

Abstract
Microbial arsenate respiration contributes to the mobilization of arsenic from the solid to the soluble phase in various locales worldwide. To begin to predict the extent to which As(V) respiration impacts arsenic geochemical cycling, we characterized the expression and activity of the Shewanella sp. strain ANA-3 arsenate respiratory reductase (ARR), the key enzyme involved in this metabolism. ARR is expressed at the beginning of the exponential phase and persists throughout the stationary phase, at which point it is released from the cell. In intact cells, the enzyme localizes to the periplasm. To purify ARR, a heterologous expression system was developed in Escherichia coli. ARR requires anaerobic conditions and molybdenum for activity. ARR is a heterodimer of ∼131 kDa, composed of one ArrA subunit (∼95 kDa) and one ArrB subunit (∼27 kDa). For ARR to be functional, the two subunits must be expressed together. Elemental analysis of pure protein indicates that one Mo atom, four S atoms associated with a bis-molybdopterin guanine dinucleotide cofactor, and four to five [4Fe-4S] are present per ARR. ARR has an apparent melting temperature of 41°C, a Km of 5 μM, and a Vmax of 11,111 μmol of As(V) reduced min−1 mg of protein−1 and shows no activity in the presence of alternative electron acceptors such as antimonite, nitrate, selenate, and sulfate. The development of a heterologous overexpression system for ARR will facilitate future structural and/or functional studies of this protein family.
Arsenic contamination of alluvial aquifers that supply drinking water to Bangladesh and West Bengal exposes tens of millions of people to chronic arsenic poisoning annually (13, 19). Field and microcosm studies of environments where arsenic contamination occurs determined that microbial activity is responsible for arsenic partitioning from the solid sediment to the aqueous phase (5). In particular, microbial respiratory reduction of arsenate, As(V), to arsenite, As(III), is thought to prevent the readsorption of arsenic onto sediment surfaces once it is transported away from iron-rich environments (2, 14). This unique metabolism is dependent on arsenate respiratory reductase (ARR) (9). To understand the contribution of As(V) respiration to arsenic geochemical cycling and mobilization, the factors that control ARR expression and activity must be determined. The first biochemical studies of ARR were performed with native ARR purified from Chrysiogenes arsenatis (8) and Bacillus selenitireducens (1). However, because neither of these strains is readily genetically tractable, detailed enzymological studies of the protein were challenging to perform.
To circumvent this limitation, an arsenate-respiring strain, Shewanella sp. strain ANA-3, was isolated that could also respire on oxygen and grow on agar plates (16). These capabilities allowed for easier laboratory manipulation of the strain and the application of standard genetic techniques. Using ANA-3, As(V) respiration was shown to be independent of the well-characterized ars detoxification system that includes a small cytoplasmic arsenate reductase, ArsC (16). Two genes, arrA and arrB, were cloned and shown to be required for As(V) respiration (17). They were predicted to encode a bis-molybdopterin guanine dinucleotide-containing member of the dimethyl sulfoxide (DMSO) reductase family that directly interacts with As(V). Using these gene sequences, it was determined that arrA is well conserved among almost all of the isolated bacterial As(V) respirers, and a set of degenerate PCR primers was designed and applied to show that arrA was expressed in Haiwee Reservoir, a site of arsenic contamination (9). In microcosm studies using a sediment analog that consisted of hydrous ferric oxide sorbed with As(V), the arr system was required for the reduction of As(V), while the ars system was not (9). In addition, studies of arrAB regulation revealed that these genes are upregulated upon the shift to anaerobic conditions and in the presence of low micromolar concentrations of As(V) or nanomolar concentrations of As(III) (18).
Because the arr system is required for As(V) reduction under environmentally relevant conditions, is highly conserved among diverse bacterial species, and is expressed in arsenic-contaminated field sites, understanding its posttranscriptional activity is necessary to be able to quantify and predict the contribution of bacterial arsenate respiration to the arsenic geochemical cycle. As a step toward this goal, we present here the characterization of ARR expression and activity in ANA-3 and in a heterologous overexpression system. The latter will permit future comparative studies between ARR proteins from different species.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial strains and plasmids used in the present study are presented in Table 1.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Genotype or markers and characteristics and uses | Source or reference |
|---|---|---|
| Strains | ||
| Shewanella sp. strain ANA-3 | As(V)-respiring gram-negative bacterium isolated from a wooden pier in Eel Pond, Woods Hole, MA | 16 |
| E. coli | ||
| UQ950 | DH5α λ(pir) lysogen of DH5α used for cloning | D. Lies, Caltech |
| C43 | Mutant of overexpression strain BL21 optimized for expression of membrane and toxic proteins | 10 |
| Plasmids | ||
| pET32H | Modified version of pET32 (Novagen) in which sequences encoding the S tag and enterokinase cleavage site were removed | 22 |
| pET32H Xa-2 | Modified version of pET32H in which the sequence encoding the thrombin cleavage site was replaced with a factor Xa cleavage site and the His6 tag was replaced with a His9 tag | This study |
| pET32HP | Modified version of pET32H in which the sequence encoding the thrombin cleavage site was replaced with a Prescission protease cleavage site | L. Dietrich, Caltech |
Activity assays.
Respiratory As(V) reduction activity was quantified with a colorimetric assay. A reaction mix containing 120 μM methyl viologen (MV), 90 μM dithionite, 50 mM morpholinepropanesulfonic acid buffer (pH 7), and 0.15 M NaCl was placed into a plastic cuvette in an anaerobic chamber. The solution was allowed to equilibrate for several seconds, and then enzyme samples and electron acceptors were added. The cuvettes were stoppered before they were removed from the anaerobic chamber, and MV oxidation was measured by monitoring loss of blue color at 600 nm using a DU7400 or a DU800 spectrophotometer (both from Beckman Coulter). Electron acceptors used in this assay included As(V), As(III), selenate, antimonate, nitrate, and DMSO at 50 mM each. Malate dehydrogenase activity was measured by reacting a 5-μl sample with 1 ml of 0.1 M potassium phosphate (pH 7.5), 1 μl of oxaloacetate, and 10 μl of 27 mM NADH in a cuvette. NADH oxidation was monitored by measuring the optical density at 340 nm for 10 min (8). Alkaline phosphatase activity was measured according to a protocol adapted from Sigma-Aldrich. A 90-μl sample was mixed with 10 μl of 4% p-nitrophenyl phosphate disodium salt (Sigma) in a microtiter plate. The appearance of p-nitrophenyl was determined by measuring the optical density at 405 nm over 3 h using a Synergy HT Multi-Detection microplate reader (Bio-Tek).
Protein expression dynamics.
To measure ARR expression in bacterial cultures over various phases of growth, ANA-3 cultures were grown anaerobically at 30°C in medium containing LB Miller broth (25 g/liter), 10 mM lactate, and 10 mM As(V) at pH 7. At each time point, 50 μl of sample was withdrawn from the cultures and used in activity assays in the presence or absence of As(V) to determine As(V)-dependent reduction activity. The optical density of the cultures was measured by placing 1 ml of culture into a cuvette and measuring the optical density using a DU7400 spectrophotometer reading at 600 nm.
Filtrate analysis.
Samples from early- and late-phase cultures of ANA-3 growing in As(V) medium were withdrawn and filtered through a nylon microcentrifuge filter with a 0.2-μm pore size (Costar Spin-X column) by centrifugation at ∼13,000 × g for 1 min. The activity was measured simultaneously in samples that had and had not been filtered. Heat denaturation was carried out by heating the samples to 95°C for 10 min.
Localization.
Initial fractionation separated soluble proteins from membrane-bound proteins in 50 mM Tris buffer (pH 8), 50 mM PIPES buffer (pH 7), and 50 mM PIPES buffer plus 0.3 M NaCl (pH 7) to account for possible differences in protein interaction due to buffer choice or salt concentration. Cell cultures were homogenized using a Mini-Beadbeater 8-Cell Disrupter (Biospec Products, Inc.) and 0.1-mm zirconia/silica beads for 60 s to release the cytoplasmic contents. Additional cycles in the Beadbeater did not result in increasing concentrations of protein as measured by a Bio-Rad protein assay. Unbroken cells were removed by centrifugation at 8,000 × g. The activity was measured in the remaining fraction before and after ultracentrifugation, as well as in resuspended membrane fractions.
To separate the periplasmic contents from the cytoplasmic contents, cells were centrifuged and washed once in buffer containing 50 mM Tris (pH 8) and resuspended in 50 mM Tris with 20% (wt/vol) sucrose. EDTA was added to a final concentration of 500 μM, and lysozyme was added to a concentration of 0.3 mg/ml. The mixture was allowed to sit on ice for 30 min, which corresponded to the time required for the formation of spheroplasts, which were visualized under a light microscope. The sample was centrifuged at 10,000 × g for 10 min to separate the supernatant containing the periplasmic contents from the pellet containing spheroplasts. The spheroplasts were homogenized by using the Beadbeater. Both the periplasmic fraction and the cytoplasmic fraction were ultracentrifuged to remove contaminating membrane components.
Construction of overexpression vectors.
Initial attempts to overexpress and purify ARR with a C-terminal His6 tag resulted in low-affinity binding to TALON Superflow metal affinity resin (Clontech). As an alternative, a truncated version of ArrA was created, in which the first 41 amino acids at the N terminus, corresponding to a predicted TAT signal cleavage domain, were deleted to prevent the loss of an affinity tag at that location. Truncated arrA was amplified by using the primers NcoarrA5-2 (5′-CAT GCC ATG GGT GCT GAA TTA CCC GCG CCT CTA-3′) and XhoarrA3-2 (5′-CCG CTC GAG TCA CAC TTT CTC AAC ACG AAC-3′), and full-length arrB was amplified by using the primers NcoarrB5 (5′-CCA TGG GTA TGA GAT TAG GAA TGG TGA TT-3′) and XhoarrB3 (5′-CCG CTC GAG TTA ATA AGC GGT TTT AAC ACC-3′). arrA and arrB were amplified together by using the primers NcoarrA5-2 and XhoarrB3. The PCR protocol was 1 cycle at 95°C for 5 min, followed by 33 cycles at 95°C for 35 s, 58°C for 40 s, and 72°C for 2 min, and finishing with 1 cycle at 72°C for 10 min. The Fail Safe PCR system using PreMix D (Epicenter) was used in all PCRs. Products were digested with NcoI and XhoI (New England Biolabs) for vector insertion.
Two derivatives of the pET32H overexpression vector (22) were used in the present study. pET32H Xa-2 encodes a thioredoxin fused to a His9 tag upstream of the multiple cloning site. A Factor Xa protease cleavage site is located between the His9 and the N terminus of the protein of interest; however, no cleavage was performed using this construct. The presence of the His9 tag resulted in stronger binding of the fusion protein to cobalt resin compared to a His6 tag. This allowed washes with higher imidazole concentrations and rapid partial purification of ARR, ArrA, and ArrB for the activity studies presented in Fig. 6. For the final purification of ARR used in kinetic studies, pET32HP was created in which the thrombin protease cleavage site in pET32 was replaced by a Prescission Protease (GE Healthcare Life Sciences) cleavage site. This allowed for more efficient cleavage of the affinity tag from the protein at 4°C, resulting in higher concentrations of pure enzyme.
FIG. 6.

Activity of individual subunits. The activities in crude lysates from C43 cells expressing ARR heterodimer (A), ArrA alone (B), ArrB alone (C), or ArrA mixed with ArrB after independent induction of each subunit (D) were determined. Columns and error bars represent averages and standard deviations of triplicate samples.
All constructs were sequenced to confirm that no mutations were introduced during PCR or other genetic manipulations.
Heterologous expression of ARR.
Overexpression of ARR was based on a previous protocol for the expression of sulfite oxidase (21). Escherichia coli strain C43 cells were initially grown overnight aerobically in Miller LB at 37°C. To induce, cultures were diluted 1 to 25 in fresh aerobic Miller LB supplemented with 20 mM lactate and 1 mM sodium molybdate at 30°C and grown with shaking until they reached an optical density of 1.0 at 600 nm in a standard 1-cm cuvette. The culture was then diluted 1 to 25 in anaerobic medium containing LB, 20 mM lactate, 1 mM sodium molybdate, 40 μM IPTG (isopropyl-β-d-thiogalactopyranoside), and 14 mM DMSO as a terminal electron acceptor. These cultures were grown at room temperature for 18 h, during which time the enzyme was expressed. Cells were harvested by centrifugation at 10,000 × g for 15 min at 4°C, and the resulting pellet was frozen at −80°C until further use.
Purification.
E. coli strain C43 cells from 8 liters of medium were resuspended in 30 ml of 50 mM Tris-0.3 M NaCl (pH 7.0) and amended with two tablets of protease inhibitor cocktail (Sigma). To lyse the cells, 1.26 g of CelLytic powder (Sigma) was added to the solution, and the mixture was passed through a French press once. The crude lysate was clarified by centrifugation at 15,000 × g for 10 min. The resulting supernatant was added to 2 ml of washed Cobalt resin slurry and incubated on ice with gentle rocking for 2 h. Resin was collected by centrifugation at 500 × g for 5 min and washed two times with 20 ml of buffer containing 5 mM imidazole followed by two washes with 10 mM imidazole. The sample was then eluted in 3 ml of buffer containing 200 mM imidazole. Then, 80 U of Prescission protease was added to the sample and allowed to cleave off the thioredoxin and His tag for 2 h at 4°C. The sample was then incubated with 1 ml of glutathione-Sepharose for 30 min, desalted, and incubated with 2 ml of cobalt resin for 1 h. A sample was collected by using a gravity flow column and concentrated in a Microcon centrifugal filter device with a molecular mass cutoff of 10 kDa by centrifugation at 4°C.
Size exclusion chromatography.
Size exclusion chromatography was performed on an AKTA FPLC using a 16/60 XK column prepacked with Superdex 200 Prep Grade resin. The mobile phase contained 50 mM Tris with 0.15 M NaCl (pH 7.0) and was run at a flow rate of 0.5 ml/min. The protein concentration was measured using UV absorbance at 280 nm. Elution volumes were plotted against the log of the molecular weight for the standards, and the best fit line was found. From this line, an equation was derived and used to estimate molecular mass based on elution volume.
Protein gel electrophoresis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done using a precast 12% denaturing gel (Bio-Rad). Unstained broad-range standards (Bio-Rad) were used. Gel images were acquired with a Chemidoc XRS gel imager (Bio-Rad) with epi illumination.
Elemental analysis.
Metal analysis was performed at the elemental analysis and speciation facility at the UCLA Molecular Instrumentation Center using an Agilent 7500ce inductively coupled plasma-mass spectrometer (ICP-MS). The ICP-MS is equipped with an Octapole reaction system for plasma and matrix based interference removal. The protein solution was diluted to 2 ml with 2% HNO3 (Optima grade nitric acid diluted with 18 MΩ deionized water). A 100-ppm multi-element standard (CPI International) was used to generate 1-, 10-, 100-, and 200-ppb standard solutions. The ICP-MS analysis parameters were as follows: forward power, 1,500 W; carrier gas, 1.05 liters/min; nebulizer pump, 0.12 rps; sample depth, 7 mm; xenon flow, 0.12 ml/min; and hydrogen flow, 4 ml/min. Three readings were taken per mass, and the average is reported.
CD spectrometry and thermal denaturation.
All circular dichroism (CD) data were collected on an Aviv 62DS spectrometer with a 1-mm path length cell. The protein sample was 0.6 μM in 50 mM Tris-HCl (pH 7.0)-300 mM NaCl. Far-UV CD spectra were collected at 25°C, with a data averaging time of 5 s. The data were collected every 1 nm from 200 to 250 nm. Thermal denaturation was followed by monitoring CD ellipticity at 222 nm from 1 to 99°C. The data were collected every 1°C, with an equilibration time of 90 s and a data averaging time of 30 s. The data were normalized to a fraction folded using the following equation:
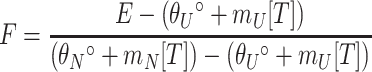 |
where F is the fraction of protein folded at a given temperature (T), E is the ellipticity at that temperature, 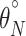 and
and 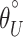 are the y intercepts of the pre- and posttransition baselines, respectively, and mN and mU are the slopes of the pre- and posttransition baselines.
are the y intercepts of the pre- and posttransition baselines, respectively, and mN and mU are the slopes of the pre- and posttransition baselines.
The apparent midpoint of the thermal denaturation (Tm) is defined as the temperature at which 50% of the protein is folded. It should be noted, however, that the protein irreversibly precipitated during denaturation, and the reported apparent Tm should only be taken as an estimate of the thermostability of the protein.
RESULTS
ARR expression dynamics.
To characterize the expression dynamics of ARR over various phases of growth, As(V)-dependent MV oxidation by ANA-3 cultures was monitored (Fig. 1). Activity was first detected by 5.75 h. This coincided with the exponential phase of growth. Activity increased past 8 h, at which time the optical density of the culture began to decrease dramatically from 0.25 to 0.03 at 13.5 h. At 12 h, the activity began to taper, although it persisted at significant levels throughout the 24-h experiment.
FIG. 1.

ARR expression dynamics. The ARR activities (•) and culture optical densities (▪) over different growth phases for ANA-3 respiring on As(V) were determined. The shaded area indicates the period at which arrA mRNA transcripts are expressed (based on the findings of Saltikov et al. [16]). The arrows indicate time points analyzed for the release of ARR (see Fig. 2). One unit (U) = 1 μmol of As(V) reduced min−1. The data are representative of duplicate cultures.
Extracellular release of ARR.
The decrease in optical density in late-phase cultures was not observed when cells were growing aerobically or in the presence of other electron acceptors. To determine the fate of cells after this decrease in optical density, samples from late-phase As(V)-respiring cultures were stained with DAPI (4′,6′-diamidino-2-phenylindole) and visualized by using fluorescence microscopy. While a small population of normal-sized cells were seen (∼5%), the majority of cells appeared to be reduced in size to ∼1 μm in length (Fig. 2A).
FIG. 2.

Cell dwarfing and release of ARR in late-phase cultures. (A) Light microscope image of late-phase As(V)-respiring cultures. The gray arrow indicates a typical-sized cell. The white arrow indicates a dwarf cell. (B) At 7 and 12.5 h, samples were collected and analyzed for ARR activity with or without filtration. Bars: ▪, total activity; ░⃞, filtrate activity. (C) Activity in 12.5-h filtrate without heat denaturation (▪) and with heat denaturation (░⃞). Columns and error bars represent the averages and standard deviations of triplicate samples.
To determine whether the disparity between activity and the optical density was due simply to the dwarfing of cells or whether the enzyme was released into the medium, samples were taken during exponential growth (7 h) and after the decrease in optical density (12.5 h) and assayed before and after passage through a 0.2-μm-pore-size filter. At 7 h, only 20.8% of the total activity was retained after filtration. At 12.5 h, 81% of the activity was retained (Fig. 2B). Filtrates were observed under a microscope to confirm that no cells were able to pass through the filter. Incubation of filtrates at 95°C for 10 min resulted in an 86% loss of the activity (Fig. 2C). This indicates that ARR was released from the cells as they aged.
Localization.
ARR localization was determined by measuring native activity within intact ANA-3 cells growing anaerobically in the absence of As(V) (Table 2). Soluble and membrane fractions were analyzed first. In all buffers and salt concentrations tested, 100% of the arsenate reductase activity was determined to be soluble. Washes were also assayed for activity to determine whether the reductase was loosely bound to the outside of the cell. Under all conditions, no activity was found in the outer fractions. We concluded that the reductase was soluble and present on the inside of the cell.
TABLE 2.
Localization of ARR in ANA-3
| Fractionation and enzyme | Fraction | Total activity (%) |
|---|---|---|
| Fractionation 1 | ||
| Arsenate reductase | Outer membrane associated | 0 |
| Soluble | 100 | |
| Membrane | 0 | |
| Fractionation 2 | ||
| Arsenate reductase | Periplasm | 90 |
| Cytoplasm | 10 | |
| Alkaline phosphatase | Periplasm | 73 |
| Cytoplasm | 27 | |
| Malate dehydrogenase | Periplasm | 23 |
| Cytoplasm | 77 |
In a second fractionation, the periplasmic contents were separated from the cytoplasmic fractions by incubating anaerobic cells in EDTA, lysozyme, and sucrose on ice until spheroplasts formed. Samples were centrifuged to separate the periplasmic contents from the spheroplasts. The spheroplasts were then homogenized to release the cytoplasmic contents, and both the periplasm and the cytoplasmic fractions were ultracentrifuged to remove contamination from membrane components. Malate dehydrogenase served as a cytoplasmic marker, and alkaline phosphatase served as a marker for the periplasm. A total of 77% of the malate dehydrogenase activity was found in the cytoplasmic fraction, and 73% of the alkaline phosphatase activity was found in the periplasmic fraction. Under these conditions, 90% of the arsenate reductase activity was found in the periplasmic fraction compared to the cytoplasmic fraction.
Heterologous expression.
Members of the DMSO reductase family all possess a molybdopterin cofactor assembled by the anaerobically expressed mod operon, involved in molybdenum uptake, and the moa operon, involved in molybdopterin biosynthesis. At the center of this cofactor is either a Mo or W atom that serves as the electron donor or acceptor in the catalyzed redox reaction. To characterize the conditions required for the assembly of active arsenate reductase and to develop an overexpression method for future enzymological studies of ARR, arrA and arrB were cloned into an overexpression vector, introduced into C43 E. coli cells, and the cells were induced under different conditions (Fig. 3). Under all of the conditions tested, ARR was expressed at roughly similar levels relative to the background proteins. Cultures grown in the presence of oxygen and molybdate showed an activity of 55 ± 14 μmol of As(V) reduced min−1 mg of protein−1. This rate increased to an activity of 152 ± 52 μmol of As(V) reduced min−1 mg of protein−1 when cells were induced under anaerobic conditions in the presence of molybdate. No activity was found in either condition when tungstate was added, nor was any activity detected in the absence of supplemental metal.
FIG. 3.

Expression of ARR under various conditions. The activity in crude lysates was measured after cells were induced under anaerobic conditions with 1 mM sodium molybdate (A), anaerobic conditions with 1 mM sodium tungstate (B), anaerobic conditions with no metal supplement (C), and aerobic conditions with sodium molybdate (D). ArrA was visualized by staining lysates run on an SDS denaturing gel. Columns and error bars represent averages and standard deviations of triplicate samples.
Purification of ArrAB, ArrA, and ArrB.
Overexpressed ARR was purified by using affinity chromatography (Fig. 4). Two subunits were present when the purified sample was run on an SDS-PAGE gel, one with a molecular mass of 95 kDa and one with a molecular mass of 27 kDa. Mass spectrometry showed that each of the visible bands only consisted of a single protein and confirmed that the identity of these subunits were ArrA and ArrB from Shewanella sp. strain ANA-3, respectively. Seventeen fragments of ArrA were identified, which covered 32% of the protein sequence. Five fragments of ArrB were identified, which covered 16% of the protein sequence. Size exclusion chromatography using a 16/60 gel filtration column and Superdex 200 Prep Grade resin estimated the molecular mass of the reductase to be 131 kDa, which is consistent with the reductase being a heterodimer composed of one ArrA subunit and one ArrB subunit. Overexpressed ArrA and ArrB were also partially purified individually to ∼50% purity for activity assays and reconstitution experiments.
FIG. 4.

SDS-PAGE of purified ARR. Protein was stained with Bio-Safe Coomassie dye (Bio-Rad). Left lane: Bio-Rad SDS-PAGE Standards, broad range (myosin, 200 kDa; β-galactosidase, 116.25 kDa; phosphorylase b, 97.4 kDa; bovine serum albumin, 66.2 kDa; ovalbumin, 45 kDa; carbonic anhydrase, 31 kDa; trypsin inhibitor, 21.5 kDa; lysozyme, 14.4 kDa; aprotinin, 6.5 kDa). Right lane: purified ARR from C43 overexpression cells.
Cofactor composition.
Elemental analysis of the purified active ARR revealed a stoichiometry of 1.35 mol of Mo: 23.5 mol of inorganic S: 16.5 mol of Fe per mol of enzyme. Analysis of the ARR induced in the presence of W revealed a stoichiometry of 0.15 mol of W per mol of enzyme, suggesting that the metal was not incorporated into the protein.
CD spectrometry and thermal denaturation.
The secondary structure and thermal stability of ARR were assessed by CD analysis. The shape of the far-UV CD spectrum of ARR indicates a folded structure (Fig. 5A). Due to the buffer conditions used in the purification, it was not possible to accurately predict the percentage of alpha or beta secondary structure in the sample. However, the minima at 209 and 222 nm suggest the presence of an alpha-helical secondary structure, and the overall spectrum is consistent with a folded protein containing both alpha and beta secondary structures. The thermal stability of ARR was assessed by monitoring the increase in ellipticity of the sample at 222 nm as the temperature was increased. The fraction of protein folded as a function of temperature is shown in Fig. 5B. The thermal denaturation was cooperative but not reversible, since ARR precipitated at high temperatures. We therefore report an apparent Tm, the temperature at which 50% of the protein is folded, instead of thermodynamic parameters such as free energy, enthalpy, and entropy. The apparent Tm of this unfolding reaction is 41°C, indicating that most of the protein is folded at the growth temperature of 30°C and the in vitro assay temperature of 25°C.
FIG. 5.

CD measurements of ARR. (A) Far-UV CD spectrum of ARR at 25°C. Each point represents a data averaging time of 5 s. (B) Fraction of ARR folded as a function of temperature as monitored by CD at 222 nm. Each point represents a data averaging time of 30 s.
Activity.
Arsenate respiratory reductase activity was measured by using a colorimetric assay that couples MV oxidation to As(V) reduction. No activity was measured when ArrA or ArrB were heterologously expressed alone or when they were combined after expression (Fig. 6). However, when ArrA and ArrB were expressed together, a Km of 5 μM and a Vmax of 11,111 μmol of As(V) reduced min−1 mg of protein−1 were measured. The presence of 50 mM As(III) or 50 mM phosphate in addition to As(V) did not affect activity. In addition, ARR showed no activity in the presence of arsenite, selenate, sulfate, nitrate, DMSO, or antimonate.
DISCUSSION
Characterization of the properties of ARR is required to predict and quantify the contribution of this enzyme to the arsenic geochemical cycle. Because the ARR enzyme family appears to be highly conserved among diverse bacterial species, development of an overexpression system for one member of this family (e.g., ARR from Shewanella sp. strain ANA-3 [the present study]) should be broadly applicable to the other members of this group.
In a previous study of gene expression during growth of ANA-3 in As(V) medium, arrA was upregulated at the beginning of the exponential phase, reached maximal expression before the peak in optical density was attained, and decreased significantly upon the onset of stationary phase and the subsequent decrease in optical density (18). Our observation of ARR activity during growth determined that enzyme activity also began at the start of exponential growth, suggesting the lack of additional extracellular signals required for posttranscriptional upregulation. The persistence of ARR activity beyond the time point at which arrA transcripts are detectable suggests that the enzyme has a half-life of at least several hours and does not require constant regeneration. Thus, in the environment, the identification of arrA mRNA transcripts implies the activity of ARR; however, ARR may also be active when arrA mRNA transcripts cannot be detected.
In late-phase cultures, cells growing on As(V) shrunk and/or lysed and released ARR into the medium. Once released, the enzyme retained activity for several hours, showing that it is stable in solution at 30°C, as expected, given that its Tm is 41°C. Together, these results imply that the enzyme may be active extracellularly in environments such as alluvial aquifers and surface sediments where arsenic mobilization occurs, although its stability in the complex natural environment is likely to be much shorter than that measured in our laboratory studies. The release of enzyme into the medium suggests that the properties of ARR in both whole cells and pure enzyme samples may be directly relevant to environmental studies. Although supernatant filtrates of other isolated As(V) respirers have not been studied, similar decreases in the optical density of these isolates growing on As(V) have been observed, suggesting that the release of ARR may be a general phenomenon. An important caveat, however, is that the release of ARR is most likely a response to the accumulation of millimolar concentrations of As(III) in the medium (Chad Saltikov, unpublished data). Thus, we would expect the release of ARR only to be important if arsenite accumulated in microenvironments surrounding the cell. The presence of cell-free ARR in the environment may be relevant if electron donors with sufficiently low redox potentials are present to allow the enzyme to catalyze As(V) reduction. Redox active small molecules, including natural products, could possibly serve this purpose, although this hypothesis has yet to be tested.
Within intact cells, the periplasmic localization of ARR allows the cell to couple As(V) reduction to energy production before As(V) can be transported to the cell cytoplasm and enter the ars detoxification pathway, an energy-requiring process. In environmental contexts where As(V) is present at micromolar or nanomolar concentrations, the activity of ARR may be sufficient to reduce all of the available As(V) without involving ArsC (the cytosolic arsenate reductase used for detoxification). Consistent with this, our previous studies of ANA-3 with or without ArrA suggest that ArsC does not contribute significantly to total As(V) reduction when soluble As(V) concentrations are in the low micromolar range (3, 9). The initial periplasmic localization of ARR also supports the idea that at least some As(V) desorbs from sediments and enters the cell prior to respiratory reduction. This transient dissociation may allow arsenic mobilization if water flow transports the newly reduced As(III) as soon as it exits the cell from environments capable of sequestering it, such as iron-rich sediments, to environments that cannot sequester it, such as those dominated by aluminum oxides, manganese oxides, or silicate and carbonate minerals (14). Such a model has been proposed for sediments in Bangladesh, in which the source of contaminating arsenic is thought to be iron-rich surface sediments where As(V) respiration and iron reductive dissolution are active and able to be inhibited by antibiotics.
Metal cofactors associated with ARR are essential to its activity. Other members of the DMSO reductase family are characterized as having a tungsten or molybdenum atom coordinated to the sulfur atoms of the molybdenum guanine dinucleotide cofactors. The ability of an enzyme to use Mo or W is thought to depend on metal availability under which the enzyme is assembled and other currently unidentified factors involved in insertion of the metal into the enzyme active site (6). In Rhodobacter capsulatus, the DMSO reductase is active with either tungsten or molybdenum in its active site, although only the molybdenum-substituted form is capable of catalyzing the reverse reaction of oxidizing dimethylsulfide (20). These cells require nanomolar concentrations of molybdenum to produce active tungstoenzyme, and this phenomenon is thought to have arisen due to (i) the requirement for Mo to be involved in the biosynthesis of the molybdopterin cofactor or (ii) the requirement for Mo to activate the molybdate/tungstate transporter repressor, ModE, that prevents tungsten from accumulating to toxic levels inside the cell. In our studies, the induction of ARR in the presence of 1 mM tungstate resulted in inactive protein that did not incorporate tungsten, but the lack of activity does not seem to result from either of the processes mentioned above. Because induction was allowed to occur for 18 h in cells growing with DMSO as an electron acceptor, active DMSO reductase was required, suggesting that the assembly of the molybdopterin cofactor proceeded normally and that tungsten did not accumulate to levels high enough to inhibit growth. Thus, other mechanisms must be involved in the exclusion of W from ARR.
The inhibition of ARR expression by oxygen likely is due to the fact that anaerobic conditions are required for the expression of enzymes needed to assemble ARR (18). This requirement has been observed for other DMSO reductase family members. The assembly of the molybdopterin cofactor, for example, is known to be dependent on the moa and mod operons, both of which are expressed under anaerobic conditions. Regardless of the mechanism involved, the inhibition of ARR expression in the presence of oxygen is relevant in environmental settings where sediments are frequently exposed to open air. Such a situation was observed in Haiwee Reservoir, where water levels frequently drop below the surface level of sediments (7). The suggestion that contaminating arsenic originates in surface sediments in Bangladesh (14) may also mean that As(V)-respiring bacteria are exposed to oxygen. The extent to which these aeration events decrease the level of As(V) reduction presents an important area of study, and aeration may serve as an effective inhibitor of respiratory As(V) reduction.
Comparison of the purified ARR from ANA-3 with homologous enzymes from C. arsenatis and B. selenitireducens reveals similarities and differences within this group (Table 3). Sequence alignment (see Fig. S1 in the supplemental material) and comparison shows that the three sequences share 47% sequence identity and 64 to 65% sequence similarity. All three of the enzymes are predicted to be heterodimeric proteins consisting of one large A subunit and one small B subunit; however, they differ in their metal composition. The stoichiometry of the metal cofactors associated with ANA-3 ARR is consistent with each enzyme molecule having one Mo atom, four S atoms associated with the bis-molybdopterin guanine dinucleotide cofactor, and four to five [4Fe-4S] clusters. These results agree with bioinformatic predictions, where four putative [4Fe-4S]-binding domains were identified in ArrB and one putative domain was identified in ArrA (17). The enzyme from C. arsenatis contains similar ratios of these three elements; however, zinc was also found to be associated with the purified enzyme (8). Molybdenum and iron were found in significantly lower ratios in the B. selenitireducens enzyme (1). The ANA-3 ARR has the lowest Km and the highest Vmax of the three studied enzymes. Its Km of 5 μM As(V) is within the concentration range of 1 to 10 μM As(V) required to upregulate arr gene expression in anaerobically growing ANA-3 cells (18), suggesting that these concentrations are physiologically relevant. The localization of ARR appears to be different among the three bacteria: ARR from ANA-3 or C. arsenatis is soluble, whereas in B. selenitireducens it is membrane associated. Moreover, while ARR from ANA-3 and C. arsenatis are both specific to arsenate, the B. selenitireducens enzyme is reported to reduce As(III), selenate, and selenite, in addition to As(V), albeit with significantly lower activity. Such variations in substrate specificity have been observed for other members of the DMSO reductase family (15). An understanding of the active site environment and mechanism of substrate binding will help to determine the cause of these differences.
TABLE 3.
Summary of properties of ARR
| Property | Shewanella sp. strain ANA-3 | Chrysiogenes arsenatis | Bacillus selenitireducens |
|---|---|---|---|
| Km (μM) | 5 | 300 | 34 |
| Vmax (U/mg of protein) | 11,100 | 7,013 | 2.5 |
| Stoichiometry | α1β1 | α1β1 | α1β1 |
| Localization | Periplasm | Periplasm | Membrane associated |
| Metal stoichiometry per mol of enzyme | 1.3 Mo: 16.5 Fe: 23.5 inorganic S | 0.89 Mo: 14 Fe: 16.4 inorganic S: 0.67 Zn | 0.18 Mo: 9 Fe |
| Substrate specificity | Arsenate only | Arsenate only | Arsenate, arsenite, selenate, selenite |
Heterologous expression of the independent subunits ArrA and ArrB resulted in no As(V) reductase activity. Taken together with the facts that (i) the two genes encoding these proteins are found adjacent to one another in all of the sequenced As(V) respirers (11, 17) and (ii) ArrA and ArrB have been associated with one another in every purification to date (1, 8), this provides evidence that the two subunits function together in vivo. The lack of activity upon mixing the subunits together further suggests that ArrA and ArrB may depend on each other for proper assembly. Such a requirement has been observed in other molybdoenzymes such as nitrogenase, in which the Fe protein is required for both the maturation of the Fe-Mo cofactor and the maturation of the Mo-Fe protein (4). Recently, cymA, a gene encoding a membrane-bound c-type cytochrome was shown to be required for As(V) respiration in Shewanella sp. strain ANA-3 and Shewanella putrefaciens strain CN-32 (11). It was suggested that CymA is the site of interaction between ARR and the rest of the electron transport chain. Since this cytochrome is also known to participate in other modes of respiration, such as Fe(III), Mn(IV), and fumarate respiration, our results suggest that ArrA and ArrB form the minimal set of unique proteins required to confer As(V)-respiring ability.
To extend the findings reported here to the environment, techniques to detect and quantify the abundance of ARR in the field are required. The heterologous expression system developed in the present study will allow the production of antibodies or other probes for this purpose. In addition, this system will enable comparative structural and/or functional studies between different ARR family members that will help elucidate the mechanism by which this enzyme class functions and possibly result in the development of specific inhibitors for As(V) respiration.
Supplementary Material
Acknowledgments
We thank Stephen Mayo (Caltech) for help with the CD analyses and members of the Newman Lab for constructive discussions.
The Packard Foundation and Howard Hughes Medical Institute are acknowledged for providing financial support.
Footnotes
Published ahead of print on 19 October 2007.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Afkar, E., J. Lisak, C. W. Saltikov, P. Basu, R. S. Oremland, and J. F. Stolz. 2003. The respiratory arsenate reductase from Bacillus selenitireducens strain MLS10. FEMS Microbiol. Lett. 226:107-112. [DOI] [PubMed] [Google Scholar]
- 2.Ahmann, D., A. L. Roberts, L. R. Krumholz, and F. M. Morel. 1994. Microbe grows by reducing arsenic. Nature 371:750. [DOI] [PubMed] [Google Scholar]
- 3.Campbell, K. M., D. Malasarn, C. W. Saltikov, D. K. Newman, and J. G. Hering. 2006. Simultaneous microbial reduction of iron(III) and arsenic(V) in suspensions of hydrous ferric oxide. Environ. Sci. Technol. 40:5950-5955. [DOI] [PubMed] [Google Scholar]
- 4.Christiansen, J., P. J. Goodwin, W. N. Lanzilotta, L. C. Seefeldt, and D. R. Dean. 1998. Catalytic and biophysical properties of a nitrogenase Apo-MoFe protein produced by a nifB deletion mutant of Azotobacter vinelandii. Biochemistry 37:12611-12623. [DOI] [PubMed] [Google Scholar]
- 5.Harvey, C. F., C. H. Swartz, A. B. Badruzzaman, N. Keon-Blute, W. Yu, M. A. Ali, J. Jay, R. Beckie, V. Niedan, D. Brabander, P. M. Oates, K. N. Ashfaque, S. Islam, H. F. Hemond, and M. F. Ahmed. 2002. Arsenic mobility and groundwater extraction in Bangladesh. Science 298:1602-1606. [DOI] [PubMed] [Google Scholar]
- 6.Johnson, M. K., D. C. Rees, and M. W. Adams. 1996. Tungstoenzymes. Chem. Rev. 96:2817-2840. [DOI] [PubMed] [Google Scholar]
- 7.Kneebone, P. E., P. A. O'Day, N. Jones, and J. G. Hering. 2002. Deposition and fate of arsenic in iron- and arsenic-enriched reservoir sediments. Environ. Sci. Technol. 36:381-386. [DOI] [PubMed] [Google Scholar]
- 8.Krafft, T., and J. M. Macy. 1998. Purification and characterization of the respiratory arsenate reductase of Chrysiogenes arsenatis. Eur. J. Biochem. 255:647-653. [DOI] [PubMed] [Google Scholar]
- 9.Malasarn, D., C. W. Saltikov, K. M. Campbell, J. M. Santini, J. G. Hering, and D. K. Newman. 2004. arrA is a reliable marker for As(V) respiration. Science 306:455. [DOI] [PubMed] [Google Scholar]
- 10.Miroux, B., and J. E. Walker. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289-298. [DOI] [PubMed] [Google Scholar]
- 11.Murphy, J. N., and C. W. Saltikov. 2007. The cymA gene encoding a tetraheme c-type cytochrome is required for arsenate respiration in Shewanella species. J. Bacteriol. 189:2283-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman, D. K., E. K. Kennedy, J. D. Coates, D. Ahmann, D. J. Ellis, D. R. Lovley, and F. M. Morel. 1997. Dissimilatory arsenate and sulfate reduction in Desulfotomaculum auripigmentum sp. nov. Arch. Microbiol. 168:380-388. [DOI] [PubMed] [Google Scholar]
- 13.Nickson, R., J. McArthur, W. Burgess, K. M. Ahmed, P. Ravenscroft, and M. Rahman. 1998. Arsenic poisoning of Bangladesh groundwater. Nature 395:338. [DOI] [PubMed] [Google Scholar]
- 14.Polizzotto, M. L., C. F. Harvey, S. R. Sutton, and S. Fendorf. 2005. Processes conducive to the release and transport of arsenic into aquifers of Bangladesh. Proc. Natl. Acad. Sci. USA 102:18819-18823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridge, J. P., K. F. Aguey-Zinsou, P. V. Bernhardt, I. M. Brereton, G. R. Hanson, and A. G. McEwan. 2002. Site-directed mutagenesis of dimethyl sulfoxide reductase from Rhodobacter capsulatus: characterization of a Y114→F mutant. Biochemistry 41:15762-15769. [DOI] [PubMed] [Google Scholar]
- 16.Saltikov, C. W., A. Cifuentes, K. Venkateswaran, and D. K. Newman. 2003. The ars detoxification system is advantageous but not required for As(V) respiration by the genetically tractable Shewanella species strain ANA-3. Appl. Environ. Microbiol. 69:2800-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saltikov, C. W., and D. K. Newman. 2003. Genetic identification of a respiratory arsenate reductase. Proc. Natl. Acad. Sci. USA 100:10983-10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saltikov, C. W., R. A. Wildman, Jr., and D. K. Newman. 2005. Expression dynamics of arsenic respiration and detoxification in Shewanella sp. strain ANA-3. J. Bacteriol. 187:7390-7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith, A. H., E. O. Lingas, and M. Rahman. 2000. Contamination of drinking-water by arsenic in Bangladesh: a public health emergency. Bull. W. H. O. 78:1093-1103. [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart, L. J., S. Bailey, B. Bennett, J. M. Charnock, C. D. Garner, and A. S. McAlpine. 2000. Dimethyl sulfoxide reductase: an enzyme capable of catalysis with either molybdenum or tungsten at the active site. J. Mol. Biol. 299:593-600. [DOI] [PubMed] [Google Scholar]
- 21.Temple, C. A., T. N. Graf, and K. V. Rajagopalan. 2000. Optimization of expression of human sulfite oxidase and its molybdenum domain. Arch. Biochem. Biophys. 383:281-287. [DOI] [PubMed] [Google Scholar]
- 22.Tochio, H., M. M. Tsui, D. K. Banfield, and M. Zhang. 2001. An autoinhibitory mechanism for nonsyntaxin SNARE proteins revealed by the structure of Ykt6p. Science 293:698-702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.