuclease I (Δxon), exonuclease VII (xseA), RecJ DNase (recJ), and SbcCD DNase (sbcCD) and exonuclease I mutant alleles xonA2 and sbcB15. The xse recJ sbcCD Δxon and xse recJ sbcCD sbcB15 quadruple mutants were cold sensitive, while the quadruple mutant with xonA2 was not. UV sensitivity increased with ssDNase deficiencies. Most triple and quadruple mutants were highly sensitive. The absence of ssDNases hardly affected P1 transductional recombinant formation, and conjugational recombinant production was decreased (as much as 94%) in several cases. Strains with sbcB15 were generally like the wild type. We determined that the sbcB15 mutation caused an A183V exchange in exonuclease motif III and identified xonA2 as a stop codon eliminating the terminal 8 amino acids. Purified enzymes had 1.6% (SbcB15) and 0.9% (XonA2) of the specific activity of wild-type Xon (Xon+), respectively, with altered activity profiles. In gel shift assays, SbcB15 associated relatively stably with 3′ DNA overhangs, giving protection against Xon+. In addition to their postsynaptic roles in the RecBCD pathway, exonuclease I and RecJ are proposed to have presynaptic roles of DNA end blunting. Blunting may be specifically required during conjugation to make DNAs with overhangs RecBCD targets for initiation of recombination. Evidence is provided that SbcB15 protein, known to activate the RecF pathway in recBC strains, contributes independently of RecF to recombination in recBCD+ cells. DNA end binding by SbcB15 can also explain other specific phenotypes of strains with sbcB15.
The heterotrimeric RecBCD enzyme (encoded by the recB, recC, and recD genes) is a central component of the main pathway of genetic recombination and recombinational DNA repair of Escherichia coli (the RecBCD pathway) and functions in the initiation of these processes (31, 34, 37). The enzyme (also termed exonuclease V [ExoV]), with its DNase and helicase activities, processively degrades duplex DNA from an end until it reaches an octanucleotide termed Chi, which is present in the E. coli genome once per 5,000 nucleotides on average. Upon contact with Chi, the duplex DNA degradation activity of the enzyme is attenuated and switched to produce a 3′ single-stranded (ss) DNA end on which it loads RecA protein, making a nucleoprotein filament ready to initiate recombination. recB or recC null mutations drastically reduce homologous recombination, increase the sensitivity of cells to DNA-damaging agents, and impair cell viability (34). In several studies extragenic suppressors of the severe effects of recBC mutations have been isolated and characterized. One group, termed sbcA mutations, was found to express recombination genes of the cryptic Rac prophage (43, 68). The other group of mutations was located in the gene for ExoI and affected ExoI activity (32, 33). ExoI is a 3′-specific processive exonuclease for ssDNA (35, 69). One class of these mutations, termed xonA mutations, suppressed the UV sensitivity and the growth defect of recBC strains; the other, termed sbcB mutations, restored recombination as well (32, 33). Recombination and recombination repair occurring in the recBC sbcB strains were attributed to the RecF pathway of recombination, because they were found to depend on a number of genes that were not essential for recombination in cells with the functional RecBCD enzyme; among these genes are recF, recO, recR, recJ, recQ, recN, and ruv (14, 37). It was also found that additional mutations, located in the sbcC gene, were present in the initial recBC sbcB mutants and that newly constructed recBC sbcB strains rapidly accumulated sbcC mutations, presumably because only together with sbcC mutations did sbcB mutations fully suppress the poor growth of recBC strains along with their recombination deficiency and UV sensitivity (36). sbcC is part of the sbcC sbcD operon (27), and mutations in either gene resulted in suppression. The SbcC and SbcD proteins constitute a DNase with multiple activities. SbcCD is both an ss endonuclease (19) and a duplex DNA exonuclease degrading the 3′ strand (15, 17), and it has hairpin-cutting (18) and ss overhang-cleaving (15) activities. Genetic and physiological studies have shown that the RecF pathway acts mainly in the repair of daughter strand gaps, while the RecBCD pathway acts in the repair of double-strand breaks, in double-strand end repair during replication, and in the recombination of linear duplex DNA molecules with the cellular genome during transduction and conjugation (34, 37). Why the sbcB but not the xonA class of mutations allows efficient recombination in recBC strains has been discussed but is not known (34, 56). In recBC sbcB sbcCD mutants, recombination is dependent on the RecQ helicase (54) and the RecJ DNase (encoded by recJ [42]). RecJ is a 5′-specific processive ssDNA exonuclease that is thought to degrade the 5′ strand after RecQ helicase action on duplex DNA, leaving a 3′ overhang for recombination initiation. RecJ is also implicated in the RecBCD pathway by postsynaptically degrading the displaced 5′ tail competing with the transferred single strand for pairing, by postsynaptically trimming 5′ tails to generate ligatable structures, and by blunting of ends for recombination initiation (50, 60, 73).
Single-strand-specific DNases have important roles in methyl-directed mismatch repair (MMR) in E. coli by removing the strand containing the mismatch base (52). In vitro studies have shown that when the incision of a newly synthesized strand occurs 5′ to the mispaired base, the 5′-specific enzymes RecJ and ExoVII (encoded by xseAB [12]) can remove the single strand, while after incision 3′ to the mismatch, the 3′-specific enzyme ExoI, ExoVII, or ExoX (encoded by exoX [74]) degrades the single strand (72). A lack of all four enzymes abolished normal repair in cell extracts (72). In vivo there is considerable redundancy between the ssDNase functions, since double and triple mutants did not show increased spontaneous mutability (28, 73). Even the quadruple mutant strain showed only weakly increased mutability, but this was attributed to the underrecovery of spontaneous mutants due to loss of viability as a result of attempted but unsuccessful MMR (11, 72). The four ssDNases also have an important role in the suppression of deletion formation between tandem repeats in DNA (24). A further 3′ ss-specific exonuclease, encoded by xni (ExoIX), was shown not to belong to the group of ssDNases of E. coli involved in MMR, recombination, and DNA repair (40).
Single-strand-specific DNases are also involved in a phenomenon termed in vivo silencing of ExoV. While ExoV is the strongest duplex DNA exonuclease in E. coli active in the degradation of restricted foreign DNA (64) or unprotected infecting phage genomes like that of T4 2− (55), its activity rapidly disappears when the enzyme interacts in vivo with linear DNA containing Chi sequences. Silencing occurs during rolling-circle replication of Chi-containing plasmids (20) or after restriction of phage DNA with Chi sites (26; B. Thoms and W. Wackernagel, unpublished data), fragmentation of the E. coli genome by gamma-irradiation of cells (6), or treatment with bleomycin (30). It was assumed that the blunt duplex DNA ends produced by these treatments allowed RecBCD to attack the DNA and rapidly encounter Chi sites, where the enzyme is silenced. In fact, 97% of the ExoV activity had disappeared from extracts prepared immediately after a brief bleomycin treatment of cells compared to the activity in extracts from untreated cells (30). Silencing of ExoV was also observed after UV irradiation of cells, which does not directly produce DNA strand breaks (71). The process required excision repair and about 2 h of postirradiation incubation, during which genomic DNA fragmentation occurred. Remarkably, the UV-induced silencing of ExoV was greatly diminished in multiple ssDNase mutants such as recJ sbcCD and xonA recJ sbcCD strains (71). These and other observations were interpreted as indicating that (i) after heavy UV irradiation the chromosome is fragmented in a uvrA-dependent process, (ii) the fragments have ss tails long enough to prevent the attack by RecBCD (58, 67), (iii) ssDNases degrade the tails, leading to blunt ends, and (iv) RecBCD then starts DNA degradation and is silenced. It was hypothesized that the ssDNases in recBCD+ cells would blunt DNA ends to make them available for RecBCD and thereby would help to direct tailed intermediates into the RecBCD pathway (71).
In this study we have combined wild-type and null alleles of four ssDNases (ExoI, ExoVII, RecJ, and SbcCD) in all possible combinations in a recBCD+ background and also with two widely studied alleles of the ExoI gene, sbcB15 and xonA2. These alleles code for proteins with low ExoI activity and are known to suppress the phenotype of recBC mutants. The 31 mutant strains plus the wild type were characterized with respect to their UV sensitivities and recombination proficiencies during transduction and conjugation. In addition, the sbcB15 and xonA2 mutations were identified, and some properties of the purified SbcB15 and XonA2 proteins were analyzed. The data provide evidence that ssDNases are more important for homologous recombination in recBCD+ cells during conjugation than during transduction and that the ExoI alleles Δxon, xonA2, and sbcB15 influence recombination frequencies differently alone and in combination with other ssDNase mutations. Genetic and biochemical evidence suggests that xonA2 is leaky and not a null allele like Δxon.
MATERIALS AND METHODS
Bacterial strains.
The bacterial strains used in this study are listed in Table 1. Most strains were newly constructed by P1 transduction using standard procedures (63), with allele sources as indicated (Table 1), and selection for the presence of an antibiotic resistance cassette in the null alleles. Strain DL733 (19) has a nonsense mutation in proC, which was separated from the closely linked ΔsbcCD::nptII by transferring ΔsbcCD::nptII into BT560 (which is like AB1157 but proA+). A Pro+ Kmr transductant (BT564) was the donor for transduction of ΔsbcCD::nptII.
TABLE 1.
E. coli strains used in this study
| Strain | Relevant genotype | Construction by P1 transduction or source (reference) |
|---|---|---|
| AB1157 | F−argE3 hisG4 leuB6 proA2 thr-1 thi-1 ara-14 galK2 lacY1 mtl-1 xyl-5 tsx-33 rpsL31 supE44 | 29 |
| BT122 | recJ284::Tn10 | P1·JC12166 × AB1157 |
| BT223 | Like JC7623 but his::Tn10 proC::Tn5 | Laboratory strain |
| BT384 | ΔsbcCD::nptII | P1·BT564 × AB1157 |
| BT386 | ΔsbcCD::nptII recJ284::Tn10 | P1·BT122 × BT384 |
| BT420 | sbcB15 his | From BT418a |
| BT421 | xonA2 ΔsbcCD::nptII | P1·BT564 × WA818 |
| BT422 | xonA2 ΔsbcCD::nptII recJ284::Tn10 | P1·BT122 × BT421 |
| BT430 | ΔxseA18::bla | P1·SMR3465 × AB1157 |
| BT431 | xonA2 ΔsbcCD::nptII recJ284::Tn10 ΔxseA18::bla | P1·BT430 × BT422 |
| BT432 | ΔsbcCD::nptII ΔxseA18::bla | P1·BT430 × BT384 |
| BT434 | sbcB15 ΔxseA18::bla | P1·BT430 × BT420 |
| BT435 | xonA2 ΔsbcCD::nptII ΔxseA18::bla | P1·BT430 × BT421 |
| BT436 | ΔxseA18::bla recJ284::Tn10 | P1·BT122 × BT430 |
| BT442 | sbcB15 ΔxseA18::bla ΔsbcCD::nptII | P1·BT564 × BT434 |
| BT443 | sbcB15 ΔxseA18::bla recJ284::Tn10 | P1·BT122 × BT434 |
| BT444 | sbcB15 ΔsbcCD::nptII | P1·BT564 × BT420 |
| BT445 | sbcB15 recJ284::Tn10 | P1·BT122 × BT420 |
| BT446 | ΔsbcCD::nptII ΔxseA18::bla recJ284::Tn10 | P1·BT122 × BT432 |
| BT447 | sbcB15 ΔsbcCD::nptII recJ284::Tn10 | P1·BT122 × BT444 |
| BT448 | ΔsbcCD::nptII ΔxseA18::bla ΔxonA300::cat | P1·BT449 × BT432 |
| BT449 | ΔxonA300::cat his+ | P1·SMR838 × AB1157 |
| BT450 | ΔxseA18::bla ΔxonA300::cat | P1·BT449 × BT430 |
| BT459 | ΔxseA18::bla ΔxonA300::cat recJ284::Tn10 | P1·BT122 × BT450 |
| BT460 | ΔsbcCD::nptII ΔxonA300::cat recJ284::Tn10 | P1·BT122 × BT543 |
| BT477 | ΔxonA300::cat recJ284::Tn10 | P1·BT122 × BT449 |
| BT497 | ΔsbcCD::nptII ΔxseA18::bla ΔxonA300::cat recJ284::Tn10 | P1·BT122 × BT448 |
| BT498 | sbcB15 ΔxseA18::bla ΔsbcCD::nptII recJ284::Tn10 | P1·BT122 × BT442 |
| BT534 | xonA2 ΔxseA18::bla | P1·BT430 × WA818 |
| BT536 | xonA2 recJ284::Tn10 ΔxseA18::bla | P1·BT430 × BT538 |
| BT538 | xonA2 recJ284::Tn10 | P1·BT122 × WA818 |
| BT543 | ΔsbcCD::nptII ΔxonA300::cat | P1·BT449 × BT384 |
| BT564 | ΔsbcCD::nptII | P1·DL733 × BT560a |
| BT568 | ΔsbcCD::nptII sbcB15 recF332::Tn3 | P1·JC10990 × BT444 |
| BT569 | ΔsbcCD::nptII ΔxonA300::cat recF332::Tn3 | P1·JC10990 × BT543 |
| BT570 | ΔxseA18::bla sbcB15 recF400::Tn5 | P1·WA576 × BT434 |
| BT571 | ΔxseA18::bla ΔxonA300::cat recF400::Tn5 | P1·WA576 × BT450 |
| BW113 | Hfr P4X | 44 |
| DL733 | ΔsbcCD::nptII | 19 |
| JC7623 | recB21 recC22 sbcB15 sbcC201 argE3 hisG4 proA2 thr-1 ara-14 galK2 lacY1 mtl-1 xyl-5 rpsL131 supE44 tsx-33 leuB6 thi-1 | 33 |
| JC8260 | xonA2 | 56 |
| JC10990 | recF332::Tn3 | A. J. Clark |
| JC12166 | recJ284::Tn10 | 41 |
| SMR838 | ΔxonA300::cat | 28 |
| SMR3465 | ΔxseA18::bla | 28 |
| WA112 | (K12s) prototrophic | 2 |
| WA576 | recF400::Tn5 | 70 |
| WA632 | recB21 | 62 |
| WA818 | xonA2 | P1·JC8260 × AB1157 (62) |
See Materials and Methods.
Two nonselective alleles (sbcB15 and xonA2) were transferred in the following ways. The sbcB15 allele was recovered from the original strain JC7623 (33) by first transducing his::Tn10 into the strain (selection for Tcr and screening for UVr; strain BT223) and then cotransducing sbcB15 with his::Tn10 (about 50% cotransduction) into AB1157, giving strain BT418. The sbcB15 allele in BT418 was identified by cotransducing it from BT418 with his::Tn10 into a recB21 sbcC201 strain (UV sensitive), which became UV resistant. sbcB15 was also verified by allele-specific PCR (see below). BT418 was made tetracycline sensitive by removal of Tn10 (45), giving strain BT420 (his). This strain had the same phenotype as BT418, and the presence of sbcB15 was verified by allele-specific PCR. The xonA2 allele from the original strain JC8260 (32) was transferred to AB1157 by cotransduction with the his+ marker to make strain WA818, which was verified as reported previously (62) and by allele-specific PCR (see below).
Media and growth conditions.
Bacterial cultures were grown in TBY (10 g of Bacto tryptone, 5 g of Bacto yeast extract, and 5 g of NaCl per 1,000 ml) at 30°C. Plates contained TBY agar (TBY solidified with 1.5% agar) and were incubated at 30°C, unless stated otherwise. If necessary, antibiotics were included in TBY: kanamycin (25 μg ml−1), tetracycline (15 μg ml−1), ampicillin (100 μg ml−1), and chloramphenicol (15 μg ml−1). M9-glucose minimal agar medium was supplemented with the required amino acids (50 μg ml−1 each), thiamine (0.1 μg ml−1), and streptomycin (40 μg ml−1) to select against the Hfr strain BW113.
Conjugation and transduction crosses.
Interrupted matings (at 37°C for 30 min unless stated otherwise) were performed with log-phase cells in TBY at an F−/Hfr cell ratio of 9:1. The BW113 Hfr cells were grown at 37°C without agitation, the F− cells at 30°C on a shaker. Matings were interrupted by strong vortexing. Transductions with P1 were performed with cells of fresh overnight cultures suspended in Tris-Ca2+ buffer (50 mM Tris-HCl [pH 7.9], 2 mM CaCl2) at a multiplicity of infection of 0.2 for 20 min at 37°C for phage adsorption. Then cells were sedimented and resuspended in 0.1 ml of Tris-Ca2+ buffer before plating. The pro+ Strr (rpsL31) exconjugants and leu+ transductants were determined on selective M9 medium. Plates were incubated at 30°C (at 37°C for strains BT497 and BT498, and at both temperatures for BT431). Recombination frequencies in conjugation experiments were expressed as recombinants per donor cell. A conjugation efficiency control by plasmid transfer was omitted because previous experiments with a strain series like that used here (all strains were AB1157 derivatives) had not shown conjugation differences (73). Transduction frequencies were expressed as recombinants per P1 PFU. In each crossing experiment, a comparison with AB1157 was included as a reference to which the results with other strains were related. In experiments at 37°C, the reference experiment was also performed at 37°C. The frequencies of recombination during conjugation and transduction were corrected for the viability of recipient cells, which was between 0.5 and 1 for all single and multiple ssDNase mutants and was 0.3 for the recB21 strain WA632.
Cloning of xon genes, overexpression of genes, and isolation of exonucleases.
The open reading frames (ORFs) of the wild-type xon (xon+), sbcB15, and xonA2 genes were amplified with Phusion DNA polymerase as detailed by the manufacturer (Finnzymes, Espoo, Finland) using primer XonA-f-BE (5′-ATC GGA TCC ATG ATG AAT GAC GGT AAG CAA TCT ACC), covering the first 27 nucleotides of the ORF plus an overhang with a BamHI and a half-EcoRV cleavage site (underlined), and primer XonA-r-E (5′-ATC TTA GAC AAT CTC TTC CGC GTA CT), covering the last 24 nucleotides (positions 1405 to 1428) of the ORF plus an overhang with a half-EcoRV cleavage site (underlined). The PCR products were cloned into the EcoRV site of pPCR-Script-Cam-SK(+) (Stratagene, La Jolla, CA), the F factor-derived single-copy-number plasmid pJE256 (22), and the N-terminal hexahistidine tag expression vector pQE30 by using strain SG13009 with pREP4 as described by the provider (Qiagen, Hilden, Germany). The sbcB15 and xonA2 genes, including the LacI-repressed T5 promoter plus the hexahistidine tag sequence from pQE30 derivatives, were also cloned into the pACYC184-derived vector pBO1 carrying the lacI gene. These plasmids (pBO1-hexa-his-sbcB15 and pBO1-hexa-his-xonA2), conferring Cmr, were introduced into strains by electroporation and selection on a medium with chloramphenicol (100 μg ml−1). Induction of cells containing pQE30 derivatives with isopropyl-β-d-thiogalactopyranoside (IPTG; 0.2 mM), further growth at 26°C (xon+ and sbcB15) or 23°C (xonA2), collection of cells, preparation of cell extracts, and affinity chromatography on nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen, Hilden, Germany) were performed as described in reference 58a. The enzymes were eluted at 100 to 200 mM imidazole. The preparations were dialyzed overnight against 20 mM Tris-HCl (pH 8.0), 0.5 mM EDTA, 5 mM 2-mercaptoethanol, and 50% glycerol (storage buffer) and were stored at −20°C. Protein concentrations were determined with a protein microassay kit (Bio-Rad Laboratories, Hercules, CA) using bovine serum albumin as a reference. The purity of the final preparations was estimated after subjecting 1 μg of each purified enzyme preparation to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10% acrylamide; 1-mm-thick gels) with six-H-tagged protein markers (Sigma, St. Louis, MO) and staining with Coomassie blue. The homogeneities of the preparations were ≥95% for Xon+, 80% for SbcB15, and 55% for XonA2.
Exonuclease I assay.
The reaction conditions of Lehman and Nussbaum (35) were used (reaction volume, 0.3 ml): 67 mM glycine buffer (pH 9.5), 6.7 mM MgCl2, 10 mM 2-mercaptoethanol, and 7 nmol heat-denatured [3H]thymidine-labeled E. coli DNA. The DNA was isolated by the genomic-tip protocol (Qiagen) and had a specific radioactivity of 7.4 × 104 cpm μg−1. Immediately before use, the DNA was heated at 100°C for 10 min in 10 mM Tris-HCl (pH 8.0)-0.5 mM EDTA, followed by quenching in ice-water. Reactions (30 min at 37°C) were terminated by addition of 0.2 ml of an ice-cold bovine serum albumin solution (25 mg ml−1) followed by 0.3 ml of ice-cold trichloracetic acid (12%, wt/vol). After 5 min on ice, acid-precipitable material was sedimented for 10 min, and 0.4 ml of the supernatant was counted in a liquid scintillation counter (acid-soluble products). One unit of enzyme releases 10 nmol of acid-soluble products from DNA under these conditions (35). The enzyme was diluted in storage buffer without glycerol and EDTA. In some experiments, the assay mixture was reduced in volume without any change in concentrations.
Gel retardation assay.
The DNA substrates with ss overhangs were obtained by annealing of oligonucleotides of 100 nucleotides at 85°C in 50 mM NaCl for 4 min, followed by 1 h at 65°C and slow cooling to room temperature. The following oligonucleotides were employed: oligonucleotide a, 5′-ATCGCCGCTC CCGATTCGCA GCGCATCGCC TTCTATCGCC TTCTTGACGA GTTCTTCTGA GCGGGACTCT GGGGTTCGAA CCAGCGGACC AAGCTACAAA; oligonucleotide b, 5′-TTCGAACCCC AGAGTCCCGC TCAGAAGAAC TCGTCAAGAA GGCGATAGAA GGCGATGCGC TGCGAATCGG GAGCGGCGAT ACCGTAAAGC ACGAGGAAGA; oligonucleotide c, 5′-TCGTCAAGAA GGCGATAGAA GGCGATGCGC TGCGAATCGG GAGCGGCGAT ACCGTTAAGC ACGAGGAGGA ACCGTAAAGC ACGAGGAAGA ACGAGGAACA; oligonucleotide d, 5′-GCTTCCTCGT GCTTTACGGT ATCGCCGCTC CCGATTCGCA GCGCATCGCC TTCTATCGCC TTCTTGACGA GTTCTTCTGA GCGGGACTCT GGGGTTCGAA; oligonucleotide e, 5′-GGCGTCGCTT GGTCCGCTCC TTCGAACCCC AGAGTCCCGG TCAGAAGAAC TCGTCAAGAA GGCGATAGAA GGCGATGCGC TGCGAATCGG GAGCGGCGAT. Oligonucleotides a plus b and d plus e gave molecules with two 20-nucleotide overhangs of 3′ and 5′ polarity, respectively. Oligonucleotides a plus c gave molecules with two 3′ overhangs of 50 nucleotides. The DNA (mostly 100 or 200 ng/assay) was incubated in 10 μl of 67 mM glycine buffer (pH 9.5)-10 mM 2-mercaptoethanol with the enzyme. The concentration of MgCl2, as well as the incubation temperature and period, is given for each experiment. The amount of enzyme added and the ratio of enzyme molecules to ssDNA ends were calculated by using 55,000 as the Mr for the three enzymes and correcting for the homogeneity of each enzyme preparation. After incubation, the samples were briefly quenched in ice, mixed with 2 μl slot marker (not containing SDS or EDTA), and electrophoresed on a 1.5 or 2% agarose gel in 89 mM Tris-borate buffer (pH 8.5) at 3°C for 3 to 4 h at 70 to 100 V. The gel was stained with ethidium bromide and photographed with a charge-coupled device camera on a UV transilluminator.
UV irradiation.
Log-phase cells were washed in phosphate buffer and irradiated with an Osram HNS10 germicidal lamp at a dose rate of 1.8 J m−2 s−1 as described elsewhere (70). Appropriate dilutions were plated onto TBY medium, and colonies were counted after 1 to 2 days of incubation at 30°C (at 37°C for strains BT497 and BT498, and at both temperatures for BT431).
Nucleotide sequence accession numbers and allele-specific PCR.
The nucleotide sequences of the xon genes were determined by the dideoxynucleotide sequencing procedure and were deposited in the EMBL database. The accession numbers are AM235175 (xon+), AM235176 (sbcB15), and AM235177 (xonA2). To identify the sbcB15 allele by PCR, primers exo-f-t (5′-AAC GCC CAC GAT GTG; bp 535 to 549 of the gene) and exo-r-c (5′-CTT CCG CGT ACT GCC; bp 1403 to 1417) were used; primer exo-f-c (5′-AAC GCC CAC GAT GCG) was used for the wild-type allele control. To identify the xonA2 allele, primers exo-f-c and exo-r-t (5′-CTT CCG CGT ACT GTC) were used; exo-r-c was used for the wild-type allele control. PCR (30 cycles) with Taq polymerase was run for 15 s at 94°C (denaturation), 30 s at 58°C (annealing), and 120 s at 68°C (synthesis) per cycle.
RESULTS
Quadruple ssDNase mutants are cold sensitive depending on the xon allele.
Strains with single and multiple null alleles of ssDNase genes were constructed in the AB1157 background by transduction (Table 1) and are presented with phenotypes arranged in Table 2. All possible double and triple combinations of the alleles recJ284::Tn10, ΔxseA18::bla, ΔsbcCD::nptII, and ΔxonA300::cat (referrred to below as recJ, xseA, sbcCD, and Δxon) were obtained. However, no quadruple mutant was obtained in several different attempts, suggesting that the quadruple mutant was not viable. We also constructed strains having all possible combinations of the recJ, xseA, and sbcCD alleles with the xonA2 or sbcB15 allele instead of Δxon (Table 1); these strains are also included in Table 2. These two alleles code for proteins with low residual DNase activity (56). In the course of these constructions, a recJ xseA sbcCD xonA2 quadruple mutant (strain BT431) was isolated without any problem, but no quadruple mutant with the sbcB15 allele was obtained, suggesting that the xonA2 allele was not as detrimental as the Δxon and sbcB15 alleles in the quadruple combination. While this work was in progress, Burdett et al. (11) showed that a recJ xseA ΔexoX Δxon quadruple mutant was cold sensitive, i.e., inviable at 30°C. All our experiments were performed at 30°C to allow better growth on media with several antibiotics. After switching the incubation temperature to 37°C, we succeeded in isolating quadruple mutants with Δxon (BT497) or sbcB15 alleles (BT498). These strains did not grow at 30°C in broth medium (survival, about 4 × 10−2 compared to that at 37°C) and therefore are cold sensitive, as was the other xon strain reported (11). We tested the assumption that the cold resistance of the quadruple mutant BT431 was in fact due to the xonA2 allele. For this purpose, xonA2 was transduced into the quadruple mutant BT498, replacing the sbcB15 allele (cotransduction with his+; the presence of the xonA2 mutation in the transductants was confirmed by allele-specific PCR [see Materials and Methods]). The resulting strain was as cold resistant as BT431, indicating that xonA2 provided an activity allowing the quadruple mutants to grow at 30°C. The idea that the xonA2 allele is a leaky and not a null allele gained support from the observations that most multiple mutants with xonA2 were less UV sensitive than those with Δxon and often gave higher recombination frequencies, as well as from biochemical data (see below). The quadruple mutants and the xse sbcCD sbcB15 strain (BT442) were slow growing and required about 20 h to form countable colonies on broth medium. The viabilities of the quadruple mutants ranged from 0.5 to 0.6, higher than that of the recB21 strain WA632 (0.3 [data not shown]).
TABLE 2.
Effects of recJ, xseA, sbcCD, Δxon, sbcB15, and xonA2 mutations on the UV survival and genetic recombination of recBCD+ strainsa
| Strain | Relevant genotype
|
Relative survival at 72 J m−2b (n) | Relative frequencyc
|
|||||
|---|---|---|---|---|---|---|---|---|
| recJ | xseA | sbcCD | xon | leu+ transduction (A) (n) | pro+ exconjugants (B) (n) | Ratio (B/A) | ||
| AB1157 | + | + | + | + | 1 (3) | 1 (13) | 1 (19) | |
| BT122 | recJ | + | + | + | 0.18 ± 0.12 (3) | 0.88 ± 0.11 (3) | 0.65 ± 0.08 (3) | 0.74 |
| BT430 | + | xseA | + | + | 1.48 ± 0.43 (2) | 1.14 ± 0.05 (3) | 1.04 ± 0.13 (3) | 0.91 |
| BT384 | + | + | sbcCD | + | 0.95 ± 0.05 (2) | 1.01 ± 0.12 (3) | 1.13 ± 0.26 (4) | 1.12 |
| WA818 | + | + | + | xonA2 | 1.25 ± 0.33 (2) | 0.90 ± 0.14 (3) | 0.26 ± 0.02 (4) | 0.28 |
| BT420 | + | + | + | sbcB15 | 3.75 ± 1.24 (2) | 1.21 ± 0.02 (3) | 1.16 ± 0.20 (3) | 0.95 |
| BT449 | + | + | + | Δxon | 1.55 ± 0.10 (2) | 0.71 ± 0.09 (3) | 0.19 ± 0.04 (3) | 0.26 |
| BT386 | recJ | + | sbcCD | + | 0.33 ± 0 (2) | 0.70 ± 0.09 (4) | 0.92 ± 0.08 (3) | 1.31 |
| BT432 | + | xseA | sbcCD | + | 1.25 ± 0.25 (2) | 1.22 ± 0.38 (3) | 1.03 ± 0.19 (3) | 0.84 |
| BT436 | recJ | xseA | + | + | 0.0035 ± 0.0003 (2) | 0.95 ± 0.09 (3) | 0.88 ± 0.14 (3) | 0.93 |
| BT538 | recJ | + | + | xonA2 | 0.28 ± 0.08 (4) | 0.49 ± 0.14 (5) | 0.143 ± 0.015 (3) | 0.29 |
| BT445 | recJ | + | + | sbcB15 | 0.10 ± 0.07 (2) | 0.86 ± 0.11 (3) | 0.33 ± 0.08 (3) | 0.38 |
| BT477 | recJ | + | + | Δxon | 0.17 ± 0.14 (3) | 0.31 ± 0.12 (3) | 0.066 ± 0.006 (3) | 0.21 |
| BT534 | + | xseA | + | xonA2 | 1.25 ± 0.50 (2) | 0.97 ± 0.20 (4) | 0.42 ± 0.04 (3) | 0.43 |
| BT434 | + | xseA | + | sbcB15 | 1.08 ± 0.30 (2) | 1.25 ± 0.09 (3) | 1.35 ± 0.42 (5) | 1.08 |
| BT450 | + | xseA | + | Δxon | 1.18 ± 0.27 (2) | 1.55 ± 0.13 (3) | 1.78 ± 0.06 (3) | 1.15 |
| BT421 | + | + | sbcCD | xonA2 | 0.70 ± 0.45 (3) | 0.82 ± 0.13 (4) | 0.33 ± 0.06 (3) | 0.40 |
| BT444 | + | + | sbcCD | sbcB15 | 0.43 ± 0.03 (2) | 1.3 ± 0.21 (3) | 1.50 ± 0.25 (3) | 1.15 |
| BT543 | + | + | sbcCD | Δxon | 0.73 ± 0.25 (2) | 0.62 ± 0.02 (3) | 0.34 ± 0.03 (3) | 0.55 |
| BT446 | recJ | xseA | sbcCD | + | 0.0038 ± 0 (2) | 0.58 ± 0.06 (3) | 0.63 ± 0.16 (3) | 1.02 |
| BT536 | recJ | xseA | + | xonA2 | 0.0016 ± 0.0009 (5) | 0.80 ± 0.3 (5) | 0.31 ± 0.08 (3) | 0.39 |
| BT443 | recJ | xseA | + | sbcB15 | 0.00014 ± 0.00003 (3) | 1.3 ± 0.23 (3) | 0.75 ± 0.20 (3) | 0.58 |
| BT459 | recJ | xseA | + | Δxon | 0.00025 ± 0.00018 (3) | 1.03 ± 0.18 (3) | 0.46 ± 0.04 (3) | 0.45 |
| BT435 | + | xseA | sbcCD | xonA2 | 0.16 ± 0.01 (2) | 1.14 ± 0.09 (4) | 0.92 ± 0.10 (3) | 0.81 |
| BT442 | + | xseA | sbcCD | sbcB15 | 0.0045 ± 0.0001 (2) | 1.11 ± 0.11 (4) | 0.90 ± 0.19 (3) | 0.81 |
| BT448 | + | xseA | sbcCD | Δxon | 0.017 ± 0.006 (2) | 1.15 ± 0.16 (4) | 1.76 ± 0.29 (3) | 1.53 |
| BT422 | recJ | + | sbcCD | xonA2 | 0.13 ± 0.05 (2) | 0.40 ± 0.07 (4) | 0.096 ± 0.013 (3) | 0.24 |
| BT447 | recJ | + | sbcCD | sbcB15 | 0.023 ± 0.012 (2) | 1.01 ± 0.17 (3) | 0.70 ± 0.19 (3) | 0.69 |
| BT460 | recJ | + | sbcCD | Δxon | 0.028 ± 0.010 (2) | 0.17 ± 0.05 (4) | 0.065 ± 0.007 (3) | 0.38 |
| BT431 | recJ | xseA | sbcCD | xonA2 | 0.0030 ± 0.0011 (3) | 0.54 ± 0.21 (5) | 0.39 ± 0.01 (3) | 0.72 |
| 0.0011 ± 0.0001 (2) | 0.61 ± 0.09 (3) | 0.45 (1)d | 0.73 | |||||
| BT498 | recJ | xseA | sbcCD | sbcB15 | 0.0011 ± 0.0005 (3) | 0.48 ± 0.16 (3) | 0.35 ± 0.02 (3)d | 0.73 |
| BT497 | recJ | xseA | sbcCD | Δxon | 0.00006 ± 0.00001 (3) | 0.76 ± 0.04 (3) | 0.41 ± 0.02 (3)d | 0.54 |
| WA632 (recB) | + | + | + | + | 0.0011 ± 0.0005 (3) | 0.0041 ± 0.0023 (3) | 0.0083 ± 0.0043 (4) | |
The recJ, xseA, sbcCD, and Δxon mutations are null alleles due to insertion or deletion. sbcB15 is a missense mutation, and xonA2 is an unsuppressed nonsense mutation. All strains are derivatives of AB1157. UV survival and genetic recombination were measured by transduction and conjugation, respectively.
Survival values are means from n independent determinations given with the deviation from the mean (when n is 2) or the standard deviation (when n is 3 to 5) and are expressed relative to the survival of strain AB1157 [(4.0 ± 3.0) × 10−2].
Transduction and exconjugant frequencies are means from n independent determinations with standard deviations and are expressed relative to those of strain AB1157. Strain AB1157 has a leu+ transduction frequency of (4.9 ± 1.3) × 10−5 and a pro+ exconjugant frequency of (1.1 ± 0.2) × 10−1.
Experiments performed at 37°C; all other experiments were performed at 30°C.
Transductional recombination is partially blocked in a recJ sbcCD Δxon strain.
Transduction by P1 transports duplex DNA fragments of the donor chromosome into the recipient cell, where they can be integrated into the recipient chromosome by homologous recombination (46, 76). Transduction of the leuB+ allele from WA112 (donor) into each of the 32 recBCD+ strains (Table 2) was unimpaired in the majority of the single and multiple ssDNase mutants; frequencies were mostly 62 to 155% of that obtained with the wild type. The double mutants BT538 (recJ xonA2) and BT477 (recJ Δxon) showed a slight Rec− phenotype (49 and 31% of the wild-type recombination frequency, respectively), and this phenotype was slightly enhanced by sbcCD (strains BT422 and BT460). A recB21 mutant (a null allele due to an insertion in recB with a polar effect on recD [1]), known to make cells recombination deficient, gave the low (0.4% of the wild type) transduction frequency expected (14) (Table 2, strain WA632). Therefore, none of the four ssDNases examined is essential on its own for recombination during transduction of recBCD+ cells, and the combination of several defective alleles also did not dramatically decrease recombination, although the combination of recJ with sbcCD and Δxon/xonA2 led to lower transduction frequencies.
Conjugational recombination.
During conjugation, ssDNA is transferred from the Hfr strain to the recipient, in which it is converted to duplex DNA that has ss overhangs (25, 65). The various mutant strains, all carrying the proA2 mutation, were recipients in conjugation experiments with the Hfr strain BW113 (P4X origin of transfer [44]) transferring the pro+ locus early. All of the DNase genes studied are located more than 42 map min downstream from the P4X transfer origin on the E. coli chromosome, and their transfer was prevented by mating interruption at 30 min. The relative frequencies of pro+ Strr (rpsL31) recombinants obtained with all mutant recipient strains are shown in Table 2. We first consider the null alleles. Among the single mutants, only the Δxon strain (BT449) showed a notable reduction in conjugational recombinant production (about 19% that of the wild type), consistent with previous observations on conjugation (73) and Chi-dependent λ recombination (60). This is a ∼3-fold-stronger phenotype than that for transduction (Table 2, last column). The recombination frequency of the recJ Δxon double mutant (BT477) was low, as reported previously (60, 73), and was not further decreased by the additional presence of the sbcCD mutation (BT460). For the latter two strains, conjugational recombination was 5-fold and 2.5-fold more strongly affected than transduction, respectively. On the other hand, the combination of Δxon with xseA (BT450) increased conjugational recombination about ninefold over that for the Δxon strain (BT449). This finding indicates that the absence of the two 3′ exonucleases ExoI and ExoVII allows highly efficient conjugational recombination in recBCD+ cells, and this phenotype is independent of SbcCD, as seen with the triple mutant BT448. For the triple mutants BT446 and BT459 and for the quadruple mutant BT497, conjugational recombination was decreased by 37 to 59%. With the exception of the Δxon xseA strain and its sbcCD derivative, the frequency of conjugational recombination was decreased in the single and multiple mutants, and conjugation was two- to five-fold more strongly affected than transduction.
The phenotype of xonA2 was less strong than that of Δxon in several of the single and multiple mutants (Table 2). For instance, compared to that for the recJ single mutant (BT122), the frequency of conjugational recombination was more strongly decreased for the recJ Δxon mutant (BT477) (ninefold) than for the recJ xonA2 mutant (BT538) (twofold). Further, for the xonA2 strain (WA818), the addition of an xseA mutation increased recombination only twofold (BT534), whereas for the Δxon strain, xseA mutation increased recombination about ninefold (BT450). Apparently, only the total absence of both 5′ exonucleases provides the normal high-recombination phenotype. For all eight single and multiple mutants with xonA2, conjugation was again more strongly affected (1.2- to 4-fold) than transduction, although not to the extent seen with Δxon. The phenotypic differences between strains with Δxon or xonA2 can be taken as evidence that xonA2 is not a null allele but provides some residual activity.
The phenotype of sbcB15 differed from those of Δxon and xonA2 in single and multiple mutants. The single mutant BT420 was fully proficient at conjugational recombination, and the double and triple mutants with this allele were not impaired or only slightly impaired, except for the recJ sbcB15 strain (BT445). This strain displayed a conjugational recombination frequency 67% lower than that of the wild type, but this reduction was clearly smaller than that observed for the Δxon and xonA2 derivatives of the recJ mutant (strains BT477 and BT538). It appears that sbcB15 plays a supporting role in the conjugational recombination of recBCD+ strains that is not provided by Δxon and xonA2 and that is particularly evident in recJ mutants. Further, the sbcCD sbcB15 strain (BT444) showed a conjugational recombination frequency about 4.5-fold higher than those of the sbcCD Δxon (BT543) and sbcCD xonA2 (BT421) strains. This observation suggests that the sbcB15 sbcCD combination, known to fully suppress the recombination deficiency of recBC strains by activation of the RecF pathway, also provides extra recombination capacity to recBCD+ strains. We tested the idea that the extra capacity resulted from the switched-on RecF pathway in a recBCD+ background by crossing a recF mutation into several strains and measuring the frequency of conjugational recombination (Table 3). For strains with any combination of sbcB15 or Δxon with sbcCD or xseA mutations, the frequency of recombination was reduced partially by recF and no more than for the wild type. Since in recBC sbcB15 sbcCD strains, a recF mutation causes a ∼240-fold decrease in conjugational recombination frequency as a result of a blocked RecF pathway (14), the results in Table 3 indicate that the RecF pathway does not play a major role in strains that have sbcB15 plus sbcCD mutations in a recBCD+ background. Strain BT498 (recJ xseA sbcCD sbcB15) showed a 65% lower conjugational recombination frequency than the wild type, a reduction similar to that for the other quadruple mutants with Δxon or xonA2. This finding indicates that the high-recombination phenotype of the sbcB15 sbcCD strain requires ExoVII and RecJ. In contrast to Δxon and xonA2, the sbcB15 allele mediated conjugational recombination in the recBCD+ background at a level close to the wild-type level, irrespective of the presence of the other ssDNases.
TABLE 3.
Effects of recF deficiency on the conjugational recombination of strains with combinations of Δxon, sbcB15, sbcCD, and xseA mutations in a recBCD+ background
| Strain | Relevant genotype | Relative frequency of pro+ exconjugantsa | Relative effect of recF |
|---|---|---|---|
| AB1157 | Wild type | 1 | 1 |
| WA576 | recF400 | 0.66 ± 0.05 | 0.66 |
| BT444 | sbcB15 sbcCD | 1.23 ± 0.06 | 1 |
| BT568 | sbcB15 sbcCD recF332 | 0.66 ± 0.12 | 0.54 |
| BT543 | Δxon sbcCD | 0.25 ± 0.05 | 1 |
| BT569 | Δxon sbcCD recF332 | 0.12 ± 0.01 | 0.52 |
| BT434 | sbcB15 xseA | 1.65 ± 0.12 | 1 |
| BT570 | sbcB15 xseA recF400 | 1.52 ± 0.30 | 0.93 |
| BT450 | Δxon xseA | 1.25 ± 0.25 | 1 |
| BT571 | Δxon xseA recF400 | 0.87 ± 0.32 | 0.7 |
Exconjugant frequencies are means from three to five independent experiments with standard deviations and are expressed relative to that for AB1157 [(1.0 ± 0.2) × 10−1]. The crosses were performed at 37°C. The conjugation time was 20 min to avoid transfer of the recF+ allele into the recipients.
SbcCD is one of the ssDNases with a role in the repair of UV damage.
The UV sensitivities (at 72 J m−2) of the various mutants are shown in Table 2. The single mutants were as resistant as the wild type except for the recJ strain (BT122; survival, 18%). Of the 12 double mutants, 10 were rather similar to the wild type, while the recJ sbcB15 strain (BT445) was more sensitive (survival, 10%) and the recJ xseA mutant (BT436) had even lower survival (0.4%). The survival of all triple mutants (16 to 0.01%) and particularly that of the quadruple mutants (0.3 to 0.006%) was clearly lower than that of the wild type and often comparable to that of the recB strain. These results confirm data previously reported on recJ, xse, and Δxon (73) and extend them by showing that sbcCD deficiency decreases the UV survival of most strains and particularly that of the xseA Δxon and recJ Δxon strains (BT448, BT460). Thus, SbcCD is one of the ssDNases that has profound effects on UV survival by serving a partially redundant function. Further, we observed that UV survival was influenced by the allele of the ExoI gene. Most multiple mutants with xonA2 (including BT422, BT435, BT536, and BT431) were less sensitive (as much as 18-fold) than the corresponding strains with Δxon (Table 2), suggesting that xonA2 contributes to UV survival more than Δxon.
Identification of the sbcB15 and xonA2 mutations.
The xon ORF (1,428 bp) was amplified by PCR and cloned into pPCR-Script-Cam-SK(+), pQE30, and pJE256. The DNA of AB1157 was the template for three independent PCR amplifications of xon+ and independent cloning procedures with the three vectors. Similarly, the DNA of JC7623 served for three independent events of sbcB15 cloning, and the DNA of JC8260 for three independent events of xonA2 cloning. DNAs from the three events of each series were sequenced and gave consistent results. The xon+ sequence was identical to that present in the E. coli K-12 genome sequence of strain MG1655 (EMBL accession no. U00096). The sbcB15 sequence contained a single nucleotide exchange at bp 548 (CG to TA), switching the GCG triplet to GTG, which led to the amino acid exchange A183V. The exchange is located within the conserved exonuclease (exo) motif III of ExoI, which contributes to the active center of the enzyme (7). In the xonA2 sequence, a single nucleotide exchange was also identified; it is located at bp 1404 (GC to AT), changing the TGG codon to TGA (W468stop). The resulting opal triplet shortens the polypeptide by 8 amino acids. The AB1157 genetic background does not provide an opal suppressor (2). The stop codon in xonA2 is consistent with the earlier observation of a slightly smaller ExoI polypeptide produced from the mutant gene (56).
The SbcB15 and XonA2 proteins have strongly decreased ExoI activities.
The proteins encoded by the xon+, sbcB15, and xonA2 genes cloned into the N-terminal hexahistidine tag overexpression vector pQE30 were partially purified from cell extracts by affinity chromatography on Ni-NTA agarose. Samples run on SDS-PAGE gels are shown in Fig. 1. The specific activity of the Xon+ enzyme (Table 4) was about 10-fold higher than that reported previously for a nonhomogeneous preparation of ExoI (35), about 3-fold higher than that reported by Ray et al. (59) for a homogeneous preparation, and about 27% of that reported by Prasher et al. (57) for a nearly homogeneous preparation. In accordance with previous reports, the rate of ssDNA degradation was about 40,000-fold higher than that of duplex DNA (35, 57; also data not shown). These observations indicate that the hexahistidine tag does not have a marked effect on the activity of ExoI.
FIG. 1.
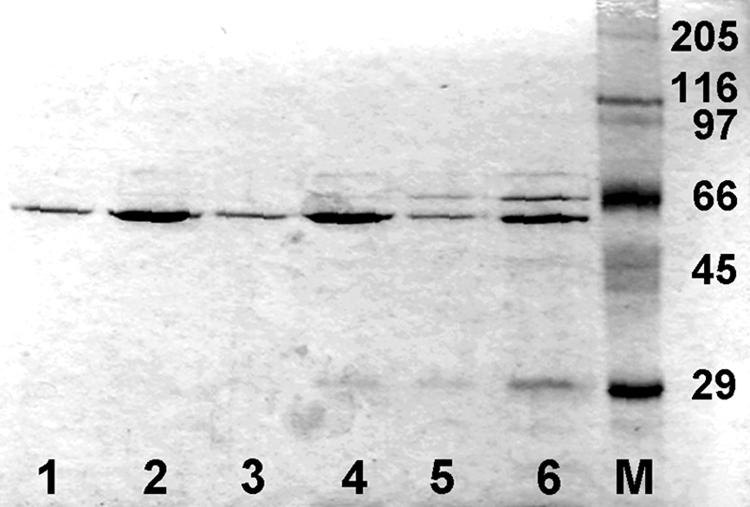
SDS-PAGE (10% acrylamide) and staining with Coomassie blue of preparations of the hexahistidine-tagged proteins Xon+, SbcB15, and XonA2 obtained by chromatography on Ni-NTA agarose. Lanes 1 and 2, Xon+ (0.2 and 1 μg, respectively); lanes 3 and 4, SbcB15 (0.2 and 1 μg, respectively); lanes 5 and 6, XonA2 (0.2 and 1 μg, respectively); lane M, molecular weight marker proteins with hexahistidine tags. Molecular weights, in thousands, are given on the right.
TABLE 4.
Specific activities of purified hexahistidine-tagged exonuclease I (Xon+) and SbcB15 and XonA2 mutant enzymes
| Enzymea | Sp act (U/mg of protein) | % Activity | % Activity correctedb |
|---|---|---|---|
| Xon+ | 1.39 × 105 | 100 | 100 |
| SbcB15 | 1.87 × 103 | 1.3 | 1.6 |
| XonA2 | 6.63 × 102 | 0.5 | 0.9 |
The enzymes were purified by Ni-NTA agarose chromatography from extracts of overexpressing cells (see Materials and Methods).
The correction considered the different levels of homogeneity of the three enzyme preparations (see Materials and Methods).
The specific activity of the SbcB15 enzyme was 1.6% of that of the Xon+ enzyme under standard conditions, and that of the XonA2 protein was 0.9% (Table 4). The markedly reduced ExoI activity of the SbcB15 protein was previously reported when crude extracts of overproducing E. coli cells were assayed, and the residual activity of the XonA2 protein was hardly detectable under those conditions (57). It was also reported that the XonA2 protein appeared less stable in cells than the wild-type enzyme (57), which fits with our observation that in independent preparations of XonA2, the protein material obtained after Ni-NTA agarose chromatography always contained two lower-molecular-weight proteins that were hardly detectable in Xon+ and SbcB15 preparations (Fig. 1). These smaller products could be fragments of the XonA2 protein that still have the hexahistidine tag.
The in vivo-synthesized, hexahistidine-tagged XonA2 and SbcB15 proteins conferred the same phenotype on cells as the corresponding natural proteins. This was shown by the following experiments. The intermediate-copy-number plasmid pBO1-hexa-his-xonA2, transferred into the recJ xseA sbcCD Δxon strain (BT497), conferred cold resistance and increased UV resistance (see Table 2) on the mutant by the background expression of the cloned gene, as did genomic xonA2. The vector plasmid had no effect. Similarly, pBO1-hexa-his-sbcB15, present in the recJ sbcCD Δxon strain (BT460), increased conjugational recombination about 12-fold, to the high level of the recJ sbcCD sbcB15 strain (BT447) (Table 2), while the vector plasmid did not. At 1 mM IPTG in the medium, either the vector alone or the vector with an insert (hexa-his-xonA2 or hexa-his-sbcB15) killed the cells.
The activity profiles of the Xon+, SbcB15, and XonA2 enzymes differ from each other.
The activities of the Xon+ and XonA2 enzymes increased two- to threefold when the MgCl2 concentration of the assay was decreased from 6.7 to 0.67 and 0.067 mM, respectively (Fig. 2). In contrast, SbcB15 activity remained at the same level at 0.67 mM MgCl2 and decreased to half at 0.067 mM. At 30°C, the Mg2+ dependence of the three enzymes was similar to that at 37°C, but the enzyme activities were lower. ExoI binds two divalent metal cations by four acidic amino acids in the three exo motifs constituting the active center (3, 7). Without added MgCl2, the Xon+ and XonA2 proteins had about 40% of the activity they displayed with 6.7 mM MgCl2, an observation consistent with results previously reported for the purified Xon+ enzyme (33). In contrast, the SbcB15 enzyme had only 4% residual activity without added MgCl2 (Fig. 2).
FIG. 2.
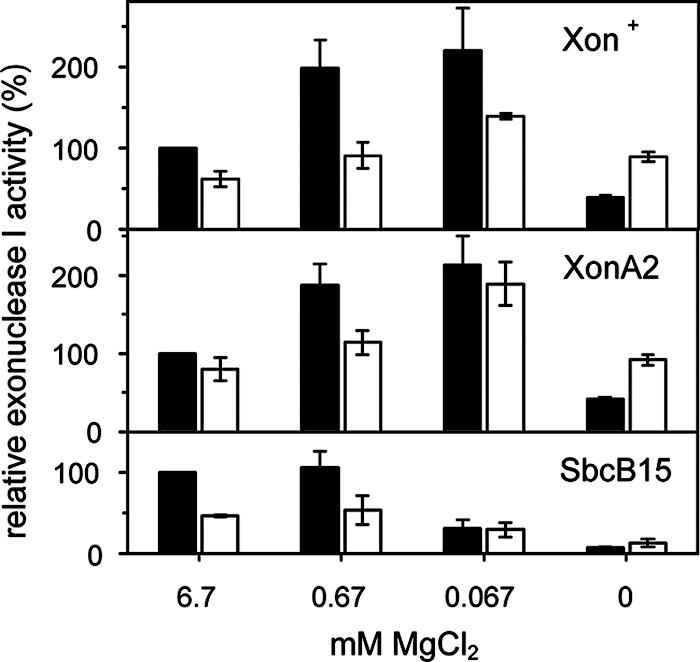
The exonuclease activities of purified Xon+, XonA2, and SbcB15 proteins were measured under standard conditions (6.7 mM MgCl) and at lower MgCl2 concentrations at 37°C (solid bars) and 30°C (open bars). Each assay mixture contained 0.15 U of Xon+, XonA2, or SbcB15 protein. Activities were expressed relative to those obtained for each enzyme at 37°C and 6.7 mM MgCl2 (100%). Data are means from three independent experiments. Error bars, standard deviations.
The activities of Xon+ and SbcB15 were rather constant between pH 9.5 and 7.5 (Fig. 3) a finding that corresponds with a previous observation made with the Xon+ enzyme (9). Results similar to those obtained at 0.67 mM MgCl2 (shown in Fig. 3) were also obtained at 6.7 mM MgCl2. Between pH 7 and 6, the activities of both enzymes declined strongly, at 37°C as well as at 30°C. In contrast, the activity of XonA2 was 4-fold higher at pH 7.5 than at pH 9.5, and even at pH 6.0 the activity was 1.4-fold that at pH 9.5. Compared to 37°C, where the pH optimum of XonA2 was 7.5, at 30°C the optimum was further shifted toward a lower pH. The data suggest that the SbcB15 enzyme, with its affected active center, differs from Xon+ in Mg2+ dependence but not in its pH activity profile, while the XonA2 enzyme displayed a Mg2+ dependence similar to that of Xon+ due to its wild-type-like active center but had a changed pH activity profile, probably as a result of the C-terminal truncation.
FIG. 3.
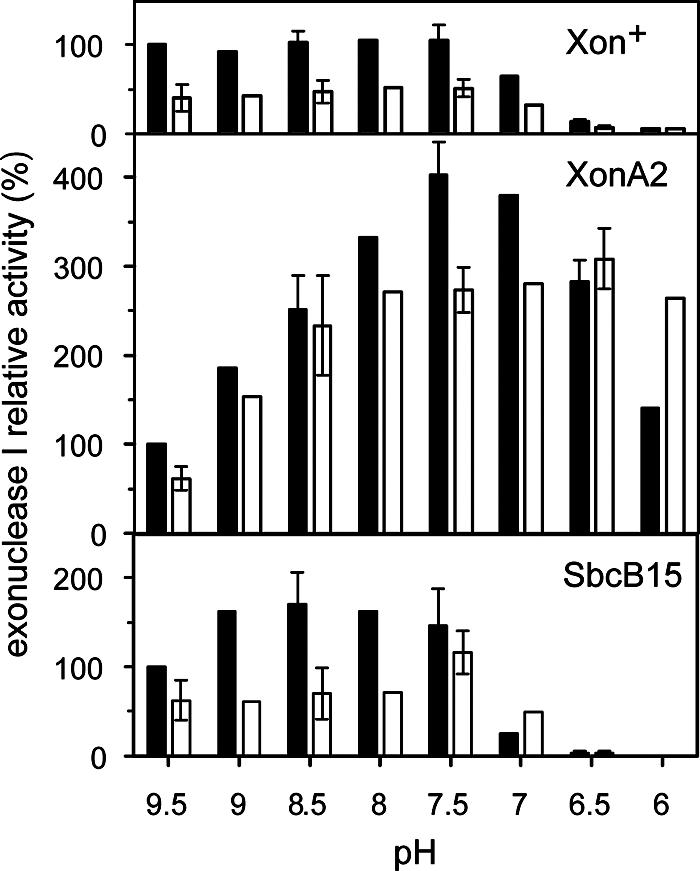
The exonuclease activities of the purified Xon+, XonA2, and SbcB15 proteins were measured at the indicated pH values of the reaction mixture (67 mM Tris-HCl, 0.67 mM MgCl2, 10 mM β-mercaptoethanol) and at two temperatures (solid bars, 37°C; open bars, 30°C). Each assay mixture contained 0.15 U of Xon+, XonA2, or SbcB15 protein. Activities were expressed relative to those obtained for each enzyme at 37°C and pH 9.5 (100%). Data are means from experiments performed in duplicate. Error bars, deviations from the means.
Extended binding of the SbcB15 enzyme to 3′ ends of ssDNA.
We used an electrophoretic assay to test for interaction of the enzymes with ssDNA. The first substrates were duplex DNAs of 80 bp with overhangs of 20 nucleotides of the same polarity at each side. Stretches of 20 nucleotides suffice for binding of ExoI and nucleolysis (8). The substrate DNA moved during agarose electrophoresis as a 120-bp duplex, indicating that the two overhangs of 20 nucleotides contributed to the electrophoretic mobility as if they were double-stranded. The substrate with 3′ overhangs was treated at 0°C for 1 min with an amount of Xon+ protein giving about 1 enzyme molecule per DNA end and was immediately analyzed by electrophoresis. The DNA moved at the position of a ∼90-bp duplex (Fig. 4A), showing that the 3′ overhangs had been shortened and the enzyme had dissociated from the DNA, probably when it approached the duplex region, as previously reported (10, 69). The same reaction product as after 1 min at 0°C was also seen after 3 min at 0°C. In separate experiments we had found that when the standard assay with heat-denatured DNA was performed at 0°C, Xon+ had 0.009% of its activity at 37°C. When the 1-min reaction at 0°C was followed by 30 s at 37°C, the DNA reached the position of a duplex of 80 bp in the gel, suggesting that the shortened overhangs had been completely removed (Fig. 4A). This observation is consistent with the previous finding that the last 6 to 8 nucleotides of an overhang before the duplex region are removed at a much lower rate, probably by enzyme molecules that have already dissociated from the substrate and then reassociated to hydrolyze a partially degraded substrate (9, 10, 69). The substrate with 5′ overhangs was not changed, confirming the 3′ specificity of the enzyme (Fig. 4A).
FIG. 4.
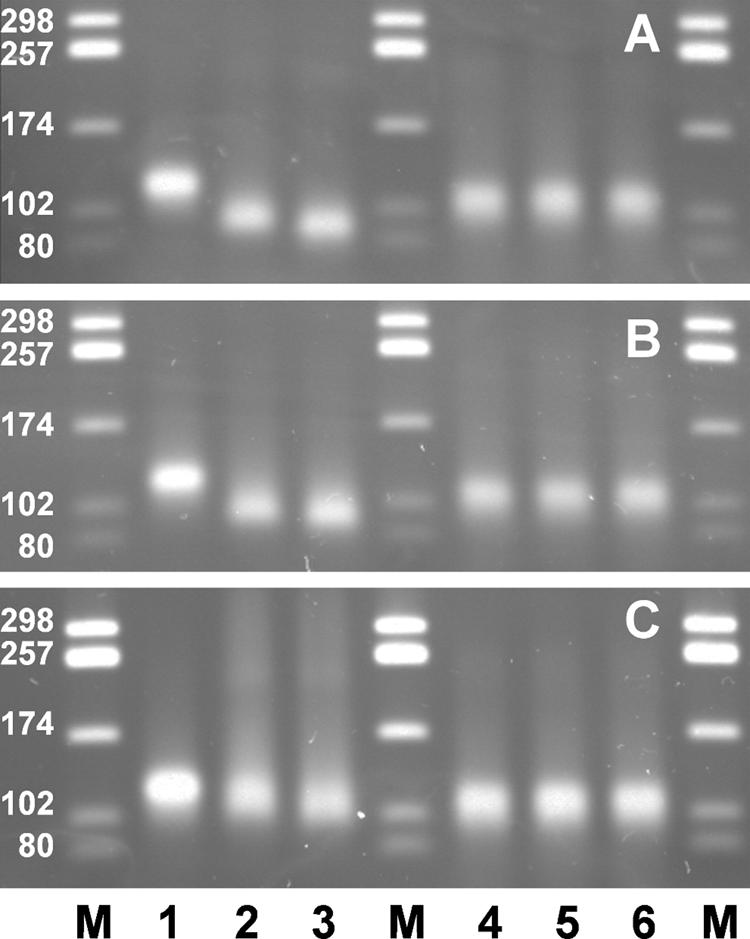
Agarose gel electrophoresis of 80-bp duplex DNA fragments with 20-nucleotide overhangs of 3′ (lanes 1 to 3) or 5′ (lanes 4 to 6) polarity at each side (100 ng per lane; 3.1 pmol ends) after incubation with 170 ng (3.1 pmol) of Xon+ (A), XonA2 (B), or SbcB15 (C) protein in reaction mixtures with added MgCl2 (6.7 mM). Lanes M, duplex DNA marker fragments; lanes 1 and 4, no protein added. Incubation was carried out for 1 min at 0°C only (lanes 2 and 5) or for 1 min at 0°C followed by 30 s at 37°C (lanes 3 and 6).
Despite its lower specific activity (Table 4), the XonA2 enzyme degraded the substrate almost as Xon+ did (Fig. 4B). This is consistent with our observation that the specific activity of XonA2 at 0°C was 62% of that of Xon+ at 0°C. The results with the SbcB15 protein differed in three respects from those with Xon+ and XonA2 (Fig. 4C). First, during the 0°C reaction, hardly any faster-moving DNA molecules were produced from the substrate. Second, a considerable fraction of DNA showed decreased mobility during gel electrophoresis, seen as a DNA smear. Third, after the additional incubation for 30 s at 37°C, more DNA moved slightly faster than the substrate DNA, indicating overhang degradation. It was concluded that SbcB15 remained associated with DNA, leading to its retardation during electrophoresis. The fact that the retarded DNA appeared as a smear could result from the detachment of SbcB15 protein from DNA during electrophoresis. There was less 3′ overhang degradation by SbcB15 than by Xon+ and XonA2. At 0°C, the specific activity of SbcB15 was about 4% of that of Xon+. The electrophoresis of substrate DNA with 5′ overhangs was not changed by either XonA2 or SbcB15 (Fig. 4B and C).
A further DNA substrate consisted of a 50-bp duplex with 3′ overhangs of 50 nucleotides on each side, moving during electrophoresis like a 150-bp duplex fragment. This substrate was reacted for 1 min at 0°C, followed by 10 s at 37°C, with increasing amounts of protein before electrophoresis (Fig. 5). At the lowest concentration of Xon+ (Fig. 5A), one part of the DNA appeared at a gel position of about 108 bp of duplex DNA, probably because one overhang was shortened to about 8 nucleotides. The other part had a position corresponding to the substrate with two largely removed overhangs (resembling a 66-bp duplex). With increasing amounts of Xon+, the latter band increased in intensity and shifted gradually toward the position of a 50-bp duplex, indicating that the residual ∼8-nucleotide overhangs per end were also degraded. The XonA2 protein degraded the DNA substrate into products similar to those from Xon+, but more slowly, and the shortest product was that corresponding to the 66-bp duplex (Fig. 5B). The rather parallel results for Xon+ and XonA2 in the experiments for which results are shown in Fig. 4 and 5 are compatible with the incubations occurring largely at 0°C, a temperature at which the activity levels of the two enzymes are not very different. The SbcB15 protein, on the other hand, caused a massive retardation of DNA, which increased with increasing protein amounts (Fig. 5C). We confirmed that DNA retardation resulted from protein association by observing the elimination of retardation when the reaction mixture was treated briefly with 0.2% SDS before gel electrophoresis (Fig. 5C). Then all DNA appeared in a broad band covering the size of the intact substrate and the sizes of partially degraded molecules. Western blot analysis of gels similar to those shown in Fig. 5 using a monoclonal antibody directed against hexahistidine tags showed that SbcB15 was present in the smear of retarded DNA but not at the position of the degraded substrate, whereas no coelectrophoresis of the Xon+ or XonA2 protein occurred with the DNA substrate (data not shown).
FIG. 5.
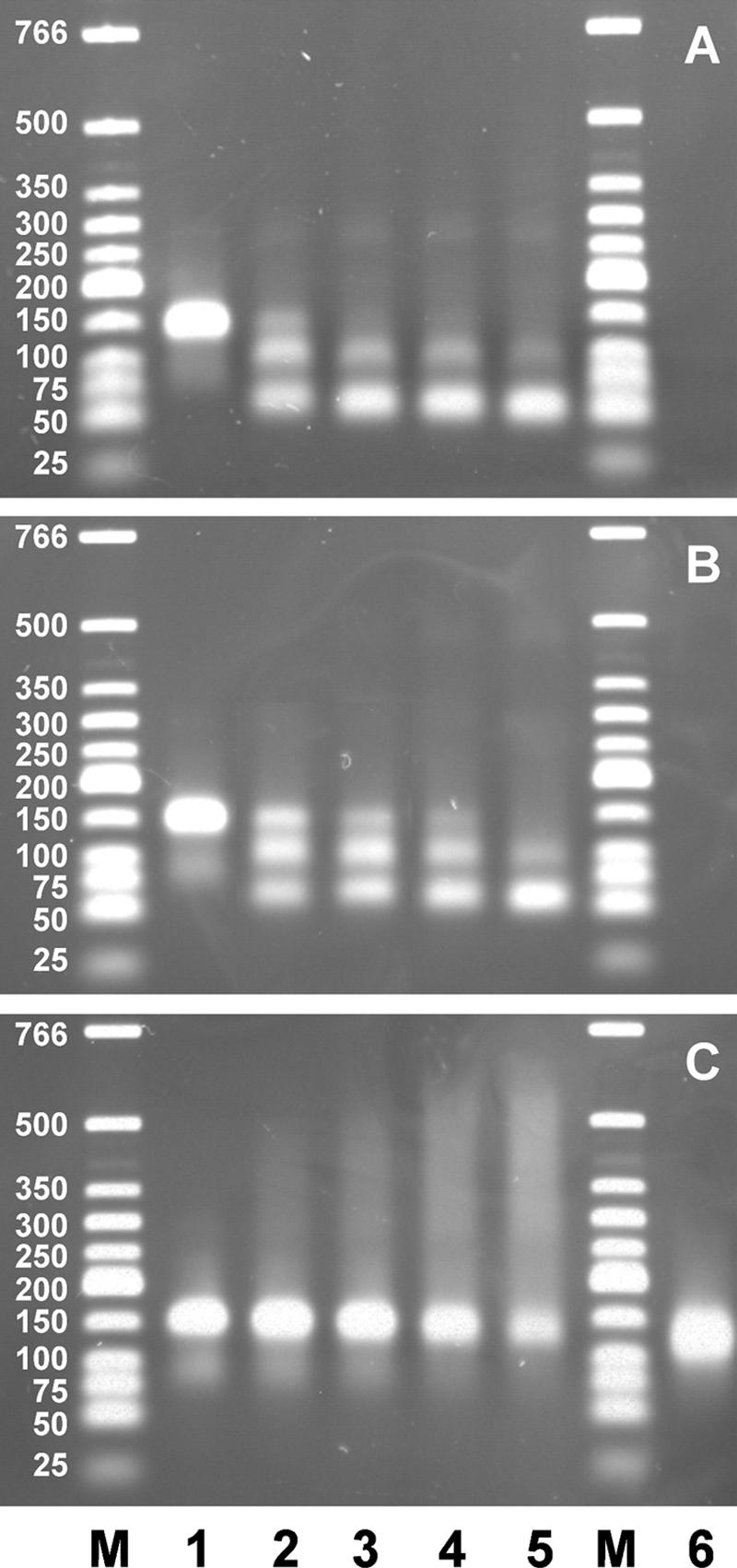
Agarose gel electrophoresis of 50-bp duplex DNA fragments with 50-nucleotide overhangs of 3′ polarity at each side (200 ng per lane; 6.2 pmol ends) after incubation for 1 min at 0°C followed by 10 s at 37°C with increasing amounts of Xon+ (A), XonA2 (B), or SbcB15 (C). Lane 1, no protein; lane 2, 170 ng (3.1 pmol); lane 3, 340 ng (6.2 pmol); lane 4, 680 ng (12.4 pmol); lane 5, 1,020 ng (18.6 pmol); lanes M, duplex DNA marker fragments. The reaction mixtures contained added MgCl2 (6.7 mM). In panel C, lane 6 contained the same reaction mixture as lane 5 except that the material was treated for 1 min at 60°C with 0.2% SDS before electrophoresis.
Kinetics of 3′ overhang degradation.
The next experiments were all performed with an excess of protein over DNA ends. Xon+ removed both 50-nucleotide overhangs completely within 5 min at 37°C, irrespective of whether MgCl2 (6.7 mM) was present or not (Fig. 6A). At shorter times (15 s and 1 min), overhang degradation was incomplete, leading to the typical intermediate products with one (108-bp product) or two (66-bp product) largely removed overhangs. XonA2 produced only partial degradation during 5 min at 37°C in the presence of added MgCl2 and even less degradation without added MgCl2 (Fig. 6B). The 50-bp product was not seen. SbcB15 produced increasing amounts of retarded DNA-protein complexes with time at 37°C in the absence of MgCl2 (Fig. 6C). The amount of retarded DNA after 5 min (about 50% of the total) was similar to that obtained with added MgCl2 within 15 s at 37°C. Further incubation at 37°C led to the formation of the typical intermediate products. A part of the DNA was still retarded in the gel, amounting to 32% after 1 min at 37°C and 25% after 5 min at 37°C, as estimated by densitometric scanning of the gel. In agreement with the experiment for which results are shown in Fig. 2, the DNA degradation without added MgCl2 by SbcB15 is less than that by Xon+ and XonA2. Incubation of DNA with SbcB15 in the absence of MgCl2 for 20 or 40 min at 37°C (Fig. 7) gave proportions of products similar to those seen after 1 or 5 min with added MgCl2, and a part of the DNA was still retarded in the gel. Thus, the enzyme reaction is slower without added MgCl2 than with added MgCl2. The removal of SbcB15 from DNA by SDS treatment before electrophoresis (Fig. 7) revealed that the smear of retarded DNA seen after a 10-min reaction at 37°C consisted largely of substrate DNA with the overhangs shortened to various extents. This observation is in agreement with the processivity of ExoI (69), which remains bound to the substrate during DNA degradation and thus would retard the electrophoresis of DNA. The parallel appearance of the typical DNA products (of about 108 and 66 bp) in the SDS-treated and untreated samples (Fig. 7) indicated that SbcB15 finally detached from the DNA when approaching the duplex region. It is concluded that at 0°C (Fig. 4) as well as at 37°C (Fig. 7), SbcB15 remained bound to the DNA substrate for extended periods while slowly degrading the overhang.
FIG. 6.
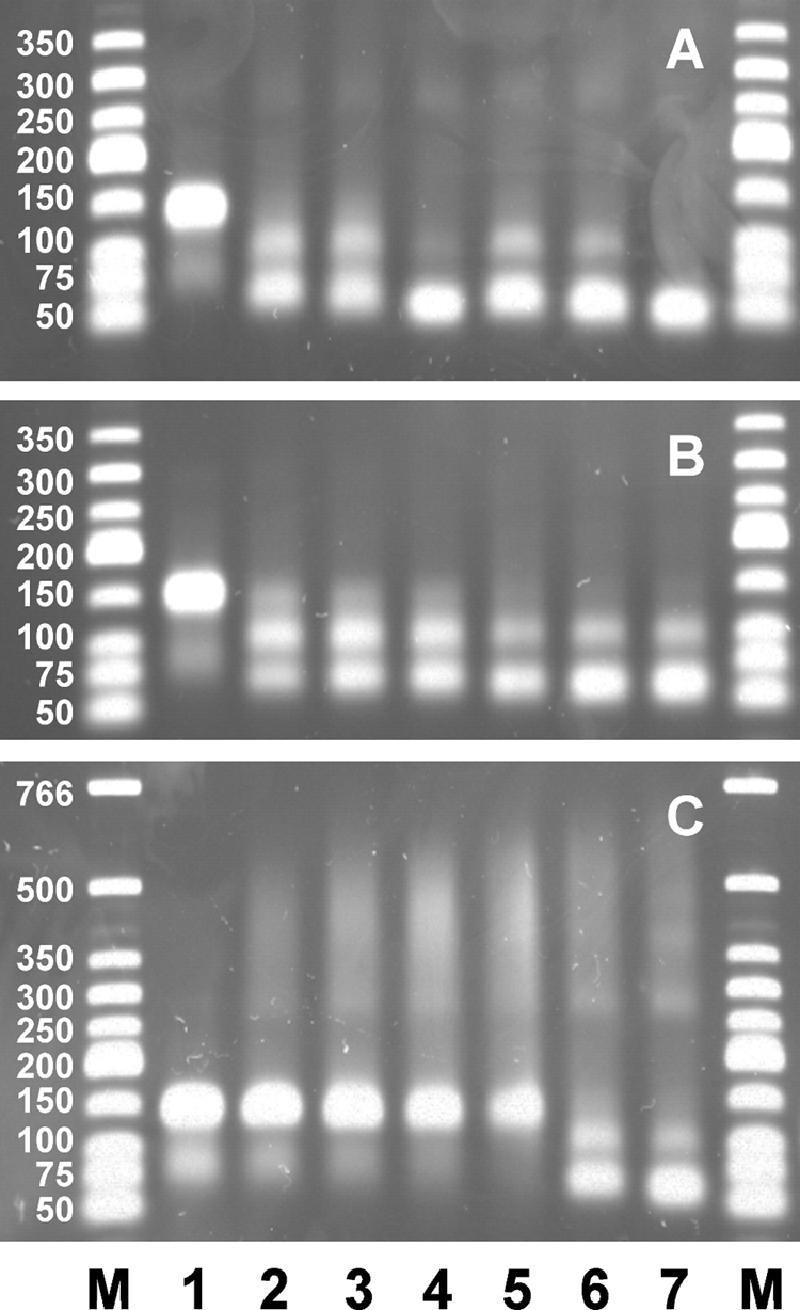
Kinetics of overhang degradation in the absence (lanes 2 to 4) or presence (lanes 5 to 7) of added MgCl2 (6.7 mM). The agarose gel shows the substrate with 3′ overhangs of 50 nucleotides (200 ng per lane; 6.2 pmol ends) after incubation with the enzyme (680 ng; 12.4 pmol) for 1 min at 0°C followed by 15 s (lanes 2 and 5), 1 min (lanes 3 and 6), or 5 min (lanes 4 and 7) at 37°C. (A) Xon+; (B) XonA2; (C) SbcB15. Lane 1, no protein; lanes M, duplex DNA marker fragments.
FIG. 7.
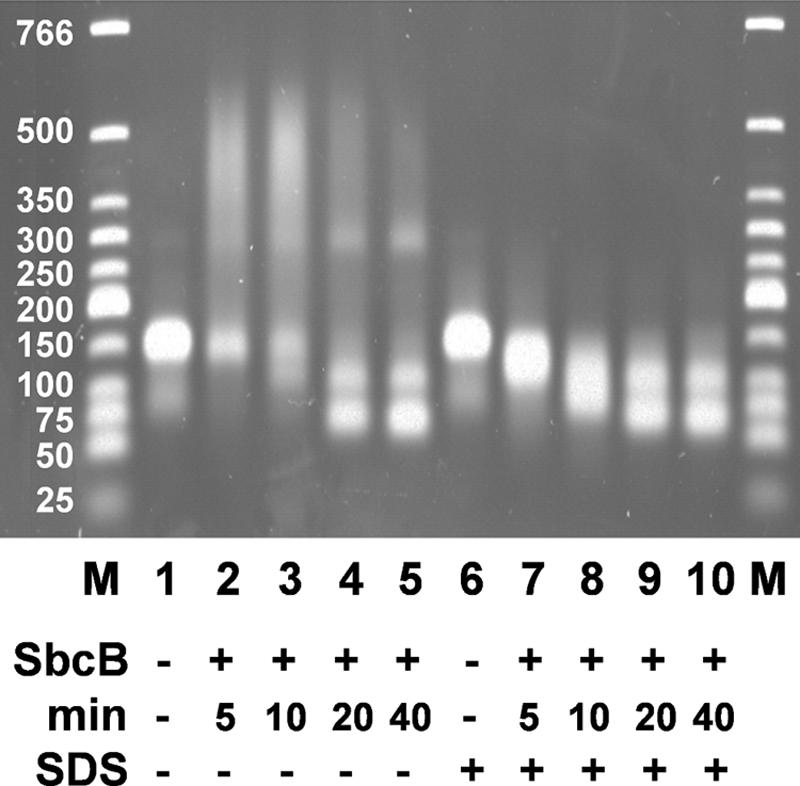
Slow and continual degradation of 3′ overhangs by SbcB15 observed by agarose gel electrophoresis. The substrate DNA with 3′ overhangs of 50 nucleotides (200 ng per lane; 6.2 pmol ends) was incubated without added MgCl2 and with the SbcB15 enzyme (1,020 ng; 18.6 pmol) for 1 min at 0°C, followed by the indicated periods at 37°C. Parallel samples were treated with SDS as for Fig. 5. M, duplex DNA marker fragments; SbcB, SbcB15 enzyme.
The SbcB15 enzyme protects 3′ DNA ends.
We asked if SbcB15 bound to a 3′ overhang would protect the end from being degraded by Xon+ and found it to be the case. We performed an experiment (Fig. 8) in which the DNA substrate with two 3′ overhangs of 50 nucleotides each was incubated for 5 min at 37°C with a threefold molar excess of SbcB15 per DNA end to allow the association of protein with DNA ends. In order to slow down the enzyme reactions, we did not add MgCl2. The association of the enzyme with DNA was evidenced by strong DNA retardation. When a parallel reaction after the 5 min at 37°C was briefly quenched on ice and then the reaction mixture was incubated with added Xon+ protein (a twofold molar excess per DNA end) for 15 s, only a very slight reduction of DNA retardation occurred, indicating that Xon+ was not acting on the DNA-SbcB15 complexes. Since the amount of Xon+ added in this experiment almost completely degraded the overhangs of the substrate not preincubated with SbcB15 within 15 s (Fig. 8, lane 5), it is clear that the bound SbcB15 protein protected the DNA ends from Xon+. In a control experiment, the simultaneous addition of the same amounts of SbcB15 and Xon+ as those used in the experiment for which results are shown in Fig. 8 led to extensive overhang degradation, indicating that the SbcB15 enzyme preparation did not inhibit Xon+ activity (data not shown) and that in a direct competition for DNA ends, Xon+ prevails. The protection of 3′ DNA ends exerted by preincubation with SbcB15 was partially overcome when the incubation at 37°C with Xon+ was extended to 5 min.
FIG. 8.
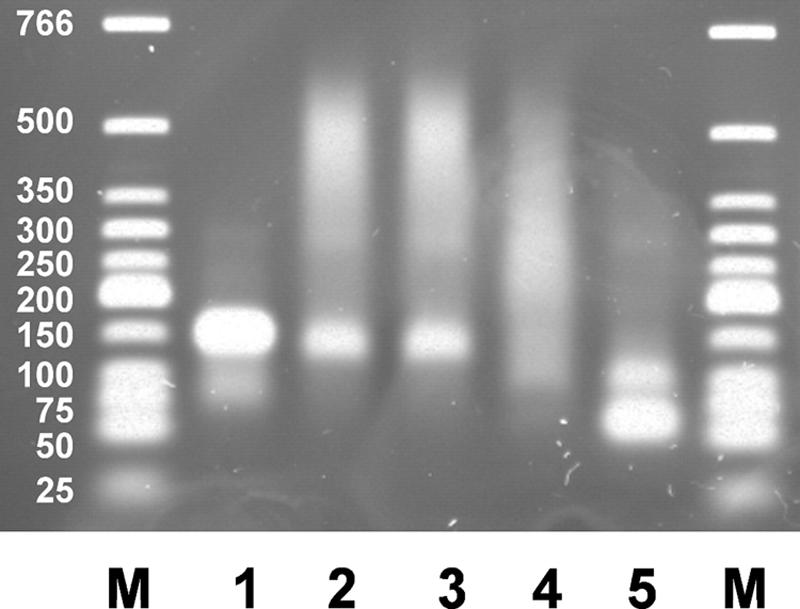
Protection of 3′ DNA overhangs against Xon+ by SbcB15 as determined by agarose gel electrophoresis. The substrate with 3′ overhangs of 50 nucleotides (200 ng per lane; 6.2 pmol ends) was incubated without added MgCl2 and with 1,020 ng of SbcB15 (18.6 pmol) for 5 min at 37°C (lane 2). A reaction mixture with SbcB15, parallel to that in lane 2, was briefly chilled on ice after 5 min at 37°C and then further incubated at 37°C for 15 s (lane 3). For a second reaction mixture with SbcB15 parallel to that in lane 2, the 15-s incubation at 37°C after brief chilling occurred in the presence of 680 ng of Xon+ (12.4 pmol) (lane 4). The DNA substrate after incubation with 680 ng of Xon+ only (12.4 pmol) for 15 s at 37°C without added MgCl2 is shown in lane 5. Lane 1, no protein; lanes M, duplex DNA marker fragments.
DISCUSSION
Evidence for a presynaptic role of ExoI and RecJ in RecBCD-dependent recombination.
ExoI and RecJ have been proposed to have overlapping postsynaptic roles in RecBCD-dependent recombination: the degradation of the displaced 3′ strand during branch migration by ExoI and the enlarging of gaps and/or the degradation of the 5′ strand remaining after transfer of its 3′ complement by RecJ (26, 50, 60, 73). In this way, recombination joints are stabilized by making strand transfer unidirectional. In accordance with the overlapping roles of these enzymes, the RecBCD-dependent recombination in transduction was rather slightly affected when only ExoI or RecJ was absent and was more pronouncedly decreased in the double-null mutant. However, conjugational recombination was more strongly decreased than transduction in single mutants and even more strongly in double mutants. In conjugation, the donor DNA strand is transferred with its 5′ end ahead and is converted into a duplex in the recipient cell (25). If the conversion to a duplex does not reach the tip of the transferred DNA, a 5′ ss overhang will remain. Similarly, when conjugation terminates (usually by breakage of the transferred strand), the priming for the complementary-strand synthesis may not occur at the very end of the transferred DNA, so that a 3′ overhang will remain. Since 5′ or 3′ overhangs on duplex DNAs of about 100 nucleotides or more strongly impede the interaction with RecBCD (58, 67), conjugationally transferred DNA would often not provide a substrate for RecBCD recombination unless its ends were blunted. It is proposed that ssDNases have a presynaptic role in conjugation: the blunting of DNA ends to make them a substrate for RecBCD to initiate recombination. This role is not required in transduction, where the transferred DNA already has blunt or nearly blunt ends due to the pacAB-encoded packase of P1 packaging DNA into phage proheads (39, 66). Consistent with the putative presynaptic role of ssDNases in addition to their postsynaptic role, the absence of ExoI decreased conjugation more strongly than transduction, the RecJ deficiency resulted in a similar although less pronounced effect, and the combination of both deficiencies decreased transduction 3-fold and conjugation 15-fold. The blunting of DNA fragments with overhangs by ssDNases including ExoI and RecJ in UV-irradiated cells was previously shown to improve the interaction with RecBCD (71). DNA blunting was recently also proposed as an explanation of the fact that in recD strains, recJ and Δxon mutations led to stronger decreases of conjugational recombination than of transduction (21). The RecBC (RecD−) enzyme (like RecBCD) binds with high affinity to blunt or nearly blunt DNA ends to initiate recombination (77). No contribution of SbcCD or ExoVII to the putative DNA blunting is apparent from our data. Rather, the sbcCD mutation increased conjugative recombination in the ExoI single mutants (Δxon, sbcB15, and xonA2) about 1.5-fold, which can be interpreted as indicating that the absence of SbcCD further improved the stability of 3′ overhangs, enhancing their chance of initiating recombination directly. xseA mutants have a hyperrecombination phenotype in recBCD+ and recD backgrounds, suggesting that ExoVII has an antirecombination function (13, 21, 23). Removal of 3′ exonuclease activity by the combined xseA and Δxon mutations increased recombination to a level higher than that in the wild type. This phenotype is in agreement with the recent finding that ExoI and ExoVII play roles in preventing frequent RecA-independent recombination events by removing displaced 3′ single strands and free ss fragments (23). The less strong phenotype of the xseA mutation in the xonA2 strain would be compatible with the residual ExoI activity provided by XonA2.
Reversed replication forks, e.g., those formed at sites of UV damage in DNA, would have ss overhangs on their duplex ends, which would need blunting to allow their processing by RecBCD (47, 48, 49, 51). An absence of blunting activities due to ssDNase mutations could therefore increase UV sensitivity. Besides their putative blunting role, the ssDNases may have other roles in the repair of UV damage, as suggested by the high UV sensitivity of multiple mutants (e.g., the recJ xseA Δxon and recJ xseA sbcB15 strains plus their sbcCD derivatives) whose recombination was hardly affected.
SbcCD and mismatch repair.
The cold sensitivities of our quadruple mutants with Δxon (BT497) or sbcB15 (BT498) are reminiscent of that of the quadruple null mutant VB31, which was constructed to study the contribution of four ssDNases to MMR and which differs from our quadruple mutants in having an exoX deficiency instead of sbcCD (11). Like VB31, BT497 and BT498 also displayed only a minor increase in the spontaneous mutation rate (72; Thoms and Wackernagel, unpublished), and their cold sensitivities were partially suppressed by the introduction of a mutS mutation, as reported for VB31 (11; Thoms and Wackernagel, unpublished). From our data, sbcCD must be added to the list of ssDNases involved in MMR and/or cell survival following attempted mismatch repair. The cold resistance of our quadruple mutant with xonA2 can be explained by the higher ExoI activity in a xonA2 mutant than in sbcB15 and Δxon strains.
Extended binding of SbcB15 to 3′ ends can explain the phenotypes of sbcB15 strains.
The identification of sbcB15 and xonA2 mutations and some characteristics of the mutant enzymes in vitro help to explain other phenotypic effects exerted by these alleles. In SbcB15, Ala183, located between His181 and Asp186 in exo motif III, is replaced by the bulkier Val. Asp186, along with two Asp residues and one Glu residue in exo motifs I and II, is necessary for the binding of two Mg2+ ions in the active center, and His181 is involved in the appropriate positioning of the DNA substrate for phosphodiester bond cleavage (7). Evidence that Val183 affects catalysis by interfering with cation binding and/or substrate positioning is seen in the low activity of SbcB15 under standard conditions and its even lower activity at decreasing concentrations of MgCl2 compared to that of Xon+. On the other hand, the amino acid exchange does not affect the positively charged large groove following the active center where the enzyme binds a stretch of about 12 nucleotides or the part of the polypeptide chain with which the enzyme completely encloses the substrate ssDNA. The latter two properties contribute to the processivity of ExoI (7) and are not expected to be influenced in SbcB15. Thus, SbcB15 can bind efficiently to the 3′ end of ssDNA, and bound SbcB15 can proceed to degrade DNA at a low rate, about 1/40 that of Xon+ (without added MgCl2 [compare Fig. 6A, lane 3, with Fig. 7, lane 5). By its DNA end binding, SbcB15 could protect 3′ DNA overhangs against other 3′-specific exonucleases and thereby would increase their chances of engaging in recombination. In a recBC strain, this recombination could occur along the RecF pathway, which requires the additional inactivation of SbcCD for full activity (14, 34, 38, 56, 75). The need for SbcCD inactivation can be explained by a competition of the SbcCD exonuclease for 3′ ends (15), by the endonuclease activity of SbcCD for ssDNA (19), and by the deprotecting action of SbcCD on protein-bound DNA ends (16). Extending previous models of recBC suppression, we propose that elimination of ExoI by Δxon or reduction of its activity by the xonA2 mutation along with sbcCD deficiency increases the persistence of 3′ overhangs long enough to allow a recombinational resetting of UV damage-stalled replication forks by the RecF pathway (in a recBC background) and thereby suppresses the UV sensitivity of recBC mutants. The full recombination proficiency of a recBC sbcB15 sbcCD strain during transduction and conjugation is proposed to require the extended overhang persistence provided through protection by SbcB15, because these recombination events need a longer time span than those required for overcoming stalled replication forks (5). In a recBCD+ background, the extended stability of 3′ overhangs due to sbcB15 and sbcCD mutations could allow direct recombination initiation by these ends, and ends with 5′ overhangs would be converted into RecBCD substrates by 5′ ssDNases. These steps could lead to the relatively high recombination frequency that was observed.
In studies on ss recombination intermediates in vivo, the sbcB15 mutation depressed Chi-specific recombination in the presence of all other ssDNases, while the Δxon mutation did not (60). It was assumed that SbcB15 prevented the action of other exonucleases on the DNA ends, which fits with our observation of end protection. The studies in fact supported the earlier report showing that SbcB15 inhibited end processing in Chi-dependent recombination when Chi was located in one DNA molecule opposite to a heterologous DNA segment in the other DNA molecule (53). In their analysis of processes at blocked replication forks, Bidnenko et al. (4) found that replication restart did not occur in a rep recBC sbcCD sbcB15 mutant (leading to inviability) but in a rep recBC sbcCD Δxon strain. This was unexpected because the RecF recombination process assumed to form a structure for replication restart from the reversed fork was expected to be more efficient in the rep recBC sbcCD sbcB15 strain than in its Δxon counterpart. To explain the inviability of the sbcB15 derivative, Bidnenko et al. (4) proposed that SbcB15 might remain bound on the 3′ end after RecF pathway-promoted fork formation and thereby would block replication in the rep mutant. Rep helicase can displace proteins from DNA (78) and therefore was assumed to be required for the restart of replication from forks formed by the RecF pathway employing SbcB15. Another study showed that in strains carrying the sbcB15 mutation alone or in combination with other rec gene mutations, the RuvABC-mediated cleavage of Holliday structures is necessary to repair replication forks blocked by DNA irradiation damage (79). It was proposed that at the duplex tail of a regressed replication fork, SbcB15 would bind to a 3′ overhang and would remain bound even after the duplex tail had been reset into a replication fork-like structure by the action of RuvAB or RecG helicase. The bound SbcB15 would then prevent the restart of DNA replication from the restored fork-like structure by blocking the 3′-OH end (79). Although we found that SbcB15 detached from the 3′ overhang when approaching the (5′) end of the duplex region (as Xon+ and XonA2 do), it is possible that SbcB15 would not detach if, during replication fork resetting, the 3′ strand with the bound SbcB15 hybridized to a complementary strand with no 5′ end in the vicinity.
The sbcB15 allele present in single, double, and triple ssDNase mutants generally provided a higher conjugational recombination frequency than Δxon and xonA2, although the much lower exonucleolytic activity of the SbcB15 enzyme than of Xon+ (56) (Table 4) is expected to hamper the putative presynaptic and postsynaptic roles in the RecBCD pathway, as do Δxon and xonA2. Apparently, another specific property of SbcB15 phenotypically makes up for the ssDNase deficiency. Recently it was demonstrated that sbcB15 is a stronger suppressor of recBC than Δxon or xonA2, leading to the conclusion that SbcB15 is favorable for the RecF pathway (80). We support this proposal by our data showing that SbcB15 not only is less active in degrading 3′ overhangs than Xon+ but also may further improve their stability by protecting them against other exonucleases. This protection possibly increases recombination initiation by these ends. If the recombination depends on the help of the RecFOR proteins, this would indicate that the RecF pathway is activated in recBCD+ cells by the sbcB15 mutation; in other words, the RecF pathway would take over in recBCD+ cells. Based on the phenotypes in recBC strains of Δxon and xonA2 on the one hand and of sbcB15 on the other hand, Zahradka et al. (80) proposed two types of RecF pathways. One type of pathway is activated by mutations eliminating all ExoI activities (such as Δxon and xonA2 mutations) and is rather strongly dependent on other RecF pathway genes (e.g., recF or recQ), while the other type occurs in the sbcB15 mutant, provides some protection of 3′ ends, and is less dependent on other RecF pathway genes. From the relatively high conjugative recombination frequency of sbcB15 mutants with a recBCD+ background, it is tempting to speculate that the presumed second type of RecF pathway is also active in the presence of RecBCD. While in the first pathway the extended 3′ ssDNA stability is the prerequisite for RecFOR-organized loading of RecA to produce a recombinogenic nucleoprotein filament, in the second pathway the bound SbcB15 protein is considered to stimulate RecA assembly on the single strand itself, thereby making recombination less dependent on RecFOR (80). Since conjugational recombination in recBCD+ strains with an sbcB15 and/or sbcCD mutation was reduced only 7 to 48% by a recF mutation (Table 3), the RecF pathway does not contribute strongly to the recombination occurring in these strains.
Residual ExoI activity in xonA2 strains is compatible with the phenotypic differences to Δxon strains.
The truncated C terminus of XonA2 is located at the surface of the protein and therefore presumably does not contribute to the active center, the DNA binding groove, or the polypeptide chain extending across the groove necessary to enclose the DNA (7). In accordance with an unaffected active center, the Mg2+ dependence of the enzyme resembled that of Xon+. Possibly XonA2 has decreased conformation stability, leading to lower activity and decreased intracellular stability (57). At a pH of 7 (which is much closer to the intracellular pH of E. coli [61] than the pH of 9.5 used in the standard assay) and at 30°C, the specific activity of XonA2 amounts to about 8% of that of Xon+ (Fig. 3). This residual activity could be the reason why xonA2 often provided higher UV survival and recombination than Δxon and why the quadruple mutant with xonA2 was cold resistant whereas quadruple mutants with Δxon or sbcB15 (about 2% of Xon+ activity under the conditions described above) were not. Apparently, the residual activity of XonA2 can partially perform the putative presynaptic function and the postsynaptic function of ExoI, since the recombination frequency for xonA2 strains was somewhat higher than that for Δxon strains (compare BT534 with BT450 or BT422 with BT460). The residual activity of XonA2 appears not to be helpful in situations when maximum 3′ ssDNA stability would provide a better chance for recombination (as in the strains lacking ExoVII activity). Further detailed characterization of the XonA2 and SbcB15 proteins is required. It is necessary to determine the levels of processivity of the enzymes and their substrate binding constants in order to better understand the phenotype of the mutants and why the xon+ allele is dominant over sbcB15 (33).
Acknowledgments
We are grateful to Susan M. Rosenberg, A. John Clark, Sidney R. Kushner, David R. Leach, and K. Brooks Low for bacterial strains.
Footnotes
Published ahead of print on 26 October 2007.
REFERENCES
- 1.Amundsen, S. K., A. F. Taylor, A. M. Chaudhury, and G. R. Smith. 1986. recD: the gene for an essential third subunit of exonuclease V. Proc. Natl. Acad. Sci. USA 835558-5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachmann, B. J. 1972. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev. 36525-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beese, L. S., and T. A. Steitz. 1991. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1025-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bidnenko, V., M. Seigneur, M. Penel-Colin, M.-F. Bouton, S. D. Ehrlich, and B. Michel. 1999. sbcB sbcC null mutations allow RecF-mediated repair of arrested replication forks in rep recBC mutants. Mol. Microbiol. 33846-857. [DOI] [PubMed] [Google Scholar]
- 5.Birge, E. A., and K. B. Low. 1974. Detection of transcribable recombination products following conjugation in rec+, recBC− and recC− strains of Escherichia coli K12. J. Mol. Biol. 83447-457. [DOI] [PubMed] [Google Scholar]
- 6.Brčić-Kostić, K., E. Salaj-Šmic, N. Maršić, S. Kajić, I. Stojiljković, and Ž. Trgovčević. 1991. Interaction of RecBCD enzyme with DNA damaged by gamma irradiation. Mol. Gen. Genet. 228136-142. [DOI] [PubMed] [Google Scholar]
- 7.Breyer, W. A., and B. W. Matthews. 2000. Structure of Escherichia coli exonuclease I suggests how processivity is achieved. Nat. Struct. Biol. 71125-1128. [DOI] [PubMed] [Google Scholar]
- 8.Brody, R. S. 1991. Nucleotide positions responsible for the processivity of the reaction of exonuclease I with oligodeoxyribonucleotides. Biochemistry 307072-7080. [DOI] [PubMed] [Google Scholar]
- 9.Brody, R. S., K. G. Doherty, and P. D. Zimmermann. 1986. Processivity and kinetics of the reaction of exonuclease I from Escherichia coli with polydeoxyribonucleotides. J. Biol. Chem. 2617136-7143. [PubMed] [Google Scholar]
- 10.Brutlag, D., and A. Kornberg. 1972. Enzymatic synthesis of deoxyribonucleic acid. 36. A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 247241-248. [PubMed] [Google Scholar]
- 11.Burdett, V., C. Baitinger, M. Viswanathan, S. T. Lovett, and P. Modrich. 2001. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc. Natl. Acad. Sci. USA 986765-6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chase, J. W., B. A. Rabin, J. B. Murphy, K. L. Stone, and K. R. Williams. 1986. Escherichia coli exonuclease VII. Cloning and sequencing of the gene encoding the large subunit (xseA). J. Biol. Chem. 26114929-14935. [PubMed] [Google Scholar]
- 13.Chase, J. W., and C. Richardson. 1977. Escherichia coli mutants deficient in exonuclease VII. J. Bacteriol. 129934-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark, A. J., and K. B. Low. 1988. Pathways and systems of homologous recombination in Escherichia coli, p. 155-215. In K. B. Low (ed.), The recombination of genetic material. Academic Press, San Diego, CA.
- 15.Connelly, J. C., E. S. de Leau, and D. R. Leach. 1999. DNA cleavage and degradation by the SbcCD protein complex from Escherichia coli. Nucleic Acids Res. 271039-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connelly, J. C., E. S. de Leau, and D. R. F. Leach. 2003. Nucleolytic processing of a protein-bound DNA end by the E. coli SbcCD (MR) complex. DNA Repair 2795-807. [DOI] [PubMed] [Google Scholar]
- 17.Connelly, J. C., E. S. de Leau, E. A. Okely, and D. R. Leach. 1997. Overexpression, purification, and characterization of the SbcCD protein from Escherichia coli. J. Biol. Chem. 27219819-19826. [DOI] [PubMed] [Google Scholar]
- 18.Connelly, J. C., L. A. Kirkham, and D. R. Leach. 1998. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. USA 957969-7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connelly, J. C., and D. R. F. Leach. 1996. The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells 1285-291. [DOI] [PubMed] [Google Scholar]
- 20.Dabert, P., S. D. Ehrlich, and A. Gruss. 1992. χ sequence protects against RecBCD degradation of DNA in vivo. Proc. Natl. Acad. Sci. USA 8912073-12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dermić, D. 2006. Functions of multiple exonucleases are essential for cell viability, DNA repair and homologous recombination in recD mutants of Escherichia coli. Genetics 1722057-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Vries, J., and W. Wackernagel. 1992. Recombination and UV resistance of Escherichia coli with the cloned recA and recBCD genes of Serratia marcescens and Proteus mirabilis: evidence for an advantage of intraspecies combination of P. mirabilis RecA and RecBCD enzyme. J. Gen. Microbiol. 13831-38. [DOI] [PubMed] [Google Scholar]
- 23.Dutra, B. E., V. A. Sutera, Jr., and S. T. Lovett. 2007. RecA-independent recombination is efficient but limited by exonucleases. Proc. Natl. Acad. Sci. USA 104216-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feschenko, V. V., L. A. Rajman, and S. T. Lovett. 2003. Stabilization of perfect and imperfect tandem repeats by single-strand DNA exonucleases. Proc. Natl. Acad. Sci. USA 1001134-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Firth, N., K. Ippen-Ihler, and R. Skurray. 1996. Structure and function of the F factor and mechanism of conjugation, p. 2377-2401. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 26.Friedman-Ohana, R., and A. Cohen. 1998. Heteroduplex joint formation in Escherichia coli recombination is initiated by pairing of a 3′-ending strand. Proc. Natl. Acad. Sci. USA 956909-6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson, F. P., D. R. Leach, and R. G. Lloyd. 1992. Identification of sbcD mutations as cosuppressors of recBC that allow propagation of DNA palindromes in Escherichia coli K-12. J. Bacteriol. 1741222-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris, S., K. J. Ross, M. J. Lombardo, and S. M. Rosenberg. 1998. Mismatch repair in Escherichia coli cells lacking single-strand exonucleases ExoI, ExoVII, and RecJ. J. Bacteriol. 180989-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howard-Flanders, P., and L. Theriot. 1966. Mutants of E. coli defective in DNA repair and genetic recombination. Genetics 531137-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Köppen, A., S. Krobitsch, B. Thoms, and W. Wackernagel. 1995. Interaction with the recombination hot spot χ in vivo converts the RecBCD enzyme of Escherichia coli into a χ-independent recombinase by inactivation of the RecD subunit. Proc. Natl. Acad. Sci. USA 926249-6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowalczykowski, S. C., D. A. Dixon, A. K. Eggleston, S. D. Lauder, and W. M. Rehrauer. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58401-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kushner, S. R., H. Nagaishi, and A. J. Clark. 1972. Indirect suppression of recB and recC mutations by exonuclease I deficiency. Proc. Natl. Acad. Sci. USA 691366-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kushner, S. R., H. Nagaishi, A. Templin, and A. J. Clark. 1971. Genetic recombination in Escherichia coli: the role of exonuclease I. Proc. Natl. Acad. Sci. USA 68824-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuzminov, A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63751-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehman, I. R., and R. Nussbaum. 1964. The deoxyribonuclease of Escherichia coli. V. On the specificity of exonuclease I (phosphodiesterase). J. Biol. Chem. 2392628-2636. [PubMed] [Google Scholar]
- 36.Lloyd, R. G., and C. Buckman. 1985. Identification and genetic analysis of sbcC mutations in commonly used recBC sbcB strains of Escherichia coli K-12. J. Bacteriol. 164836-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lloyd, R. G., and K. B. Low. 1996. Homologous recombination, p. 2236-2255. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 38.Lloyd, R. G., and A. Thomas. 1984. A molecular model for conjugal recombination in Escherichia coli K12. Mol. Gen. Genet. 197328-336. [DOI] [PubMed] [Google Scholar]
- 39.Lobocka, M. B., D. J. Rose, G. Plunkett III, M. Rusin, A. Samojedny, H. Lehnherr, M. B. Yarmolinsky, and F. R. Blattner. 2004. Genome of bacteriophage P1. J. Bacteriol. 1867032-7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lombardo, M.-J., I. Aponyi, M. P. Ray, M. Sandigursky, W. A. Franklin, and S. M. Rosenberg. 2003. xni-deficient Escherichia coli are proficient for recombination and multiple pathway repair. DNA Repair 21175-1183. [DOI] [PubMed] [Google Scholar]
- 41.Lovett, S. T., and A. J. Clark. 1984. Genetic analysis of the recJ gene of Escherichia coli K-12. J. Bacteriol. 157190-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lovett, S. T., and R. D. Kolodner. 1989. Identification and purification of a single-stranded DNA-specific exonuclease encoded by the recJ gene of Escherichia coli. Proc. Natl. Acad. Sci. USA 862627-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Low, B. 1973. Restoration by the rac locus of recombinant forming ability in recB− and recC− merozygotes of Escherichia coli K-12. Mol. Gen. Genet. 122119-130. [DOI] [PubMed] [Google Scholar]
- 44.Low, K. B. 1996. Hfr strains of Escherichia coli K-12, p. 2402-2405. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 45.Maloy, S. R., and W. D. Nunn. 1981. Selection for loss of tetracycline resistance by Escherichia coli. J. Bacteriol. 1451110-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masters, M. 1996. Generalized transduction, p. 2421-2441. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 47.McGlynn, P., and R. G. Lloyd. 2002. Recombination repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 3859-870. [DOI] [PubMed] [Google Scholar]
- 48.Michel, B., M.-J. Flores, E. Viguera, G. Grompone, M. Seigneur, and V. Bidnenko. 2001. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. USA 988181-8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel, B., G. Grompone, M.-J. Flores, and V. Bidnenko. 2004. Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. USA 10112783-12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miesel, L., and J. R. Roth. 1996. Evidence that SbcB and RecF pathway functions contribute to RecBCD-dependent transductional recombination. J. Bacteriol. 1783146-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miranda, A., and A. Kuzminov. 2003. Chromosomal lesion suppression and removal in Escherichia coli via linear DNA degradation. Genetics 1631255-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Modrich, P. 1991. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 25229-253. [DOI] [PubMed] [Google Scholar]
- 53.Myers, R. S., M. M. Stahl, and F. W. Stahl. 1995. χ recombination activity in phage λ decays as a function of genetic distance. Genetics 141805-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama, H., K. Nakayama, R. Nakayama, N. Irino, Y. Nakayama, and P. C. Hanawalt. 1984. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol. Gen. Genet. 195474-480. [DOI] [PubMed] [Google Scholar]
- 55.Oliver, D. B., and E. B. Goldberg. 1977. Protection of parental T4 DNA from a restriction exonuclease by the product of gene 2. J. Mol. Biol. 116877-881. [DOI] [PubMed] [Google Scholar]
- 56.Phillips, G. J., D. C. Prasher, and S. R. Kushner. 1988. Physical and biochemical characterization of cloned sbcB and xonA mutations from Escherichia coli K-12. J. Bacteriol. 1702089-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prasher, D. C., L. Conarro, and S. R. Kushner. 1983. Amplification and purification of exonuclease I from Escherichia coli K12. J. Biol. Chem. 2586340-6343. [PubMed] [Google Scholar]
- 58.Prell, A., and W. Wackernagel. 1980. Degradation of linear and circular DNA with gaps by the recBC enzyme of Escherichia coli. Eur. J. Biochem. 105109-116. [DOI] [PubMed] [Google Scholar]
- 58a.Qiagen. 1999. The QIA expressionist. Qiagen, Hilden, Germany.
- 59.Ray, R. K., R. Reuben, I. Molineux, and M. Gefter. 1974. The purification of exonuclease I from Escherichia coli by affinity chromatography. J. Biol. Chem. 2495379-5381. [PubMed] [Google Scholar]
- 60.Razavy, H., S. K. Szigety, and S. M. Rosenberg. 1996. Evidence for both 3′ and 5′ single-strand DNA ends in intermediates in Chi-stimulated recombination in vivo. Genetics 142333-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richard, H., and J. W. Foster. 2004. Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol. 1866032-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rinken, R., B. Thoms, and W. Wackernagel. 1992. Evidence that recBC-dependent degradation of duplex DNA in Escherichia coli recD mutants involves DNA unwinding. J. Bacteriol. 1745424-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 64.Simmon, V. F., and S. Lederberg. 1972. Degradation of bacteriophage lambda deoxyribonucleic acid after restriction by Escherichia coli K-12. J. Bacteriol. 112161-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith, G. R. 1991. Conjugational recombination in E. coli: myths and mechanisms. Cell 6419-27. [DOI] [PubMed] [Google Scholar]
- 66.Sternberg, N., and J. Coulby. 1987. Recognition and cleavage of the bacteriophage P1 packaging site (pac). II. Functional limits of pac and location of pac cleavage termini. J. Mol. Biol. 194469-479. [DOI] [PubMed] [Google Scholar]
- 67.Taylor, A. F., and G. R. Smith. 1985. Substrate specificity of the DNA unwinding activity of the RecBC enzyme of Escherichia coli. J. Mol. Biol. 185431-443. [DOI] [PubMed] [Google Scholar]
- 68.Templin, A., S. R. Kushner, and A. J. Clark. 1972. Genetic analysis of mutations indirectly suppressing recB and recC mutations. Genetics 72105-115. [PMC free article] [PubMed] [Google Scholar]
- 69.Thomas, K. R., and B. M. Olivera. 1978. Processivity of DNA exonucleases. J. Biol. Chem. 253424-429. [PubMed] [Google Scholar]
- 70.Thoms, B., and W. Wackernagel. 1987. Regulatory role of recF in the SOS response of Escherichia coli: impaired induction of SOS genes by UV irradiation and nalidixic acid in a recF mutant. J. Bacteriol. 1691731-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thoms, B., and W. Wackernagel. 1998. Interaction of RecBCD enzyme with DNA at double-strand breaks produced in UV-irradiated Escherichia coli: requirement for DNA end processing. J. Bacteriol. 1805639-5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Viswanathan, M., V. Burdett, C. Baitinger, P. Modrich, and S. T. Lovett. 2001. Redundant exonuclease involvement in Escherichia coli methyl-directed mismatch repair. J. Biol. Chem. 27631053-31058. [DOI] [PubMed] [Google Scholar]
- 73.Viswanathan, M., and S. T. Lovett. 1998. Single-strand DNA-specific exonucleases in Escherichia coli: roles in repair and mutation avoidance. Genetics 1497-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Viswanathan, M., and S. T. Lovett. 1999. Exonuclease X of Escherichia coli. A novel 3′-5′ DNase and DnaQ superfamily member involved in DNA repair. J. Biol. Chem. 27430094-30100. [DOI] [PubMed] [Google Scholar]
- 75.Wang, T.-C. V., and K. C. Smith. 1985. Mechanism of sbcB-suppression of the recBC-deficiency in postreplication repair in UV-irradiated Escherichia coli K-12. Mol. Gen. Genet. 201186-191. [DOI] [PubMed] [Google Scholar]
- 76.Willetts, N. S., and D. W. Mount. 1969. Genetic analysis of recombination-deficient mutants of Escherichia coli K-12 carrying rec mutations cotransducible with thyA. J. Bacteriol. 100923-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wong, C. J., A. L. Lucius, and T. M. Lohman. 2005. Energetics of DNA end binding by E. coli RecBC and RecBCD helicases indicate loop formation in the 3′-single-stranded DNA tail. J. Mol. Biol. 352765-782. [DOI] [PubMed] [Google Scholar]
- 78.Yancey-Wrona, J. E., and S. W. Matson. 1992. Bound Lac repressor protein differentially inhibits the unwinding reactions catalyzed by DNA helicases. Nucleic Acids Res. 206713-6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zahradka, D., K. Zahradka, M. Petranović, D. Dermić, and K. Brčić-Kostić. 2002. The RuvABC resolvase is indispensable for recombinational repair in sbcB15 mutants of Escherichia coli. J. Bacteriol. 1844141-4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zahradka, K., S. Šimić, M. Buljubašić, M. Petranović, D. Dermić, and D. Zahradka. 2006. sbcB15 and ΔsbcB mutations activate two types of RecF recombination pathways in Escherichia coli. J. Bacteriol. 1887562-7571. [DOI] [PMC free article] [PubMed] [Google Scholar]