

Abstract
Upon contact with intestinal epithelial cells, Salmonella enterica serovar Typhimurium injects a set of effector proteins into the host cell cytoplasm via the Salmonella pathogenicity island 1 (SPI1) type III secretion system (T3SS) to induce inflammatory diarrhea and bacterial uptake. The master SPI1 regulatory gene hilA is controlled directly by three AraC-like regulators: HilD, HilC, and RtsA. Previous work suggested a role for DsbA, a periplasmic disulfide bond oxidase, in SPI1 T3SS function. RtsA directly activates dsbA, and deletion of dsbA leads to loss of SPI1-dependent secretion. We have studied the dsbA phenotypes by monitoring expression of SPI1 regulatory, structural, and effector genes. Here we present evidence that loss of DsbA independently affects SPI1 regulation and SPI1 function. The dsbA-mediated feedback inhibition of SPI1 transcription is not due to defects in the SPI1 T3SS apparatus. Rather, the transcriptional response is dependent on both the flagellar protein FliZ and the RcsCDB system, which also affects fliZ transcription. Thus, the status of disulfide bonds in the periplasm affects expression of the SPI1 system indirectly via the flagellar apparatus. RcsCDB can also affect SPI1 independently of FliZ. All regulation is through HilD, consistent with our current model for SPI1 regulation.
Salmonella enterica serovars cause disease in humans ranging from mild gastroenteritis to lethal enteric fever (17). Salmonella pathogenicity island 1 (SPI1) encodes a type III secretion system (T3SS) that is an important virulence apparatus through which the pathogen injects effector proteins directly into the cytoplasm of host epithelial cells. The resulting physiological response leads to the gastrointestinal symptoms of infection as well as cellular uptake of the bacteria that is necessary to initiate systemic disease (23).
Intricate regulation of SPI1 gene expression presumably restricts production of the T3SS to when the bacteria are in a specific location within the host and coordinates the assembly process of the secretion apparatus (20, 38). The master SPI1 regulatory gene hilA is controlled directly by three AraC-like regulators: HilD, HilC, and RtsA (19, 53, 58). HilC and HilD are encoded in the SPI1 locus, while RtsA is encoded elsewhere in the chromosome in an operon with RtsB, which negatively regulates expression of flhDC and therefore the entire flagellar regulon (19). HilC, HilD, and RtsA are each capable of activating expression of hilC, hilD, and rtsA, creating a complex regulatory loop that controls hilA expression (Fig. 1) (15). HilA directly activates transcription of the inv/spa and prg/org promoters on SPI1 (3). Encoded in the inv operon, the AraC-like protein InvF can activate transcription of other genes located within and outside of the SPI1 locus, including sopB, encoding a secreted effector protein (24). The expression of effector genes requires both InvF and the secretion chaperone SicA, which apparently coordinates transcription of effector protein genes with assembly of the SPI1 T3SS (12).
FIG. 1.
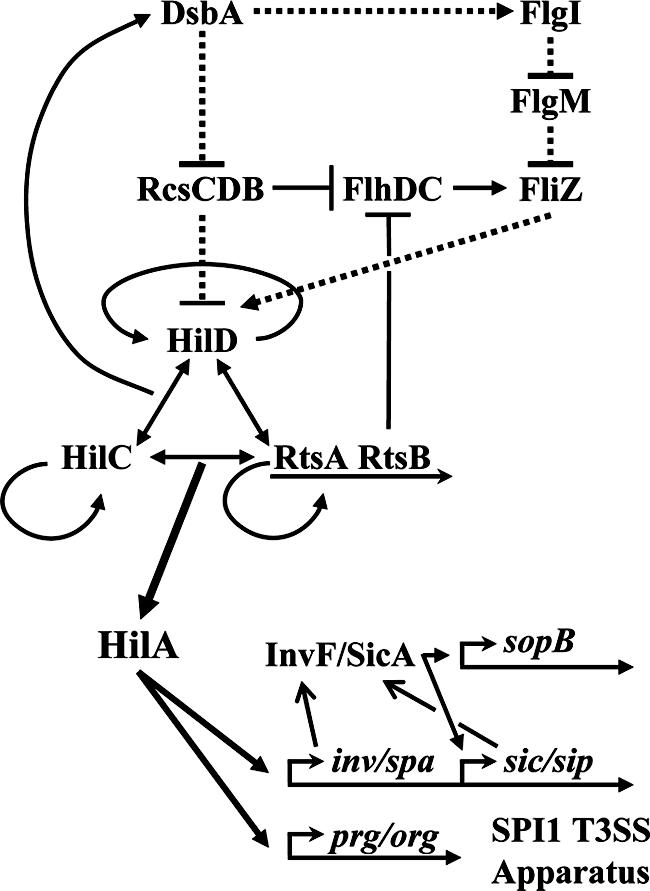
Genetic model for the regulation of SPI1 by DsbA. Solid lines indicate direct transcriptional regulation. Dashed lines indicate posttranslational effects, although the molecular mechanism is not necessarily understood. Arrows indicate positive effects. Lines with blunt ends designate negative effects. For simplicity, genes are not shown in most cases nor are all members of the various pathways given. See reference 15 for further details.
DsbA, a monomeric 21-kDa periplasmic protein, plays an important role in the folding of cell envelope proteins by catalyzing disulfide bond formation using a catalytic CXXC motif (for a review, see reference 35). After DsbA reacts with a target protein, it is reoxidized by the inner membrane protein DsbB, which then passes the electrons to quinones in the cytoplasmic membrane (4). The Salmonella virulence plasmid-encoded SrgA is a DsbA paralog whose activity is also dependent on a functional DsbB (6).
DsbA is required for full virulence in a number of pathogenic organisms. For example, a functional DsbA is required for proper assembly of cholera toxin in Vibrio cholerae (54), heat-stable toxin in enterotoxigenic Escherichia coli (52), and invasin in Yersinia pseudotuberculosis (42). DsbA has also been shown to be required for the proper function of the T3SS in Yersinia pestis (34), Shigella flexneri (64), and Pseudomonas aeruginosa (28). SrgA or DsbA is also required for folding of SpiA, the outer membrane component of the Salmonella pathogenicity island 2 (SPI2)-encoded T3SS (48). We previously provided evidence that DsbA is required for both SPI1- and SPI2-dependent type III secretion in Salmonella strains (18).
Mutations or treatments that interfere with disulfide bond formation in the periplasm are among the signals that activate the RcsCDB phosphorelay system, composed of the sensor RcsC, the response regulator RcsB, and the phosphotransfer protein RcsD (YojN) (45). RcsB, alone or together with RcsA, either positively or negatively regulates transcription of target genes, including activating those for capsule synthesis and biofilm formation (45). An rcsC constitutive mutant exhibits decreased transcription of invasion genes including hilA, invF, sipC, and invG in a process that is RcsB dependent (50).
RcsCDB can also negatively regulate the flagellar regulon (21), which contains more than 60 genes grouped into three classes according to their transcriptional hierarchy (22). Class I is composed of the flhDC master operon, which encodes transcriptional activators essential for the expression of class II operons. The protein products of class II operons include the alternative sigma factor FliA, the anti-sigma factor FlgM, and those required for the structure and assembly of the flagellar hook-basal body. Proper assembly of this structure allows the export of FlgM, freeing the FliA sigma factor to activate the class III operons (36, 51). The fliA and flgM genes are in separate operons, each of which is expressed from both class II and class III promoters (25, 32). FliZ is encoded in the fliA operon and is reported to be an enhancer of class II flagellar gene expression (31). FliZ activates expression of hilA, one of the links between regulation of SPI1 and motility (33, 44).
Our previous work suggested a role for DsbA in SPI1 T3SS assembly or function (18). In Salmonella enterica serovar Typhimurium, RtsA, HilD, and HilC directly activate dsbA and deletion of dsbA leads to loss of SPI1 function. Transcription of effector proteins was also significantly decreased in the dsbA mutant background. These results led us to propose a model in which RtsA coordinates expression of SPI1 and DsbA, required for functional assembly of the SPI1 machinery. We proposed that loss of the machine caused feedback inhibition of effector gene expression. In this work, we present evidence that dsbA-mediated feedback inhibition is not simply through the SPI1 T3SS apparatus. Rather, we show that SPI1 responds to the disulfide bond status of the periplasm through the RcsCDB system and the flagellar system as reflected in the level of FliZ (Fig. 1). These signals feed into the SPI1 regulatory network at HilD. We present further evidence that, independently of the regulatory effect, DsbA is required for assembly and/or function of the SPI1 apparatus.
MATERIALS AND METHODS
Media, reagents, and enzymatic assays.
Luria-Bertani (LB) medium was used for routine growth of bacteria (46). Motility agar contained 0.3% Bacto agar, 1% tryptone, and 0.5% NaCl. Bacterial strains were grown at 37°C, except for those containing the temperature-sensitive plasmids pCP20 and pKD46 (7, 13), which were grown at 30°C. Antibiotics were used at the concentrations indicated: ampicillin, 100 μg/ml; chloramphenicol, 10 μg/ml; kanamycin, 50 μg/ml; apramycin, 50 μg/ml. Enzymes were purchased from Invitrogen or New England Biolabs and were used according to the manufacturers' recommendations. Primers were purchased from IDT Inc. β-Galactosidase assays were performed using a microtiter plate assay as previously described (60). For most of these assays, strains were cultured overnight in LB and then subcultured in LB medium containing 1% NaCl and 2 mM dithiothreitol (DTT) and grown statically overnight. β-Galactosidase activities were calculated as (micromoles of o-nitrophenol formed per minute) × 106/(optical density at 600 nm × milliliters of cell suspension). The values are means ± standard deviations, where n = 4.
Bacterial strains.
All bacterial strains are isogenic derivatives of Salmonella enterica serovar Typhimurium strain 14028 and are listed in Table 1. Deletions of various genes and concomitant insertion of an antibiotic resistance cassette were constructed using λ Red-mediated recombination (13, 65) as described previously (16). The endpoints of each deletion are indicated in Table 1. The appropriate insertion of the antibiotic resistance marker was verified by P22 linkage to known markers and/or PCR analysis. In each case, the constructs resulting from this procedure were moved into a wild-type background (strain 14028) by P22 transduction. In some cases, antibiotic resistance cassettes were removed by using the temperature-sensitive plasmid pCP20 carrying the FLP recombinase (13). All bacterial strains were rebuilt at least once, and experiments were repeated with the independent constructs.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Genotypea or relevant characteristics | Endpoints of deletion (strain) or clone (plasmid)b | Source or referencec |
|---|---|---|---|
| Strains | |||
| 14028 | Wild-type serovar Typhimurium | ATCCd | |
| JS251 | ΔhilA112::Cm | 3019885-3021480 | 19 |
| JS252 | ΔhilC113::Cm | 3012135-3012976 | 19 |
| JS279 | Φ(hilA-lac+)112 | 19 | |
| JS326 | ΔdsbA100::Cm | 4204198-4204820 | 18 |
| JS379 | Φ(hilA-lac+)112 ΔdsbA100::Cm | 18 | |
| JS563 | ΔhilD119::Km | 3018210-3018730 | |
| JS696 | Φ(fliZ′-lac+)8042 | ||
| JS739 | ΔrtsA5::Km | 4561755-4560884 | 19 |
| JS740 | ΔinvG101 | 3041597-3043282 | |
| JS741 | ΔsrgA1::Km | 6670-7315 (plasmid pSLT) | |
| JS742 | ΔcpxRA101::Km | 4268376-4270452 | |
| JS743 | ΔaraBAD1021::Km | 117190-121300 | |
| JS744 | ΔaraE1022::Cm | 3175792-3177395 | |
| JS745 | ΔrcsBC101::Km | 2369981-2373608 | |
| JS746 | ΔfliZ8042::Tc | 2044136-2044684 | |
| JS747 | ΔflgI8043::Cm | 1263345-1264453 | |
| JS748 | ΔdsbA100::Cm ΔsrgA1::Km | ||
| JS749 | attλ::pDX1::hilA′-lacZ | ||
| JS750 | attλ::pDX1::invF′-lacZ | ||
| JS751 | attλ::pDX1::sopB′-lacZ | ||
| JS752 | attλ::pDX1::sicA′-lacZ | ||
| JS753 | attλ::pDX1::prgH′-lacZ | ||
| JS754 | ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS755 | ΔdsbA100::Cm attλ::pDX1::invF′-lacZ | ||
| JS756 | ΔdsbA100::Cm attλ::pDX1::sicA′-lacZ | ||
| JS757 | ΔdsbA100::Cm attλ::pDX1::sopB′-lacZ | ||
| JS758 | ΔdsbA100::Cm attλ::pDX1::prgH′-lacZ | ||
| JS759 | ΔsrgA1::Km attλ::pDX1::hilA′-lacZ | ||
| JS760 | ΔsrgA1::Km attλ::pDX1::sopB′-lacZ | ||
| JS761 | ΔdsbA100::Cm ΔsrgA1::Km attλ::pDX1::hilA′-lacZ | ||
| JS762 | ΔdsbA100::Cm ΔsrgA1::Km attλ::pDX1::sopB′-lacZ | ||
| JS763 | ΔcpxRA101::Km attλ::pDX1::hilA′-lacZ | ||
| JS765 | ΔcpxRA101::Km ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS767 | ΔinvG101 attλ::pDX1::hilA′-lacZ | ||
| JS768 | ΔinvG101 attλ::pDX1::invF′-lacZ | ||
| JS769 | ΔinvG101 attλ::pDX1::sicA′-lacZ | ||
| JS770 | ΔinvG101 attλ::pDX1::sopB′-lacZ | ||
| JS771 | ΔinvG101 ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS772 | ΔinvG101 ΔdsbA100::Cm attλ::pDX1::invF′-lacZ | ||
| JS773 | ΔinvG101 ΔdsbA100::Cm attλ::pDX1::sicA′-lacZ | ||
| JS774 | ΔinvG101 ΔdsbA100::Cm attλ::pDX1::sopB′-lacZ | ||
| JS775 | ΔrcsBC101 attλ::pDX1::hilA′-lacZ | ||
| JS776 | ΔrcsBC101 ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS777 | ΔrcsBC101 ΔdsbA100::Cm ΔsrgA1::Km attλ::pDX1::hilA′-lacZ | ||
| JS778 | ΔfliZ8042::Tc attλ::pDX1::hilA′-lacZ | ||
| JS779 | ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS780 | ΔfliZ8042::Tc ΔdsbA100 attλ::pDX1::hilA′-lacZ | ||
| JS781 | ΔfliZ8042::Tc ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS782 | ΔrcsBC101::Km ΔfliZ8042::Tc attλ::pDX1::hilA′-lacZ | ||
| JS783 | ΔrcsBC101::Km ΔfliZ8042::Tc ΔdsbA100 attλ::pDX1::hilA′-lacZ | ||
| JS784 | ΔrcsBC101::Km ΔfliZ8042::Tc ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS785 | ΔdsbA100 Φ(fliZ′-lac+)8042 | ||
| JS787 | ΔrcsBC101::Cm Φ(fliZ′-lac+)8042 | ||
| JS788 | ΔrcsBC101::Cm ΔdsbA100 Φ(fliZ′-lac+)8042 | ||
| JS790 | ΔflgI8043::Cm attλ::pDX1::hilA′-lacZ | ||
| JS791 | ΔflgI8043::Cm Φ(fliZ′-lac+)8042 | ||
| JS792 | ΔrcsBC101::Cm attλ::pDX1::hilA′-lacZ | ||
| JS793 | ΔflgI8043::Cm ΔrcsBC101 attλ::pDX1::hilA′-lacZ | ||
| JS794 | ΔrcsBC101 Φ(fliZ′-lac+)8042 | ||
| JS795 | ΔflgI8043::Cm ΔrcsBC101 Φ(fliZ′-lac+)8042 | ||
| JS796 | ΔflgI8043::Cm ΔrcsBC101 ΔdsbA100 attλ::pDX1::hilA′-lacZ | ||
| JS797 | ΔflgI8043::Cm ΔrcsBC101 ΔdsbA100 Φ(fliZ′-lac+)8042 | ||
| JS798 | pWKS30 attλ::pDX1::hilA′-lacZ | ||
| JS799 | pFliZ attλ::pDX1::hilA′-lacZ | ||
| JS800 | pWKS30 ΔfliZ8042::Tc attλ::pDX1::hilA′-lacZ | ||
| JS801 | pWKS30 ΔhilD114::Km attλ::pDX1::hilA′-lacZ | ||
| JS802 | pWKS30 ΔrtsA5::Km attλ::pDX1::hilA′-lacZ | ||
| JS803 | pWKS30 ΔhilC113::Cm attλ::pDX1::hilA′-lacZ | ||
| JS804 | pFliZ ΔfliZ8042::Tc attλ::pDX1::hilA′-lacZ | ||
| JS805 | pFliZ ΔhilD114::Km attλ::pDX1::hilA′-lacZ | ||
| JS806 | pFliZ ΔrtsA5::Km attλ::pDX1::hilA′-lacZ | ||
| JS807 | pFliZ ΔhilC113::Cm attλ::pDX1::hilA′-lacZ | ||
| JS808 | ΔhilD114::Km attλ::pDX1::hilA′-lacZ | ||
| JS809 | ΔrcsBC101::Cm ΔhilD114::Km attλ::pDX1::hilA′-lacZ | ||
| JS810 | ΔdsbA100::Cm ΔinvG101 | ||
| JS811 | pHilA pLacY(A177C) ΔaraBAD1021 ΔaraE1022 attλ::pDX1::sopB′-lacZ | ||
| JS812 | pHilA ΔhilD114::Km pLacY(A177C) ΔaraBAD1021 ΔaraE1022 attλ::pDX1::sopB′-lacZ | ||
| JS813 | pHilA ΔhilD114::Km ΔdsbA100 pLacY(A177C) ΔaraBAD1021 ΔaraE1022 attλ::pDX1::sopB′-lacZ | ||
| JS816 | pWKS30 ΔfliZ8042::Tc ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS817 | pWKS30 ΔfliZ8042::Tc ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS818 | pWKS30 ΔfliZ8042::Tc ΔrcsBC101::Km attλ::pDX1::hilA′-lacZ | ||
| JS819 | pWKS30 ΔfliZ8042::Tc ΔrcsBC101::Km ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS820 | pWKS30 ΔfliZ8042::Tc ΔrcsBC101::Km ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS821 | pFliZ ΔfliZ8042::Tc ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS822 | pFliZ ΔfliZ8042::Tc ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS823 | pFliZ ΔfliZ8042::Tc ΔrcsBC101::Km attλ::pDX1::hilA′-lacZ | ||
| JS824 | pFliZ ΔfliZ8042::Tc ΔrcsBC101::Km ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| JS825 | pFliZ ΔfliZ8042::Tc ΔrcsBC101::Km ΔdsbA100 ΔsrgA1 attλ::pDX1::hilA′-lacZ | ||
| JS826 | ΔflgI8043::Cm ΔdsbA100 attλ::pDX1::hilA′-lacZ | ||
| JS827 | ΔflgI8043::Cm ΔdsbA100 Φ(fliZ′-lac+)8042 | ||
| JS828 | ΔhilA112 attλ::pDX1::hilA′-lacZ | ||
| JS829 | ΔhilA112 ΔdsbA100::Cm attλ::pDX1::hilA′-lacZ | ||
| Plasmids | |||
| pKD46 | bla PBADgam bet exo pSC101 oriTS | 13 | |
| pCP20 | bla cat cI857 λPRflp pSC101 oriTS | 7 | |
| pAH125 | lacZ tL3 λattP oriR6K Kan tmgB | 29 | |
| pLacY(A177C) | 49 | ||
| pDX1 | lacZ tL3 λattP oriR6K aacIV tmgB | ||
| pWKS30 | pSC101 ori; Apr | 63 | |
| pDX2 | pWKS30::fliZ+ | 2044118-2044703 | |
| pBAD33 | cat araC PBAD pACYC184 ori | 27 | |
| pDX3 | pBAD33::hilA+ | 3019828-3021526 |
All Salmonella strains are isogenic derivatives of serovar Typhimurium strain 14028.
Numbers indicate the base pairs that are deleted or cloned (inclusive) as defined in the S. enterica serovar Typhimurium LT2 genome sequence (National Center for Biotechnology Information).
This study unless specified otherwise.
ATCC, American Type Culture Collection.
Plasmid construction.
The pAH125 lacZ reporter system is a CRIM plasmid that confers kanamycin resistance (29). The replacement of the kanamycin cassette with the apramycin cassette from a related plasmid, pAE2 (W. W. Metcalf and A. C. Eliot, unpublished data), was accomplished using λ Red-mediated recombination (13), creating plasmid pDX1. The transcription start sites of hilA, invF, sicA, sopB, and prgH have all been mapped (12, 43, 57). We amplified by PCR and cloned PhilA (−497→+420 bp), PinvF (−153→+202 bp), PsicA (−271→+130 bp), PsopB (−356→+297 bp), and PprgH (−265→+116 bp) upstream of the promoterless lacZ gene in pDX1. The resulting constructs were confirmed by DNA sequence analysis. These promoter-lacZ reporter plasmids were stably integrated in single copy on the Salmonella chromosome at the λattB site using λInt, thus creating single-copy transcriptional fusions without disrupting the native gene. PCR analysis was used to confirm the presence of reporters in single copy.
The hilA gene (−27→+1671 bp) was amplified using primers carrying XbaI and SphI sites and Pfu DNA polymerase (Stratagene), digested, and cloned into the arabinose-inducible vector pBAD33 (27) to generate pHilA. The fliZ gene (−14→+571 bp) was amplified using primers carrying EcoRI and BamHI sites and then cloned into vector pWKS30 (63).
Western blot analysis of secreted proteins.
Western blot detection of secreted SopB in pHilA background strains was performed after diluting overnight cultures 1/100 in 10 ml of LB medium without NaCl containing chloramphenicol and 0.01% l-arabinose. These cultures were grown with shaking at 225 rpm on a platform shaker for 4 h at 37°C. The hilA+ strains used for detection of SopB were grown statically overnight in LB containing 1% NaCl. In either case, 10 ml of culture was centrifuged at 5,000 × g. The cell pellet was washed and resuspended in 200 μl of 50 mM Tris (pH 8). Fifteen microliters of 2× sodium dodecyl sulfate (SDS) loading buffer (39) was added to 15 μl of the suspension (approximately 2 × 108 cells). This was considered the whole-cell extract. The original culture supernatant was centrifuged again and then filter sterilized using a 0.2-μm-diameter syringe filter and concentrated to 1 ml using an Amicon-15 filter. Proteins were precipitated with ice-cold trichloroacetic acid at a final concentration of 10% by incubating them on ice for 30 min and collected by centrifugation at 15,000 × g for 30 min at 4°C. The supernatant was then removed, and the trichloroacetic acid precipitate was washed with 1 ml of ice-cold 95% ethanol to which 100 μl saturated sodium acetate and 50 μl 0.2% phenol red indicator were added. The samples were then centrifuged for 20 min at 15,000 × g. This procedure was repeated until the solution was neutralized (66). The pellet was washed a final time in 95% ethanol and allowed to air dry. The pellet was then resuspended in 15 μl of 50 mM Tris (pH 8), and 15 μl of 2× SDS loading buffer was added. Proteins in both the whole-cell extracts and supernatant samples were separated by electrophoresis in 7.5% SDS-polyacrylamide gels (39) and blotted onto nitrocellulose (BioTrace; Pall Corporation) using a Bio-Rad MiniProtean 3 electrophoresis system for 2 h at ∼300 mA. The blots were blocked with 5% nonfat dried milk in phosphate-buffered saline containing 0.1% Tween 20. The primary antibody was rabbit anti-SopB antibody. The secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (Zymed, South San Francisco, CA) and was detected using 1/10,000 ECL substrate and ECL Hyperfilm (Amersham, Piscataway, NJ).
RESULTS
Loss of DsbA affects transcription of SPI1 genes.
We previously showed that the SPI1 regulators RtsA, HilC, and HilD activate dsbA from a novel promoter in serovar Typhimurium and that DsbA is required for SPI1 secretion of effector proteins (18). In the dsbA mutant, transcription of invF and genes downstream in the regulatory circuit was significantly reduced. This led us to propose that misfolding of a machine component(s) in the dsbA mutant resulted in feedback regulation of SPI1 gene transcription. According to our original data, transcription of hilA was unaffected by a ΔdsbA mutation. This observation suggested that the feedback regulation was acting between HilA and invF transcription. However, HilA is known to directly activate invF transcription (3), and our current understanding of SPI1 regulation could not account for this result.
To further determine where dsbA-mediated feedback regulation fits into the regulatory scheme, we constructed hilA-, invF-, sicA-, prgH-, and sopB-lacZ transcriptional fusions and integrated the fusion constructs into the Salmonella chromosome in single copy at the λ attachment site. In contrast to strains used in our previous studies, the SPI1 locus is intact in these constructs. HilA directly activates invF (and sicA via readthrough) and prgH, while InvF and SicA activate sicA and sopB. We measured β-galactosidase activity from these fusions in both wild-type and dsbA backgrounds. Consistent with our previous data, deletion of dsbA significantly decreased the expression of invF and sopB (Fig. 2A). As expected, the ΔdsbA mutation also reduced sicA expression in the same pattern as invF. However, in contrast to our previous results, loss of DsbA also affected the transcription of hilA. Consistent with the decreased expression of hilA, prgH transcription was also decreased in the ΔdsbA mutation. To further clarify this discrepancy, we directly compared the activity produced from our original hilA-lacZ fusion at the chromosomal locus [Φ(hilA-lac+)112] compared to the newly constructed fusion integrated at the λatt site (attλ::pDX1::hilA′-lacZ). β-Galactosidase activity was determined after overnight growth of isogenic dsbA+ and dsbA mutant strains in LB with 2 mM DTT (see below). As shown in Fig. 2B, only a low level of β-galactosidase activity is produced from the original fusion. The level of activity was slightly decreased (<2-fold) in the dsbA mutant grown in the presence of DTT. In contrast, the β-galactosidase expression from the newly constructed fusion was much higher and loss of dsbA caused a significant (>10-fold) decrease in expression. The original hilA-lacZ fusion is a hilA null construct. To ensure that HilA was not required for the observed phenotype, we deleted hilA in the newly constructed fusion background. Consistent with recent reports (14), deletion of hilA increased expression from the hilA promoter, but loss of DsbA led to a significant decrease in this background. Thus, the low level of β-galactosidase activity produced from our original fusion and the conditions (see below) under which we performed the experiment in our original report led us to conclude incorrectly that hilA transcription was unaffected in the dsbA background. Taken together, our current data confirm that loss of DsbA leads to decreased transcription of the SPI1 genes but suggest that this potential feedback regulation feeds into the circuit upstream of HilA.
FIG. 2.

Effects of dsbA, srgA, and cpxRA mutations on transcription of SPI1 T3SS genes. Strains contained the lac transcriptional fusions indicated below the graphs. Panel B compares the activity of two different hilA-lac fusion constructs. The Φ(hilA-lac+)112 fusion is from reference 19, whereas the attλ::pDX1::hilA′-lacZ construct is used throughout this study. Data in panel C are shown as relative β-galactosidase activity where the level of activity of each fusion in a wild-type background is considered 100%. This allows direct comparison of the two fusions. The strains used were JS749 to JS758 (A); JS279, JS379, JS749, JS754, JS828, and JS829 (B); JS749, JS751, JS754, JS757, and JS759 to JS762 (C); and JS749, JS754, JS763, and JS765 (D). WT, wild type.
SPI1 responds to the disulfide bond status in the periplasm.
DsbA catalyzes disulfide bond formation in the periplasm and thereby facilitates protein folding. To better understand the signals giving rise to the SPI1 regulatory phenotype, we examined the effects of environment and the roles of additional periplasmic proteins that control disulfide bond formation or general protein folding. A phenotype similar to loss of DsbA could be achieved by the addition of 10 mM DTT to cell cultures (data not shown), consistent with the hypothesis that loss of DsbA confers a phenotype due to a failure to form disulfide bonds. Indeed, the addition of 2 mM DTT to LB medium significantly enhanced the dsbA phenotype while having only a minor effect on hilA transcription in a wild-type background (data not shown). It is known that free cystine in rich media can partially compensate for loss of DsbA (4). Based on these observations, we added 2 mM DTT to the cultures throughout our studies.
We asked if SrgA, an additional periplasmic disulfide bond oxidase encoded on the Salmonella virulence plasmid (6), also affected SPI1. Deletion of srgA had no effect on expression of hilA or sopB, but a ΔdsbA ΔsrgA double mutation had a stronger effect than the ΔdsbA single mutation did (Fig. 2C), suggesting that SrgA is additive to DsbA with respect to expression of SPI1 genes.
The cytoplasmic membrane protein DsbB reoxidizes DsbA (4), whereas DsbC is a periplasmic protein disulfide bond isomerase that can reshuffle disulfide bonds (56). We tested whether these proteins also affected SPI1 regulation. In the presence of 2 mM DTT, deletion of dsbB decreased transcription of the hilA-lacZ fusion to approximately the same extent as seen in a dsbA mutant background, consistent with the known role of DsbB. In contrast, loss of DsbC led to a slight increase in hilA gene expression (data not shown). Thus, initial formation of disulfide bonds is apparently more important than isomerization of preexisting disulfide bonds in the SPI1 phenotype.
In E. coli, DsbA is primarily under the control of the two-component regulatory system CpxRA, which is activated by a variety of envelope stresses that cause protein misfolding in the periplasm (11). In serovar Typhimurium, dsbA is controlled by the hilA regulators RtsA, HilD, and HilC, and expression of DsbA under SPI1-inducing conditions is independent of CpxRA (18). We asked if the Cpx regulon had any role in the SPI1 phenotype conferred by loss of DsbA. As shown in Fig. 2D, deletion of CpxRA only slightly affected hilA expression. Moreover, the dsbA phenotype was evident in this background. These results are consistent with the expression of dsbA being independent of CpxRA under these conditions. Overproduction of OmpX, which can induce periplasmic stress by increasing activity of Sigma E (47), also did not affect hilA expression (data not shown). The lack of a significant phenotype in the cpx mutant or under conditions that induce the Sigma E regulon suggests that the observed defect in the dsbA mutant is due directly to a lack of disulfide bond formation, rather than general protein misfolding in the periplasm.
Feedback inhibition is not dependent on the SPI1 secretion machine.
Our original hypothesis was that, in the absence of DsbA, the SPI1 apparatus was not assembled appropriately and this led to feedback inhibition at the transcriptional level (18). If true, then mutations that disrupt the secretion machinery should confer a phenotype similar to that conferred by a dsbA deletion. It is not clear what, if any, defect in the secretion machinery is caused by loss of DsbA (48). Therefore, we constructed a series of in-frame deletions removing genes encoding various parts of the needle complex, all of which are required for secretion by the SPI1 apparatus (23, 37). These included deletion of prgH through orgA, removing the inner membrane, periplasmic, and needle components of the apparatus; deletion of invG, encoding the outer membrane secretin component of the machine; deletion of invJ, whose homolog in the Shigella secretion apparatus, Spa32, is a primary target of DsbA in that system (64); and deletion of invH, which encodes an outer membrane lipoprotein required for proper assembly of the InvG secretin (8, 9). All of these deletions conferred essentially identical phenotypes. The results for the invG deletion are detailed below.
The in-frame deletion of invG was introduced into strains containing hilA-, invF-, sicA-, or sopB-lacZ fusions. Deletion of invG had no significant effect on the transcription of hilA and invF (Fig. 3). There was a slight decrease in sicA and sopB transcription in the invG mutant that could be due to SicA-mediated secretion-dependent transcriptional regulation (12). However, introduction of the ΔdsbA mutation into the ΔinvG mutant background resulted in the expected decrease in expression of hilA, invF, sicA, and sopB. These results show that the feedback regulation conferred by loss of DsbA is not simply a result of either the physical absence or the dysfunction of the secretion machinery.
FIG. 3.
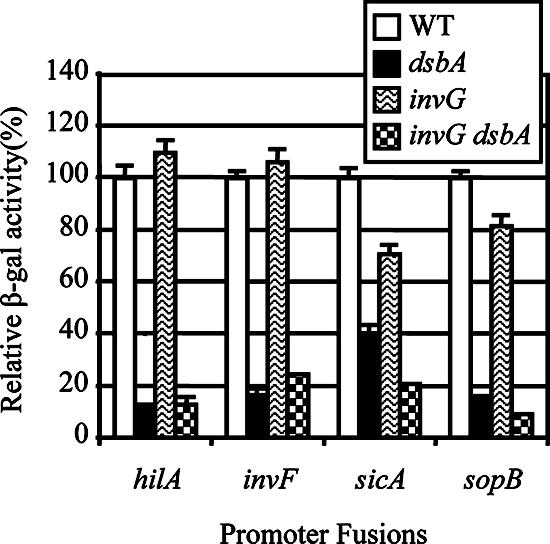
Differential effects of dsbA and invG mutations on transcription of SPI1 genes. Strains contained the promoter-lac fusions indicated below the graph and were otherwise wild type or contained a deletion of dsbA, invG, or invG dsbA. Data are shown as relative β-galactosidase activities where the level of activity of each fusion in a wild-type background is considered 100%. This allows direct comparison of the fusions. The strains used were JS749 to JS752, JS754 to JS757, and JS767 to JS774. WT, wild type.
The DsbA effect is partially dependent on the RcsCDB system.
The inability to form disulfide bonds in the periplasm appears to activate the RcsCDB system (45), which has been shown to repress expression of SPI1 (50). Indeed, dsbA srgA double mutants are mucoid (data not shown), indicating that capsule synthesis genes, controlled by RcsCDB, are being transcribed. We determined if the DsbA effect on SPI1 is mediated through the RcsCDB system. As shown in Fig. 4, in the wild-type background, hilA expression was reduced ∼8-fold by loss of DsbA and ∼34-fold by loss of both DsbA and SrgA. Deletion of RcsBC caused an increase in hilA expression in an otherwise wild-type background. Moreover, in the ΔrcsBC background, deletion of dsbA reduced hilA expression only ∼3-fold, while deletion of both dsbA and srgA showed a ∼15-fold reduction. These data suggest that the DsbA effect is partially dependent on the RcsCDB system but that additional factors also lead to decreased SPI1 expression in the dsbA mutant.
FIG. 4.
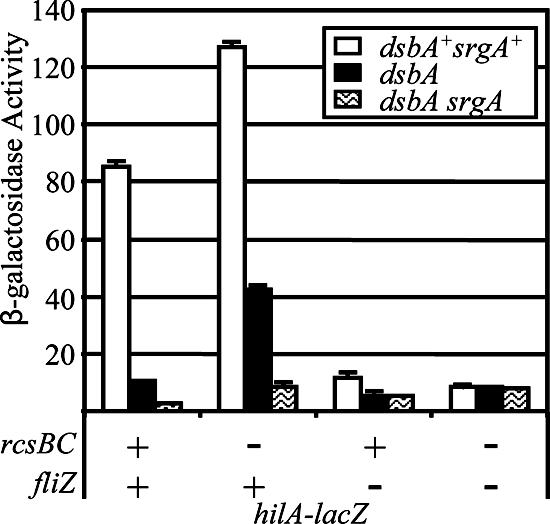
The dsbA effect on hilA expression is dependent on both rcsBC and fliZ. Strains contained the hilA-lacZ fusion with the indicated mutations. The strains used were JS749, JS754, and JS775 to JS784.
The DsbA effect is partially dependent on FliZ.
The interaction between the flagellar and SPI1 secretion systems is complex (see Discussion). An important link is FliZ, an enigmatic regulator expressed in an operon with fliA, encoding the class III-specific sigma factor. Genetically, FliZ activates expression of hilA (33, 44) and enhances class II flagellar gene expression (40). The DsbAB system is required for proper flagellar assembly in Escherichia coli (10). Figure 5 shows that loss of DsbA and SrgA in serovar Typhimurium leads to a severe motility defect. We reasoned that the loss of DsbA leads to defective assembly of the flagellar apparatus and this could result in decreased expression of fliZ and, consequently, decreased expression of hilA. This led us to test the role of FliZ in the DsbA effect. Deletion of fliZ caused a slight motility phenotype (Fig. 5), consistent with its reported role in transcriptional activation of class II promoters (32, 40). As shown in Fig. 4, the fliZ deletion also reduced hilA expression ∼7-fold. However, in the absence of FliZ, hilA expression was reduced only ∼2-fold in the dsbA mutant or dsbA srgA double mutant. These data are highly reproducible and suggest that, like RcsCDB, FliZ is partially responsible for the DsbA effect on hilA expression. We then asked if these two systems account for the entire effect on hilA expression by deleting both rcsBC and fliZ in the hilA-lac fusion strain. In the double deletion background, expression of hilA was no longer affected by loss of DsbA and SrgA (Fig. 4). Taken together, these results indicate that the DsbA effect on hilA expression is dependent on both the RcsCDB system and FliZ.
FIG. 5.

Motility phenotypes of the fliZ, rcsBC, flgI, dsbA, and dsbA srgA mutants. Single colonies were stabbed onto a motility agar plate and incubated for 5 h (left) or 9 h (right). The strains used were 14028, JS326, and JS745 to JS748. WT, wild type.
The fliZ gene is regulated in response to periplasmic disulfide bond status by both RcsCDB-dependent and -independent mechanisms.
Although the data in Fig. 4 suggest that RcsCDB and FliZ independently affect SPI1 expression in the dsbA mutant, this interpretation is complicated by the fact that RcsB and cofactor RcsA can negatively regulate the flagellar regulon (including FliZ) by binding directly to the flhDC promoter region (21). Indeed, the rcsBC mutation confers a phenotype in a motility assay, particularly if the cells are incubated for a relatively long time (Fig. 5). We wanted to determine whether the RcsCDB repression of SPI1 in the dsbA mutant is partially mediated by FliZ. As shown in Fig. 6A, we tested the effect of the dsbA mutation on expression of a fliZ-lac fusion. Deletion of dsbA decreased fliZ expression 13-fold. Deletion of rcsBC caused a slight increase in fliZ transcription in the dsbA+ background and partially relieved the repression of fliZ in the dsbA deletion background. However, a decrease in fliZ transcription caused by deletion of dsbA was still observed in the ΔrcsBC background. Thus, the regulatory effect on fliZ is partially dependent on the RcsCDB system, but additional factors also lead to decreased fliZ transcription in the dsbA mutant.
FIG. 6.
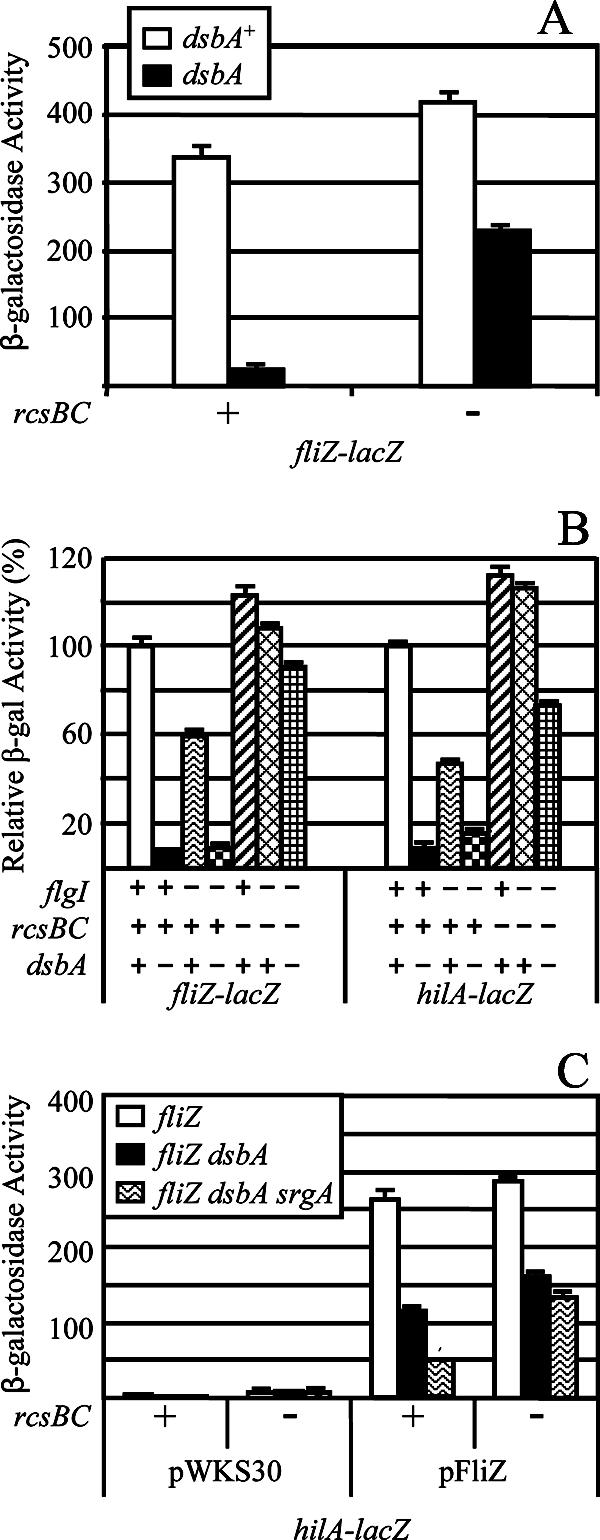
Relationship between fliZ and hilA expression. (A) The strains contained the fliZ-lacZ fusion with the indicated mutations. The strains used were JS696, JS785, JS787, and JS788. (B) The strains contained the lac fusions listed below the graph and flgI, rcsBC, and/or dsbA mutations as indicated. Data are shown as relative β-galactosidase activity where the level of activity of each fusion in a wild-type background is considered 100%. This allows direct comparison of the fusions. The strains used were JS696, JS749, JS754, JS785, JS790 to JS797, JS826, and JS827. (C) The hilA-lacZ fusion strains contained pWKS30 or pFliZ in the indicated backgrounds. The strains used were JS800, JS804, and JS816 to JS825.
Regulation of hilA mirrors regulation of fliZ.
What is the mechanism of decreased fliZ transcription caused by loss of DsbA in the rcsBC background? Based on published data, we propose the following model. The fliAZ operon is controlled by both class II and class III promoters (40). Loss of DsbA prevents proper assembly of the flagellar apparatus, at least affecting FlgI assembly and stability (10, 30). Under these conditions, the anti-sigma factor FlgM should not be exported and should block class III transcription (36, 51) and decrease FliZ expression. If true, then disabling the flagellar apparatus should confer a phenotype similar to that conferred by a dsbA mutation. To test this model, we deleted flgI. Figure 5 shows that, as expected, the ΔflgI mutant of serovar Typhimurium is completely nonmotile.
To distinguish phenotypes resulting from the loss of the flagellar apparatus from phenotypes resulting from other effects of dsbA, we specifically tested the effect of the ΔflgI mutation on expression of fliZ-lacZ and hilA-lacZ fusions. Deletion of flgI decreased both fliZ and hilA expression, consistent with the above model, but not to the extent seen in the dsbA mutant (Fig. 6B). Knocking out both dsbA and flgI conferred a phenotype almost identical to that seen in the dsbA mutant. We then asked if this additional effect of the dsbA mutation was dependent on RcsCDB. As seen above, deletion of rcsBC caused an increase in fliZ expression, presumably from the class II promoter. In the rcsBC background, however, the ΔflgI mutation had little effect on fliZ- or hilA-lacZ transcription. This result suggests that the increase in fliZ expression caused by loss of RcsCDB largely compensates for the decrease in expression caused by loss of the functional flagellar apparatus.
The data above suggest that, although loss of the flagellar apparatus can affect FliZ expression, it does not explain the decrease in transcription observed in the rcsBC background when dsbA is also mutated. Indeed, deletion of dsbA in the flgI rcsBC background led to a decrease in fliZ expression, albeit <2-fold, and a slightly greater decrease in hilA transcription (Fig. 6B). It seems unlikely that the loss of DsbA confers a defect in the flagellar apparatus that is more severe than that conferred by deletion of flgI (Fig. 5). Therefore, this result suggests that some regulatory circuit, in addition to RcsCDB, has a minor effect on expression of the flagellar regulon in response to disulfide bond formation. Taken together, these results show that transcriptional regulation of fliZ in response to disulfide bond status in the periplasm is multifactorial and complex. However, it is striking that hilA transcription closely mirrors fliZ expression, suggesting that the DsbA effect on SPI1 is largely mediated through FliZ.
DsbA can affect hilA expression independently of RcsCDB and the flagellar system.
Figure 6B shows that loss of DsbA led to a decrease in hilA expression in the flgI rcsBC mutant background that was slightly greater than the decrease in fliZ expression in this background. It is possible that DsbA can affect the SPI1 regulatory circuit independently of RcsBDC and FliZ. Note that this hypothesis is in apparent conflict with the results shown in Fig. 4. In the rcsBC fliZ mutant, loss of DsbA has no significant effect on hilA expression. However, expression of hilA is low in this background and subtle effects on expression might be difficult to discern. To test this idea, we introduced plasmid pWKS30 (vector control) and pFliZ into strains containing the hilA-lacZ fusion. In the latter plasmid, fliZ is expressed from the lac promoter and should no longer be affected by DsbA. As shown in Fig. 6C, production of FliZ from the plasmid increased expression of hilA approximately 6.7-fold. In this background, loss of DsbA decreased the expression of hilA 2.5-fold; expression was decreased 5.2-fold in the dsbA srgA double mutant. To test if this effect is solely dependent on RcsCDB, we tested the loss of dsbA srgA in an rcsBC strain containing the pFliZ plasmid. Under these conditions, loss of DsbA and SrgA still led to a modest, yet obvious, twofold decrease in expression. Quantitative PCR analysis showed that fliZ mRNA production from the plasmid was identical in the dsbA+ and dsbA mutant strains (data not shown). These results are consistent with those shown in Fig. 4 and show that the RcsCDB system can affect hilA expression independently of fliZ expression. But these results also show that loss of DsbA and SrgA can slightly affect hilA expression independently of both FliZ and the RcsCDB system.
FliZ and RcsCDB function through HilD.
Our results prove that the transcriptional effect mediated by loss of DsbA is independent of the SPI1 secretion apparatus. It follows that loss of DsbA must be affecting hilA transcription via the known SPI1 regulatory system. We have recently shown that HilD, HilC, and RtsA act in a complex regulatory loop to control the transcription of the hilA gene. Moreover, in every case where it has been explicitly tested, all known regulatory systems that affect SPI1 regulation do so through HilD (15, 20). We asked if FliZ and RcsCDB also function through HilD to control SPI1. First, we introduced pFliZ or the vector control (pWKS30) into hilA-lacZ fusion strains in which the chromosomal fliZ, hilD, hilC, or rtsA gene was deleted. As shown in Fig. 7A, production of FliZ from the plasmid was fully able to complement the effect of a ΔfliZ mutation on hilA expression and induced transcription of hilA fivefold in an otherwise wild-type background. Introduction of a hilD deletion into the hilA-lacZ fusion strain resulted in the expected decrease in hilA expression. Moreover, the presence of a hilD mutation blocked FliZ induction of hilA. In contrast, neither RtsA nor HilC was required for FliZ induction of hilA expression, although the absolute level of expression was reduced in both mutant backgrounds, consistent with our model of SPI1 regulation (15). Thus, FliZ-mediated induction of hilA expression is through HilD, while RtsA and HilC act as amplifiers of the signal.
FIG. 7.
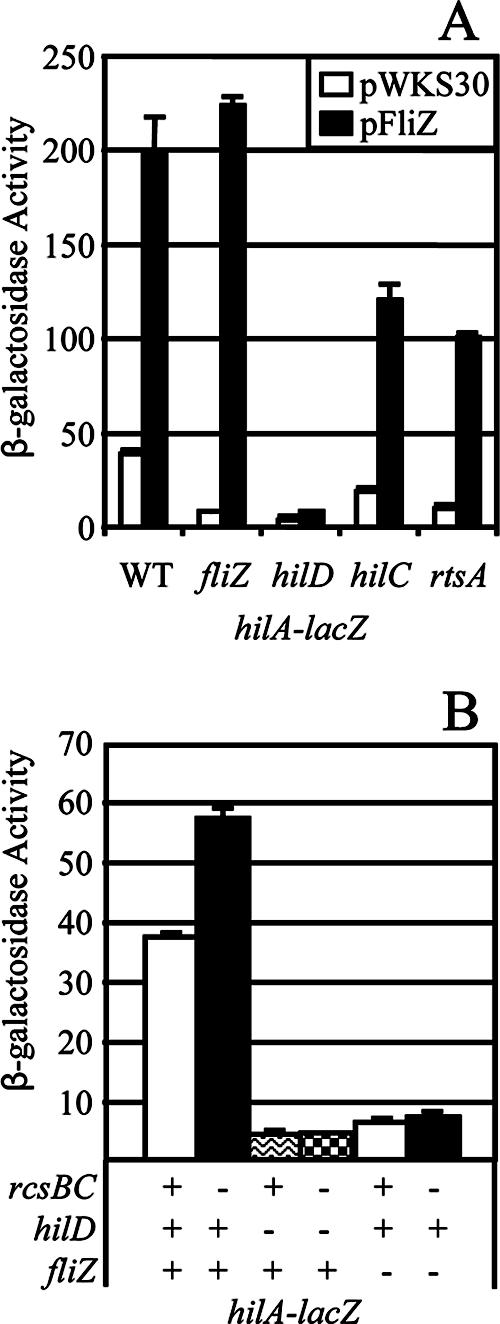
FliZ and RcsBC affect hilA expression through HilD. (A) The hilA-lacZ fusion strains with mutations indicated below the graph contained pWKS30 or pFliZ. WT, wild type. The strains used were JS798 to JS807. (B) The hilA-lacZ fusion strains contained deletions of genes indicated below the graph. The strains used were JS749, JS778, JS782, JS792, JS808, and JS809.
We then asked if the inhibition of hilA expression by RcsCDB also works through HilD. As shown in Fig. 7B (and above), deletion of rcsBC in the wild-type background caused an increase in hilA expression. However, this effect was not evident in either the ΔhilD or the ΔfliZ background. These data are consistent with the hypothesis that the RcsCDB effect on hilA expression also works through HilD, at least partially by affecting expression of fliZ.
DsbA is required for function of the SPI1 secretion apparatus.
Given that the DsbA effect is mediated through the SPI1 regulatory system and is apparently independent of the SPI1 T3SS apparatus itself, we considered the possibility that loss of DsbA has no effect on the apparatus per se but rather that the defect in SPI1-mediated secretion observed in a dsbA mutant is solely due to a decrease in the synthesis of the machine components. In order to test if loss of DsbA had a direct effect on the assembly or function of the apparatus, we contrived a situation in which the synthesis of the machine was independent of the normal signal transduction pathway. To accomplish this, we first cloned hilA onto a pBAD vector under the control of an arabinose-dependent promoter. To avoid the problem of nonuniform induction inherent in the arabinose system (49, 59), we constructed a Salmonella strain that had deletions of arabinose transporters (araE araFGH) and catabolic genes (araBAD) and contained a plasmid encoding a mutant LacY(A177C) that transports arabinose proportionally to the external concentration (49). Our background strain also contained a sopB-lac fusion integrated at the λ attachment site. We could then delete hilD to disconnect SPI1 gene expression from the regulatory effects of removing DsbA or any other known regulatory system. With the introduction of the pBAD-hilA plasmid, we titrated the arabinose concentration to achieve expression of the SPI1 system approximately equivalent to that observed in a wild-type strain under our normal inducing conditions, as monitored by expression of the sopB-lac fusion (Fig. 8).
FIG. 8.
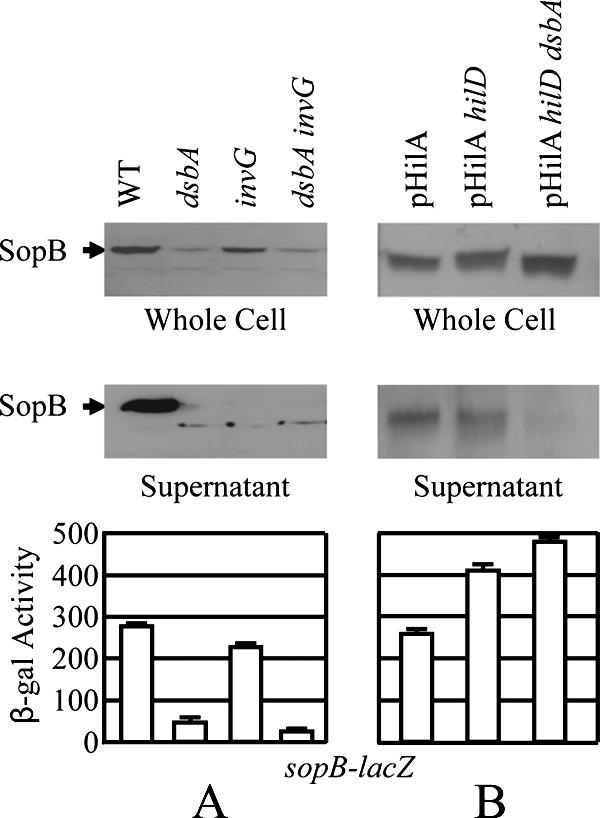
Effect of a dsbA mutation on secretion of SopB into the culture supernatant. Western blot analyses of whole-cell extracts (top panels) or culture supernatants (bottom panels) with anti-SopB antibody. (A) The strains were wild type (WT) or contained deletions of dsbA, invG, or dsbA invG as indicated above the lanes. The strains used were 14028 (wild type), JS326, JS740, and JS810. (B) The strains are pLacY(A177C) ΔaraBAD ΔaraE ΔaraFGH and contain the pHilA plasmid and mutations indicated above the lanes. The strains used were JS811 to JS813. Culture supernatants and whole-cell extracts were prepared as described in Materials and Methods. Equivalent amounts of culture supernatant or whole-cell extract from each strain were separated by 7.5% SDS-polyacrylamide gel electrophoresis. The resulting gels were blotted, and proteins were detected by using anti-SopB and horseradish peroxidase-labeled rabbit anti-mouse secondary antibody. The bar graph shows the β-galactosidase activity produced from a sopB-lacZ fusion in each of the strains assayed from an aliquot of each culture.
We monitored SPI1-mediated secretion by Western blotting, using antibodies directed against the SopB effector protein. In control experiments, we assayed the production and secretion of SopB from the wild-type strain, as well as ΔdsbA, ΔinvG, and ΔdsbA ΔinvG strains. As expected, deletion of dsbA in the wild-type background decreased production of SopB and the protein was not secreted into the culture supernatant at detectable levels (Fig. 8A). The ΔinvG mutation completely blocked secretion of SopB into the supernatant while slightly affecting expression of SopB. In the pBAD-hilA background described above, SodB was expressed and secreted normally. Although there was some variability in the level of arabinose induction, deletion of hilD, as predicted, did not affect expression or secretion, given that hilA is controlled by arabinose. Deletion of dsbA in this background now had no effect on production of SopB in whole-cell extracts. However, secretion of SopB into the supernatant was significantly decreased in the dsbA strain (Fig. 8B). These data show that, independently of the transcriptional effect on the SPI1 regulatory circuit, loss of DsbA affects either the assembly or the function of the SPI1 secretion apparatus.
DISCUSSION
The inability to form disulfide bonds in the periplasm affects the assembly of complex machinery in the cell envelope. Indeed, loss of DsbA, the primary disulfide bond oxidase in the periplasm, has been shown to block type III secretion in serovar Typhimurium, S. flexneri, P. aeruginosa, and Y. pestis and motility in E. coli and serovar Typhimurium (10, 18, 28, 34, 48, 64) (Fig. 5). But does loss of function indicate a simple defect in assembly? Here we have shown that, in addition to a structural or functional defect conferred by mutations in dsbA, there is a distinct regulatory response. The SPI1 T3SS regulatory circuit responds to the state of disulfide bonds in the periplasm and adjusts expression of the machine components. Our results suggest that loss of DsbA affects SPI1 through four distinct yet overlapping pathways (Fig. 1). First, DsbA inhibits activation of the RcsCDB system (45). Activation of RcsCDB in a dsbA mutant inhibits hilA expression (50) through HilD. Activated RcsCDB also represses flhDC, encoding the master regulators of the flagellar regulon (21). This leads to decreased production of FliZ, which activates SPI1 (33, 44), also through HilD. Thus, under conditions where disulfide bonds are not properly formed in the periplasm, activation of RcsCDB leads to decreased production of at least two complex machines, the flagellar apparatus and the SPI1 T3SS, that require assembly in the cell envelope.
Second, DsbA is required for proper assembly of the flagellar apparatus; FlgI has been identified as at least one specific target (10). In the absence of a functional flagellar apparatus, the anti-sigma factor FlgM is not exported but rather binds the FliA sigma factor, preventing expression of class III flagellar genes (36, 51). The fliAZ operon is controlled by both class II and class III flagellar promoters, and expression of this operon is significantly decreased in both dsbA and flgI mutant backgrounds. Thus, in the dsbA background, fliZ expression from both the class II (via RcsCDB) and class III (via FlgM) promoters is decreased, resulting in decreased expression of the invasion genes. Third, loss of DsbA leads to a slight decrease in fliZ expression independent of RcsCDB or the known flagellar feedback regulation, but this pathway seems relatively minor. We presume that this regulation is through flhDC, which is subject to a variety of environmental inputs (55). Fourth, loss of DsbA causes a slight decrease in SPI1 expression that is independent of RcsBCD or FliZ, but again this effect is minor. Indeed, our data point to FliZ as a primary link between expression of SPI1 and flagella.
A large number of regulatory systems and environmental signals have been implicated in SPI1 (reviewed in reference 1). The results presented here are consistent with our current model in which HilD, HilC, and RtsA act in a complex regulatory loop to control the transcription of the hilA gene (15). Although in every case where it has been tested, the identified regulatory systems feed into SPI1 through HilD, the actual mechanism of regulation is understood in only a few instances and involves posttranslational blocking of HilD function (20). Our data (not shown) suggest that FliZ also acts posttranslationally to affect HilD function. We do not mean to imply that FliZ is directly affecting HilD; the actual mechanism will require further investigation. The flhDC operon is also controlled by a number of other regulators including H-NS, cyclic AMP receptor protein, EnvZ/OmpR, QseBC, LrhA, and RcsCDB (55). Given our data, it seems likely that some of the signals that affect regulation of SPI1 could function, at least partially, by affecting FliZ production. Similarly, RcsCDB responds to a number of stimuli, and it is not clear which of, and under what conditions, these various signals are most important (reviewed in reference 45). However, our data are consistent with disulfide bond status being an important physiological signal for RcsCDB. In conjunction with FliZ, RcsCDB could be the intermediate in the SPI1 response to a variety of environmental conditions.
We have shown that, in addition to regulatory feedback on transcription, DsbA is required for appropriate assembly or function of SPI1. However, the molecular requirement for DsbA is unclear. None of the protein components of purified SPI1 needle complexes contain more than one cysteine residue (37, 39). However, the mature InvH, which acts as the pilot protein for the secretin InvG (8, 9, 41), does have two cysteine residues and is a likely candidate for the primary defect in the dsbA mutant. All indications are that, under the conditions that we are examining, dsbA is primarily controlled by RtsA, HilC, and HilD (18); CpxRA, which controls dsbA in E. coli, seems irrelevant to either expression of dsbA or assembly and function of the SPI1 T3SS or flagella in serovar Typhimurium (Fig. 2C). Thus, coregulation of dsbA with hilA apparently ensures that periplasmic disulfide bond formation is not an issue when SPI1 is being expressed and assembled.
Our data suggest that FliZ plays a major role in the regulation of SPI1 in response to periplasmic disulfide bond status. However, this is only one aspect of the complex regulatory interaction between the flagellar regulon and the invasion genes, and a number of regulators have been implicated in controlling both systems. RcsCDB negatively regulates both systems (21, 50). Likewise, FimZY, the regulator of type I fimbriae, has also been shown to negatively coregulate flagella and SPI1 (5). HilA negatively regulates itself (14), although the mechanism is not clear. A recent study suggests that HilA might also repress flhDC (62). In contrast, other systems differentially regulate the two machines. RtsA activates hilA, whereas the coregulated protein, RtsB, represses flhDC (19). SirA (indirectly via the CsrABC system [2]) also activates SPI1 but represses flhDC (26, 61). It is not clear why this relationship exists between flagellar and invasion systems, nor is it known under which natural conditions these various regulatory schemes become important. In particular, we do not know the state of these various regulatory networks during Salmonella colonization of the intestine when SPI1 expression is necessary. More studies are required to fully understand the physiological role of this complex relationship.
Acknowledgments
This work was supported by Public Health Service grant AI063230 from the National Institute of Allergy and Infectious Diseases.
We thank Brett Finlay for SopB antibody, Thomas Silhavy for plasmid pOmpX, John Cronan for plasmid pLacYA177C, and James Bardwell and Stephen Farrand for helpful discussions.
Footnotes
Published ahead of print on 19 October 2007.
REFERENCES
- 1.Altier, C. 2005. Genetic and environmental control of Salmonella invasion. J. Microbiol. 43(Spec. No.)85-92. [PubMed] [Google Scholar]
- 2.Altier, C., M. Suyemoto, and S. D. Lawhon. 2000. Regulation of Salmonella enterica serovar Typhimurium invasion genes by csrA. Infect. Immun. 686790-6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajaj, V., C. Hwang, and C. A. Lee. 1995. HilA is a novel OmpR/ToxR family member that activates the expression of Salmonella typhimurium invasion genes. Mol. Microbiol. 18715-727. [DOI] [PubMed] [Google Scholar]
- 4.Bardwell, J. C., J. O. Lee, G. Jander, N. Martin, D. Belin, and J. Beckwith. 1993. A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. USA 901038-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baxter, M. A., and B. D. Jones. 2005. The fimYZ genes regulate Salmonella enterica serovar Typhimurium invasion in addition to type 1 fimbrial expression and bacterial motility. Infect. Immun. 731377-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouwman, C. W., M. Kohli, A. Killoran, G. A. Touchie, R. J. Kadner, and N. L. Martin. 2003. Characterization of SrgA, a Salmonella enterica serovar Typhimurium virulence plasmid-encoded paralogue of the disulfide oxidoreductase DsbA, essential for biogenesis of plasmid-encoded fimbriae. J. Bacteriol. 185991-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1589-14. [DOI] [PubMed] [Google Scholar]
- 8.Crago, A. M., and V. Koronakis. 1998. Salmonella InvG forms a ring-like multimer that requires the InvH lipoprotein for outer membrane localization. Mol. Microbiol. 3047-56. [DOI] [PubMed] [Google Scholar]
- 9.Daefler, S., and M. Russel. 1998. The Salmonella typhimurium InvH protein is an outer membrane lipoprotein required for the proper localization of InvG. Mol. Microbiol. 281367-1380. [DOI] [PubMed] [Google Scholar]
- 10.Dailey, F. E., and H. C. Berg. 1993. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc. Natl. Acad. Sci. USA 901043-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danese, P. N., and T. J. Silhavy. 1997. The sigma(E) and the Cpx signal transduction systems control the synthesis of periplasmic protein-folding enzymes in Escherichia coli. Genes Dev. 111183-1193. [DOI] [PubMed] [Google Scholar]
- 12.Darwin, K. H., and V. L. Miller. 2001. Type III secretion chaperone-dependent regulation: activation of virulence genes by SicA and InvF in Salmonella typhimurium. EMBO J. 201850-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Keersmaecker, S. C., K. Marchal, T. L. Verhoeven, K. Engelen, J. Vanderleyden, and C. S. Detweiler. 2005. Microarray analysis and motif detection reveal new targets of the Salmonella enterica serovar Typhimurium HilA regulatory protein, including hilA itself. J. Bacteriol. 1874381-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellermeier, C. D., J. R. Ellermeier, and J. M. Slauch. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 57691-705. [DOI] [PubMed] [Google Scholar]
- 16.Ellermeier, C. D., A. Janakiraman, and J. M. Slauch. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290153-161. [DOI] [PubMed] [Google Scholar]
- 17.Ellermeier, C. D., and J. M. Slauch. 2006. The genus Salmonella, p. 123-158. In M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (ed.), The prokaryotes, 3rd ed. Springer, New York, NY.
- 18.Ellermeier, C. D., and J. M. Slauch. 2004. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J. Bacteriol. 18668-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellermeier, C. D., and J. M. Slauch. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 1855096-5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellermeier, J. R., and J. M. Slauch. 2007. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr. Opin. Microbiol. 1024-29. [DOI] [PubMed] [Google Scholar]
- 21.Francez-Charlot, A., B. Laugel, G. A. Van, N. Dubarry, F. Wiorowski, M. P. Castanie-Cornet, C. Gutierrez, and K. Cam. 2003. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 49823-832. [DOI] [PubMed] [Google Scholar]
- 22.Frye, J., J. E. Karlinsey, H. R. Felise, B. Marzolf, N. Dowidar, M. McClelland, and K. T. Hughes. 2006. Identification of new flagellar genes of Salmonella enterica serovar Typhimurium. J. Bacteriol. 1882233-2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galan, J. E. 2001. Salmonella interactions with host cells: type III secretion at work. Annu. Rev. Cell Dev. Biol. 1753-86. [DOI] [PubMed] [Google Scholar]
- 24.Galyov, E. E., M. W. Wood, R. Rosqvist, P. B. Mullan, P. R. Watson, S. Hedges, and T. S. Wallis. 1997. A secreted effector protein of Salmonella dublin is translocated into eukaryotic cells and mediates inflammation and fluid secretion in infected ileal mucosa. Mol. Microbiol. 25903-912. [DOI] [PubMed] [Google Scholar]
- 25.Gillen, K. L., and K. T. Hughes. 1993. Transcription from two promoters and autoregulation contribute to the control of expression of the Salmonella typhimurium flagellar regulatory gene flgM. J. Bacteriol. 1757006-7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodier, R. I., and B. M. Ahmer. 2001. SirA orthologs affect both motility and virulence. J. Bacteriol. 1832249-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J. Bacteriol. 1774121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha, U. H., Y. Wang, and S. Jin. 2003. DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect. Immun. 711590-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haldimann, A., and B. L. Wanner. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 1836384-6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hizukuri, Y., T. Yakushi, I. Kawagishi, and M. Homma. 2006. Role of the intramolecular disulfide bond in FlgI, the flagellar P-ring component of Escherichia coli. J. Bacteriol. 1884190-4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikebe, T., S. Iyoda, and K. Kutsukake. 1999. Promoter analysis of the class 2 flagellar operons of Salmonella. Genes Genet. Syst. 74179-183. [DOI] [PubMed] [Google Scholar]
- 32.Ikebe, T., S. Iyoda, and K. Kutsukake. 1999. Structure and expression of the fliA operon of Salmonella typhimurium. Microbiology 1451389-1396. [DOI] [PubMed] [Google Scholar]
- 33.Iyoda, S., T. Kamidoi, K. Hirose, K. Kutsukake, and H. Watanabe. 2001. A flagellar gene fliZ regulates the expression of invasion genes and virulence phenotype in Salmonella enterica serovar Typhimurium. Microb. Pathog. 3081-90. [DOI] [PubMed] [Google Scholar]
- 34.Jackson, M. W., and G. V. Plano. 1999. DsbA is required for stable expression of outer membrane protein YscC and for efficient Yop secretion in Yersinia pestis. J. Bacteriol. 1815126-5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadokura, H., F. Katzen, and J. Beckwith. 2003. Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72111-135. [DOI] [PubMed] [Google Scholar]
- 36.Karlinsey, J. E., S. Tanaka, V. Bettenworth, S. Yamaguchi, W. Boos, S. I. Aizawa, and K. T. Hughes. 2000. Completion of the hook-basal body complex of the Salmonella typhimurium flagellum is coupled to FlgM secretion and fliC transcription. Mol. Microbiol. 371220-1231. [DOI] [PubMed] [Google Scholar]
- 37.Kimbrough, T. G., and S. I. Miller. 2000. Contribution of Salmonella typhimurium type III secretion components to needle complex formation. Proc. Natl. Acad. Sci. USA 9711008-11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimbrough, T. G., and S. I. Miller. 2002. Assembly of the type III secretion needle complex of Salmonella typhimurium. Microbes Infect. 475-82. [DOI] [PubMed] [Google Scholar]
- 39.Kubori, T., A. Sukhan, S. I. Aizawa, and J. E. Galan. 2000. Molecular characterization and assembly of the needle complex of the Salmonella typhimurium type III protein secretion system. Proc. Natl. Acad. Sci. USA 9710225-10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kutsukake, K., T. Ikebe, and S. Yamamoto. 1999. Two novel regulatory genes, fliT and fliZ, in the flagellar regulon of Salmonella. Genes Genet. Syst. 74287-292. [DOI] [PubMed] [Google Scholar]
- 41.Lario, P. I., R. A. Pfuetzner, E. A. Frey, L. Creagh, C. Haynes, A. T. Maurelli, and N. C. Strynadka. 2005. Structure and biochemical analysis of a secretin pilot protein. EMBO J. 241111-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leong, J. M., P. E. Morrissey, and R. R. Isberg. 1993. A 76-amino acid disulfide loop in the Yersinia pseudotuberculosis invasin protein is required for integrin receptor recognition. J. Biol. Chem. 26820524-20532. [PubMed] [Google Scholar]
- 43.Lostroh, C. P., V. Bajaj, and C. A. Lee. 2000. The cis requirements for transcriptional activation by HilA, a virulence determinant encoded on SPI-1. Mol. Microbiol. 37300-315. [DOI] [PubMed] [Google Scholar]
- 44.Lucas, R. L., C. P. Lostroh, C. C. DiRusso, M. P. Spector, B. L. Wanner, and C. A. Lee. 2000. Multiple factors independently regulate hilA and invasion gene expression in Salmonella enterica serovar Typhimurium. J. Bacteriol. 1821872-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majdalani, N., and S. Gottesman. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59379-405. [DOI] [PubMed] [Google Scholar]
- 46.Maloy, S. R., V. J. Stewart, and R. K. Taylor. 1996. Genetic analysis of pathogenic bacteria: a laboratory manual. Cold Spring Harbor Laboratory Press, Plainview, NY.
- 47.Mecsas, J., R. Welch, J. W. Erickson, and C. A. Gross. 1995. Identification and characterization of an outer membrane protein, OmpX, in Escherichia coli that is homologous to a family of outer membrane proteins including Ail of Yersinia enterocolitica. J. Bacteriol. 177799-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miki, T., N. Okada, and H. Danbara. 2004. Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 27934631-34642. [DOI] [PubMed] [Google Scholar]
- 49.Morgan-Kiss, R. M., C. Wadler, and J. E. Cronan, Jr. 2002. Long-term and homogeneous regulation of the Escherichia coli araBAD promoter by use of a lactose transporter of relaxed specificity. Proc. Natl. Acad. Sci. USA 997373-7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mouslim, C., M. Delgado, and E. A. Groisman. 2004. Activation of the RcsC/YojN/RcsB phosphorelay system attenuates Salmonella virulence. Mol. Microbiol. 54386-395. [DOI] [PubMed] [Google Scholar]
- 51.Ohnishi, K., K. Kutsukake, H. Suzuki, and T. Lino. 1992. A novel transcriptional regulation mechanism in the flagellar regulon of Salmonella typhimurium: an antisigma factor inhibits the activity of the flagellum-specific sigma factor, sigma F. Mol. Microbiol. 63149-3157. [DOI] [PubMed] [Google Scholar]
- 52.Okamoto, K., T. Baba, H. Yamanaka, N. Akashi, and Y. Fujii. 1995. Disulfide bond formation and secretion of Escherichia coli heat-stable enterotoxin II. J. Bacteriol. 1774579-4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olekhnovich, I. N., and R. J. Kadner. 2002. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J. Bacteriol. 1844148-4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peek, J. A., and R. K. Taylor. 1992. Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 896210-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pruss, B. M., C. Besemann, A. Denton, and A. J. Wolfe. 2006. A complex transcription network controls the early stages of biofilm development by Escherichia coli. J. Bacteriol. 1883731-3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rietsch, A., D. Belin, N. Martin, and J. Beckwith. 1996. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. USA 9313048-13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schechter, L. M., S. M. Damrauer, and C. A. Lee. 1999. Two AraC/XylS family members can independently counteract the effect of repressing sequences upstream of the hilA promoter. Mol. Microbiol. 32629-642. [DOI] [PubMed] [Google Scholar]
- 58.Schechter, L. M., and C. A. Lee. 2001. AraC/XylS family members, HilC and HilD, directly bind and derepress the Salmonella typhimurium hilA promoter. Mol. Microbiol. 401289-1299. [DOI] [PubMed] [Google Scholar]
- 59.Siegele, D. A., and J. C. Hu. 1997. Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc. Natl. Acad. Sci. USA 948168-8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slauch, J. M., and T. J. Silhavy. 1991. cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J. Bacteriol. 1734039-4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teplitski, M., R. I. Goodier, and B. M. Ahmer. 2003. Pathways leading from BarA/SirA to motility and virulence gene expression in Salmonella. J. Bacteriol. 1857257-7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thijs, I. M., S. C. De Keersmaecker, A. Fadda, K. Engelen, H. Zhao, M. McClelland, K. Marchal, and J. Vanderleyden. 2007. Delineation of the Salmonella enterica serovar Typhimurium HilA regulon through genome-wide location and transcript analysis. J. Bacteriol. 1894587-4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100195-199. [PubMed] [Google Scholar]
- 64.Watarai, M., T. Tobe, M. Yoshikawa, and C. Sasakawa. 1995. Disulfide oxidoreductase activity of Shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc. Natl. Acad. Sci. USA 924927-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 975978-5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zurawski, D. V., and M. A. Stein. 2003. SseA acts as the chaperone for the SseB component of the Salmonella pathogenicity island 2 translocon. Mol. Microbiol. 471341-1351. [DOI] [PubMed] [Google Scholar]