

Abstract
The toxicity of nucleoside analogs used for the treatment of human immunodeficiency virus infection is due primarily to the inhibition of replication of the mitochondrial genome by the human mitochondrial DNA polymerase (Pol γ). The severity of clinically observed toxicity correlates with the kinetics of incorporation versus excision of each analog as quantified by a toxicity index, spanning over six orders of magnitude. Here we show that the rate of excision of dideoxycytidine (zalcitabine; ddC) was reduced fourfold (giving a half-life of ∼2.4 h) by the addition of a physiological concentration of deoxynucleoside triphosphates (dNTPs) due to the formation of a tight ternary enzyme-DNA-dNTP complex at the polymerase site. In addition, we provide a more accurate measurement of the rate of excision and show that the low rate of removal of ddCMP results from both the unfavorable transfer of the primer strand from the polymerase to the exonuclease site and the inefficient binding and/or hydrolysis at the exonuclease site. The analogs ddC, stavudine, and ddATP (a metabolite of didanosine) each bind more tightly at the polymerase site during incorporation than normal nucleotides, and this tight binding contributes to slower excision by the proofreading exonuclease, leading to increased toxicity toward mitochondrial DNA.
Nucleoside analogs continue to be essential components of highly active antiretroviral therapy designed for the treatment of human immunodeficiency virus (HIV) infection. Each of the eight approved nucleoside analogs is phosphorylated intracellularly by host cell kinases to create a nucleoside triphosphate (or nucleotide) analog which then acts as a chain terminator to stop viral genome replication by HIV reverse transcriptase. Unfortunately, the activated triphosphate analogs also act as substrates for host cell DNA polymerases. Toxicity results in duration-dependent mitochondrial dysfunction with various degrees of severity, depending on many factors, including uptake, transport, metabolic activation, and degradation (2-4, 11, 12, 20, 21). The incorporation of nucleoside analogs by the mitochondrial DNA polymerase γ (Pol γ) has been implicated as the primary cause underlying the mitochondrial toxicity seen clinically (1, 6, 22), with the possible exception of zidovudine (AZT) (reviewed in reference 18). While both HIV reverse transcriptase and Pol γ have 5′-to-3′ DNA polymerase activity, only Pol γ has 3′-to-5′ exonuclease proofreading activity, which can reduce toxicity by the removal of nucleoside analogs after their incorporation. Understanding the structural features of nucleoside analogs that afford efficient removal by Pol γ would provide valuable information for the design of less toxic drugs.
We have previously characterized the fidelity of DNA replication catalyzed by a reconstituted recombinant human Pol γ holoenzyme by using transient-state kinetic methods (13, 14, 16) and showed that the kinetics of incorporation of nucleoside analogs by Pol γ are correlated with the toxic side effects seen clinically (8, 15, 18). Of the nucleoside analogs examined, ddCTP (the activated triphosphate form of dideoxycytidine [zalcitabine; ddC]) was the best substrate for Pol γ (7). However, the toxicity of nucleoside analogs is also dependent upon the rate of excision by the proofreading exonuclease. In previous studies, the rate of excision of ddC was reported to be too low to measure under the conditions used, which led to the conclusion that ddC is effectively an irreversible inhibitor of mitochondrial DNA replication (7). In this study, we more rigorously explored the kinetics of exonuclease removal of ddC in an attempt to resolve the mechanisms underlying selectivity during proofreading by the mitochondrial DNA polymerase.
Our understanding of the mechanistic basis for error correction by polymerases is based largely on data from studies using T7 DNA polymerase that were subsequently extended to Pol γ (5, 13). Detailed studies inspecting the removal of a mismatched nucleotide versus the incorporation of the next correct nucleotide to bury the mismatch have shown that the polymerase stalls after a mismatch is incorporated. This stalling inverts the kinetic partitioning that normally favors polymerization over excision by allowing time for the melting and transfer of the primer strand into the exonuclease active site. Primer strand transfer is followed by fast hydrolysis and then transfer back to the polymerase site without dissociation of the DNA from the enzyme.
Given that ddC is the most toxic nucleoside analog approved by the Food and Drug Administration (FDA), it was our goal to more precisely assess the rate of ddC removal by Pol γ and extend our studies to resolve contributions from strand transfer and excision. The present study has revealed that the rate of hydrolysis of ddCMP is much lower than that of the natural nucleotide but that the rate is easily measurable. Moreover, the lower rate results from unfavorable partitioning between the exonuclease site and the polymerase site, and the binding of the next correct nucleotide further reduces the rate of excision.
MATERIALS AND METHODS
Protein expression and purification.
The overexpression and purification of the recombinant human Pol γ catalytic subunit and accessory protein were carried out as previously described (10, 16). All kinetic studies were conducted using a wild-type Pol γ holoenzyme reconstituted with a fivefold molar excess of the accessory subunit relative to the catalytic subunit. The following nucleoside analogs were used: stavudine (d4T, 2′,3′-didehydro-2′,3′-dideoxythymidine); AZT (3′-azido-2′,3′-dideoxythymidine); FIAU [1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil]; CBV (carbocylic 2′,3′-didehydro-dideoxyguanosine); PMPA [(R)-9-(2-phosphonylmethoxypropyl)-adenine]; (−)3TC [β-l-(−)-2′,3′-dideoxy-3′-thiacytidine]; and (−)FTC [(−)-β-l-2′-3′-dideoxy-5-fluoro-3′-thiacytidine].
Preparation of DNA.
Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). The 26-mer primer sequence was 5′-GCCTCGCAGCCGTCCAACCAACTCAX-3′, where the X signifies the position where either dCMP or ddCMP was positioned. The template (45-mer) sequence that was correctly base paired with the primer strand was 5′-GGACGGCATTGGATCGACAGTGAGTTGGTTGGACGGCTGCGAGGC-3′. The template sequence for the DNA containing eight different bases to create a frayed primer was 5′-GGACGGCATTGGATCGACAATATCAAGGTTGGACGGCTGCGAG GC-3′. The primer was 5′ end labeled with 32P by using T4 polynucleotide kinase according to the instructions of the manufacturer (Life Technologies, Gaithersburg, MD). The reaction was terminated by incubation at 95°C for 5 min, and excess nucleotide was removed using a Bio-Spin 6 column (Bio-Rad, Hercules, CA). The primer was annealed to the 45-mer template by combination at an equimolar ratio, heating to 95°C, and slow cooling to room temperature.
Exonuclease reaction conditions.
For reactions too fast to measure by manual mixing and quenching, a quench-flow apparatus (RFQ-3) from KinTek Corporation was used. Exonuclease assays were performed at 37°C in buffer containing 50 mM Tris-Cl, pH 7.5, 100 mM NaCl, and 2.5 mM MgCl2. The holoenzyme was reconstituted by incubating the two protein subunits for 5 min on ice in reaction buffer lacking magnesium. The radiolabeled duplex DNA was then added to the holoenzyme, and the mixture was incubated for an additional 5 min on ice. Reactions were initiated by the addition of MgCl2 to a final concentration of 2.5 mM in the same reaction buffer at 37°C and then quenched with 0.5 M EDTA. All concentrations given are concentrations after mixing. Products were separated on 15% denaturing polyacrylamide sequencing gels, imaged on a Molecular Dynamics Storm 860, and quantified using ImageQuant software (Amersham Biosciences, Uppsala, Sweden). All experiments were performed at least twice.
Data analysis.
The concentration of the 26-mer substrate was plotted against time and fit to a single ([26-mer] = A1·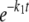 + C) or double ([26-mer] = A1·
+ C) or double ([26-mer] = A1·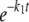 + A2·
+ A2·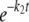 + C) exponential equation, where A is the amplitude, t is time, k1 and k2 are the rates of exonuclease removal, and C is the endpoint.
+ C) exponential equation, where A is the amplitude, t is time, k1 and k2 are the rates of exonuclease removal, and C is the endpoint.
RESULTS AND DISCUSSION
dCMP removal from correctly paired or frayed DNA.
To examine the kinetics of the exonuclease reaction, we prepared synthetic oligonucleotides to create defined primer and template strands of 26 and 45 bases, respectively. We designed one primer-template duplex to be complementary throughout the double-stranded region and one with eight mismatches on the 3′ end of the primer to create a frayed duplex. The rationale for using a frayed duplex is based upon observations suggesting that the rate of excision is usually limited by the rate of transfer of the primer strand from the polymerase site to the exonuclease site and that a frayed duplex favors partitioning into the exonuclease site so that the observed rate more closely approaches the intrinsic rate of hydrolysis (5, 13). We chose a length of eight mismatched base pairs at the end of the frayed duplex because previous work had shown that the effects of fraying on the kinetics of excision were saturated after six to eight nucleotides.
We first examined the rates of removal of the natural nucleotide (dCMP) from the primer 3′ terminus in reactions with mixtures containing either a correctly paired or frayed DNA substrate. The wild-type Pol γ holoenzyme was preincubated with either correctly paired or frayed DNA, under conditions in which the enzyme was in molar excess relative to the DNA, and rapidly mixed with Mg2+ in a chemical quench-flow to start the reaction. The exonuclease reaction was quenched at various times with 0.5 M EDTA. The concentration of the starting material (full-length primer) was plotted as a function of time (Fig. 1). The removal of dCMP from correctly base-paired DNA was biphasic, with rates of 0.86 ± 0.35 s−1 and 0.016 ± 0.004 s−1 and amplitudes of 21 ± 3 nM and 70 ± 3 nM, respectively. Because the experiment was performed with the enzyme in excess compared to the DNA, we can eliminate models invoking multiple turnovers to account for the slow phase. Rather, the amplitude of the fast phase may represent the fraction of primer strand that was in the exonuclease site of Pol γ when the reaction was initiated, while the slow, larger-amplitude phase may represent the rate at which the remaining primer partitions into the exonuclease site. The exonuclease reaction observed using the frayed DNA substrate was also biphasic, with rates of 1.6 ± 0.5 s−1 and 0.1 ± 0.07 s−1 and amplitudes of 60 ± 10 nM and 25 ± 10 nM, respectively.
FIG. 1.

Exonuclease removal of dCMP with primers with or without frayed termini. The wild-type holoenzyme (100 nM) was preincubated with 90 nM 26-mer-45-mer terminated with dCMP correctly base paired (○) or frayed at the 3′ terminus (eight mismatches; •) and mixed with Mg2+ to start the reaction. The concentration of the substrate (26-mer primer) as a function of time was fit to a double exponential equation. Reactions of both the correctly base paired and frayed DNA were biphasic. The correctly paired dCMP DNA was excised at rates of 0.86 ± 0.35 s−1 and 0.016 ± 0.004 s−1 with amplitudes of 21 ± 3 nM and 70 ± 3 nM, respectively. The frayed dCMP-terminated DNA was excised at rates of 1.6 ± 0.5 s−1 and 0.1 ± 0.07 s−1 with amplitudes of 60 ± 10 nM and 25 ± 10 nM, respectively.
In comparing the results for the frayed duplex to those for the fully complementary duplex, the rate of the fast phase was found to increase only slightly but the amplitude for the fast phase increased significantly, consistent with our working model. The rate of excision for the slow phase of the reaction with frayed DNA was an order of magnitude higher than the rate of excision for the slow phase with the correctly paired DNA, suggesting that the primer strand melting and/or transfer was faster with DNA comprising a region of 3′ mismatches. We interpret these data to mean that the intrinsic rate of hydrolysis of dCMP from the 3′ end of a primer is greater than or equal to 1.6 s−1, while the rate of excision of dCMP from properly base-paired DNA is limited by the transfer of the primer from the polymerase site at a rate 100-fold lower (0.016 s−1).
ddCMP removal from correctly paired or frayed DNA.
The removal of ddCMP was examined using a DNA sequence identical to the one in the procedure described above except that the primer was terminated with ddCMP at the 3′ end. As described above, two reactions were performed, one with correctly paired DNA and the other with frayed DNA, both with the enzyme in excess relative to the DNA. The concentration of the substrate was plotted as a function of time (Fig. 2). Unlike the removal of the natural nucleotide, the removal of ddCMP occurred in a single phase and could be fitted to a single exponential equation. The rate of the removal of ddCMP from correctly paired DNA was (3 ± 0.4) × 10−4 s−1, about 50-fold lower than the rate of dCMP removal in the slow phase. The removal of ddCMP from frayed DNA was only 15-fold faster than the removal from correctly paired DNA and occurred at a rate of (4.4 ± 0.5) × 10−3 s−1. Both reactions went to completion at rates much lower than the rate of dissociation of DNA from the enzyme active site (∼0.02 s−1) measured previously (16), implying that the vast majority of DNA dissociates from the enzyme and rebinds multiple times during the time course of excision. Nonetheless, by examining the reaction with the enzyme in excess, the observed rate is limited only by the reactions occurring at the active site. Our data suggest that at equilibrium, the majority of the correctly paired ddCMP-terminated primer remains in the polymerase active site. Moreover, the rate of hydrolysis to excise ddCMP, defined by the removal from the frayed primer-template combinations, is about 360-fold lower than that seen for dCMP. Therefore, it appears that the 3′-hydroxyl group on the primer strand plays a significant role in the binding and/or alignment of the terminal nucleotide at the exonuclease site. Moreover, as described previously, ddCTP binds more tightly than dCTP at the polymerase site, and it is reasonable to suggest that similar tighter binding at the polymerase site after incorporation shifts the partitioning away from the exonuclease site. However, the binding of the next correct nucleotide will alter the distribution among various DNA states bound to the enzyme.
FIG. 2.

Exonuclease removal of ddCMP primers with or without frayed termini. The wild-type holoenzyme (100 nM) was preincubated with 90 nM 26-mer-45-mer terminated with ddC correctly base paired (○) or frayed at the 3′ terminus (eight mismatches; •) and mixed with Mg2+ to start the reaction. The concentration of the substrate (26-mer primer) as a function of time was plotted and fit to a single exponential equation. The reactions were not double exponentials as in Fig. 1 and were dramatically slower, at a rate of 3 ± 0.4 × 10−4 s−1 for the correctly paired ddC-terminated primer and 4.4 ± 0.5 × 10−3 s−1 for the ddC-terminated frayed DNA. Both reactions had full amplitudes of about 90 nM.
Removal of ddC in the presence of dNTPs.
The binding of the next correct deoxynucleoside triphosphate (dNTP) to a primer-template lacking a 3′-OH molecule forms a ternary complex, stabilizing the primer at the polymerase site (17), thereby attenuating the migration of the primer to the exonuclease site and reducing the observed hydrolysis rate (5). Therefore, after ddCMP is incorporated, the rate of excision may be lowered by the presence of dNTPs. As shown in Fig. 3, ddCMP removal from correctly paired DNA in the presence of 100 μM dNTPs was slowed by almost fourfold to a rate of (8.0 ± 0.7) × 10−5 s−1. The rate of removal of ddCMP from frayed DNA in the presence of dNTPs [(6.3 ± 0.3) × 10−3 s−1] was comparable to that seen in their absence [(4.4 ± 0.5) × 10−3 s−1]. Therefore, the tight ternary complex cannot be formed by the binding of the next correct nucleotide to a frayed duplex. The rationale for adding all four dNTPs at 100 μM was based upon the fact that the inclusion of only the next correct base would have made the data analysis unnecessarily complex. For example, if only the next correct nucleotide was present during the excision reaction, the removal of ddCMP would have likely been followed by the misincorporation of the single dNTP present in the solution and would have led to the same length of primer with which the reaction was initiated. The presence of the other three nucleotides allowed rapid polymerization of the primer following the slow excision of ddCMP. Therefore, the disappearance of the primer was easily quantified in this format. In any event, the presence of all four nucleotides would be a more accurate representation of the true physiological circumstance than the presence of only one nucleotide and therefore provides the best estimate for the in vivo rate of excision to date. Results of previous studies suggest that the effect is specific to the next correct nucleotide (13). Table 1 provides a summary of the data obtained for the removal of dCMP and ddCMP under the various conditions examined.
FIG. 3.

Exonuclease removal of ddCMP from primer strands with or without frayed termini in the presence of dNTPs. The wild-type holoenzyme (100 nM) was preincubated with 90 nM 26-mer-45-mer terminated with ddC correctly base paired (○) or frayed at the 3′ terminus (eight mismatches; •) and mixed with Mg2+ and 100 μM dNTPs to start the reaction. The concentration of the substrate (26-mer primer) as a function of time was plotted and fit to a single exponential equation. The correctly paired ddC-terminated DNA was excised at a rate of 8 ± 0.7 × 10−5 s−1. The frayed ddC-terminated DNA was excised at a rate of 6.3 ± 0.3 × 10−3 s−1. Both reactions had amplitudes of about 90 nM.
TABLE 1.
Summary of kinetic parameters for the removal of dCMP or ddCMP from either correctly paired or frayed DNA with and without dNTPsa
| Reaction mixture contents | High rate (s−1) | High-rate amplitude (nM) | Low rate (s−1) | Low-rate amplitude (nM) |
|---|---|---|---|---|
| dCMP and correctly paired DNA | 0.86 ± 0.35 | 21 ± 3 | 0.016 ± 0.004 | 70 ± 3 |
| dCMP and frayed DNA | 1.6 ± 0.5 | 60 ± 10 | 0.1 ± 0.07 | 25 ± 10 |
| ddCMP and correctly paired DNA | (3 ± 0.4) × 10−4 | 85 ± 3 | ||
| ddCMP and frayed DNA | (4.4 ± 0.5) × 10−3 | 88 ± 2 | ||
| ddCMP, correctly paired DNA, and dNTPs | (8 ± 0.7) × 10−5 | 90 ± 0.5 | ||
| ddCMP, frayed DNA, and dNTPs | (6.3 ± 0.3) × 10−3 | 91 ± 2 |
Rates of removal of the natural nucleotide, dCMP, and the nucleoside analog, ddCMP, from either correctly paired or frayed DNA substrates (denoted in the left-hand column) are shown. In two experiments, 100 μM dNTPs were added, and this addition is also denoted in the left-hand column. There are two rates given for the removal of dCMP because excision occurred in a biphasic manner, while the removal of ddCMP was monophasic.
Of the eight FDA-approved nucleoside analogs, ddC is the best substrate for incorporation by Pol γ and is removed with the least efficiency by the proofreading exonuclease, and these combined effects lead to the marked toxicity seen clinically. The individual contributions from incorporation and exonuclease removal to the in vivo toxicity of ddC can be evaluated by computing a toxicity index as described previously (15, 18). The toxicity index expresses the increase in time required to replicate a given segment of DNA by comparing the rate of incorporation of the analog leading to chain termination to the rate of excision leading to rescue and the resumption of synthesis. The toxicity index is defined by the following equation: toxicity index = (kpol/kexo)([AnaTP]/[dNTP]/4D), where D is the discrimination (the ratio of specificity constants) against the analog triphosphate (AnaTP), kpol is the rate of incorporation of the normal correct nucleotide (dNTP), and kexo is the rate of excision of the analog monophosphate from the primer. The toxicity index for ddC is approximately 50,000 when [AnaTP] equals [dNTP], implying that it would take 50,000-fold longer to replicate the human mitochondrial genome if the concentration of ddCTP were equal to that of dCTP in the mitochondria. Although the toxicity in vivo will be attenuated by the actual [AnaTP]/[dNTP] ratio in the mitochondria, this high calculated toxicity index clearly points to the high potential for mitochondrial toxicity and explains the toxicity of ddC seen clinically.
These data correct our previous attempt to measure the rate of excision of ddC (7), in which we failed to observe any hydrolysis after 12 h of incubation and therefore placed only a lower limit on the rate of excision. It is not known why the previous measurements failed, but it is possible that the enzyme, used at a lower concentration than that in the present study, degraded over the long time course of the experiment or that the large excess of DNA may have bound to the enzyme such that hydrolysis was inhibited. Although the steady-state approach should have provided a valid measurement in this case, the single-turnover experiments reported here with the enzyme in slight excess relative to the DNA circumvent many of the possible difficulties.
From our new data, it appears that both the transfer of the primer from the polymerase to the exonuclease site and the low rate of hydrolysis at the exonuclease site contribute to the slow removal of ddCMP. The rate of hydrolysis of ddCMP is about 360-fold lower than that of the natural nucleotide, suggesting that the 3′-hydroxyl plays a major role in the binding and/or alignment of the 3′ terminus at the exonuclease active site. In vivo, ddC would be correctly base paired in most instances; therefore, the rate of excision would be hindered due to the fact that the rate of primer strand melting and/or transfer from the polymerase site to the exonuclease site was also low. Moreover, the addition of the next correct nucleotide stabilized binding at the polymerase site and lowered the rate of excision fourfold to yield a half-life of about 2.4 h.
The rate of removal of the eight FDA-approved analogs varies from 0.0003 s−1 for ddC to 0.015 s−1 for (−)3TC, as summarized in Fig. 4. For comparison, the rate of removal of correctly paired dCMP is 0.016 s−1. Conclusions about structure-function relationships pertaining to the rates of removal of these nucleoside analogs are highly speculative because there currently is no crystal structure available for Pol γ, but several notable generalizations can be drawn. There is only a 2.3-fold difference between the highest and lowest rates of excision of ddC, d4T, AZT, ddATP, and PMPA, but the rates of removal of CBV, (−)FTC, and (−)3TC are significantly higher. The analogs (−)FTC and (−)3TC are the most efficiently removed; each has a sulfur atom in the ribose ring and an unnatural l-(−) nucleoside configuration, which may destabilize binding at the polymerase site. For example, it is known that mutant forms of HIV reverse transcriptase resistant to 3TC have a structural substitution that is thought to lead to steric blocking of binding at the polymerase site (9).
FIG. 4.

Overview of rates of exonuclease removal by Pol γ. Shown in the bar graph are the relative rates of exonuclease removal of the eight FDA-approved nucleoside analogs (ddATP [ddA] is the metabolically active form of dideoxyinosine) in addition to the natural nucleotide dCMP (dC). The rate of excision of ddC was obtained in the experiment with a correctly paired DNA substrate and no dNTPs present so that parallel comparisons could be made. The numerical value for the rate of exonuclease removal (s−1) is plotted on the x axis and given below the name of each nucleoside analog. Note that the chemical structures have been abbreviated for clarity and that the only nucleoside analog with a modified nucleobase is (−)FTC, which has fluorine on C-5, denoted in the structure as C5F. The most notable differences between each of the structures are in the ribose ring, and the highest rate of removal is 33-fold higher than the lowest rate. The rates of removal of d4T, AZT, ddATP, PMPA, and CBV are from reference 15, that of (−)3TC is from reference 7, and that for (−)FTC is from reference 8.
It is notable that FIAU, which does contain a 3′-hydroxyl, was removed at a rate of 0.06 s−1, six times faster even than (−)3TC (7, 15). Unfortunately, FIAU caused severe lactic acidosis and the deaths of several patients during the initial clinical trial (19). Although the presence of a fluorine atom in the ribose ring inhibits the extension on top of FIAU approximately 500-fold, the rate of polymerization of the next correct nucleotide is still higher than the excision rate (15), leading to the stable incorporation of FIAU.
Efforts at designing more effective nucleoside analogs can incorporate the notion that the rate of removal of these chain terminators by Pol γ may play a significant role in their overall toxicities. Ideally, nucleoside analogs would be designed to mimic a mismatch so that they would be well discriminated against by Pol γ but also be removed at a high rate because primer strand melting and/or transfer would be faster. Furthermore, it appears that a better 3′-OH mimic could increase the actual rate of hydrolysis. It is noteworthy that each of the three most toxic nucleotide analogs, ddCTP, ddATP (the active metabolite of dideoxyinosine), and d4TTP, bind more tightly during polymerization than the corresponding natural nucleotides, and this tighter binding contributes significantly to their specificity constants governing incorporation, leading to increased toxicity (15). Moreover, these three analogs are the slowest to be excised by the proofreading exonuclease. It is reasonable to suppose that their greater affinities at the polymerase site reduce the rate and/or equilibrium constants governing partitioning to the exonuclease site. If so, this one feature, tighter binding at the polymerase site, contributes doubly to their net toxicity by increasing rates of incorporation while decreasing rates of excision. Correlations of toxicity with structure imply that discrimination by the mitochondrial DNA polymerase requires a more substantial steric handle than that afforded by these three simple nucleoside analogs. Other less toxic analogs differ substantially enough in their structural mimic of the deoxyribose moiety and allow significant discrimination by the mitochondrial polymerase, while retaining sufficient similarity to allow efficient incorporation by HIV reverse transcriptase. The ongoing search for new drugs must continue to find a balance between these two opposing structural constraints underlying effectiveness versus toxicity. We remain confident that the assays developed here and in our earlier work (15, 18) provide accurate, quantifiable parameters to assess the effectiveness and toxicity and thus the therapeutic index of new drugs under development.
Acknowledgments
This study was supported by the National Institutes of Health (GM 044613) and the Welch Foundation (F-1604). Kenneth A. Johnson is the president of KinTek Corporation, which provided the RQF-3 rapid quench-flow instruments used in this study.
Footnotes
Published ahead of print on 5 November 2007.
REFERENCES
- 1.Adkins, J. C., D. H. Peters, and D. Faulds. 1997. Zalcitabine. An update of its pharmacodynamic and pharmacokinetic properties and clinical efficacy in the management of HIV infection. Drugs 53:1054-1080. [DOI] [PubMed] [Google Scholar]
- 2.Cammack, N., P. Rouse, C. L. Marr, P. J. Reid, R. E. Boehme, J. A. Coates, C. R. Penn, and J. M. Cameron. 1992. Cellular metabolism of (−) enantiomeric 2′-deoxy-3′-thiacytidine. Biochem. Pharmacol. 43:2059-2064. [DOI] [PubMed] [Google Scholar]
- 3.Chang, C. N., S. L. Doong, J. H. Zhou, J. W. Beach, L. S. Jeong, C. K. Chu, C. H. Tsai, Y. C. Cheng, D. Liotta, and R. Schinazi. 1992. Deoxycytidine deaminase-resistant stereoisomer is the active form of (+/−)-2′,3′-dideoxy-3′-thiacytidine in the inhibition of hepatitis B virus replication. J. Biol. Chem. 267:13938-13942. [PubMed] [Google Scholar]
- 4.Chang, C. N., V. Skalski, J. H. Zhou, and Y. C. Cheng. 1992. Biochemical pharmacology of (+)- and (−)-2′,3′-dideoxy-3′-thiacytidine as anti-hepatitis B virus agents. J. Biol. Chem. 267:22414-22420. [PubMed] [Google Scholar]
- 5.Donlin, M. J., S. S. Patel, and K. A. Johnson. 1991. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30:538-546. [DOI] [PubMed] [Google Scholar]
- 6.Dubinsky, R. M., R. Yarchoan, M. Dalakas, and S. Broder. 1989. Reversible axonal neuropathy from the treatment of AIDS and related disorders with 2′,3′-dideoxycytidine (ddC). Muscle Nerve 12:856-860. [DOI] [PubMed] [Google Scholar]
- 7.Feng, J. Y., A. A. Johnson, K. A. Johnson, and K. S. Anderson. 2001. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J. Biol. Chem. 276:23832-23837. [DOI] [PubMed] [Google Scholar]
- 8.Feng, J. Y., E. Murakami, S. M. Zorca, A. A. Johnson, K. A. Johnson, R. F. Schinazi, P. A. Furman, and K. S. Anderson. 2004. Relationship between antiviral activity and host toxicity: comparison of the incorporation efficiencies of 2′,3′-dideoxy-5-fluoro-3′-thiacytidine-triphosphate analogs by human immunodeficiency virus type 1 reverse transcriptase and human mitochondrial DNA polymerase. Antimicrob. Agents Chemother. 48:1300-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao, H. Q., P. L. Boyer, S. G. Sarafianos, E. Arnold, and S. H. Hughes. 2000. The role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol. 300:403-418. [DOI] [PubMed] [Google Scholar]
- 10.Graves, S. W., A. A. Johnson, and K. A. Johnson. 1998. Expression, purification, and initial kinetic characterization of the large subunit of the human mitochondrial DNA polymerase. Biochemistry 37:6050-6058. [DOI] [PubMed] [Google Scholar]
- 11.Gray, N. M., C. L. Marr, C. R. Penn, J. M. Cameron, and R. C. Bethell. 1995. The intracellular phosphorylation of (−)-2′-deoxy-3′-thiacytidine (3TC) and the incorporation of 3TC 5′-monophosphate into DNA by HIV-1 reverse transcriptase and human DNA polymerase gamma. Biochem. Pharmacol. 50:1043-1051. [DOI] [PubMed] [Google Scholar]
- 12.Hart, G. J., D. C. Orr, C. R. Penn, H. T. Figueiredo, N. M. Gray, R. E. Boehme, and J. M. Cameron. 1992. Effects of (−)-2′-deoxy-3′-thiacytidine (3TC) 5′-triphosphate on human immunodeficiency virus reverse transcriptase and mammalian DNA polymerases alpha, beta, and gamma. Antimicrob. Agents Chemother. 36:1688-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson, A. A., and K. A. Johnson. 2001. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 276:38097-38107. [DOI] [PubMed] [Google Scholar]
- 14.Johnson, A. A., and K. A. Johnson. 2001. Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J. Biol. Chem. 276:38090-38096. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, A. A., A. S. Ray, J. Hanes, Z. Suo, J. M. Colacino, K. S. Anderson, and K. A. Johnson. 2001. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 276:40847-40857. [DOI] [PubMed] [Google Scholar]
- 16.Johnson, A. A., Y. Tsai, S. W. Graves, and K. A. Johnson. 2000. Human mitochondrial DNA polymerase holoenzyme: reconstitution and characterization. Biochemistry 39:1702-1708. [DOI] [PubMed] [Google Scholar]
- 17.Kati, W. M., K. A. Johnson, L. F. Jerva, and K. S. Anderson. 1992. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267:25988-25997. [PubMed] [Google Scholar]
- 18.Lee, H., J. Hanes, and K. A. Johnson. 2003. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 42:14711-14719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenzie, R., M. W. Fried, R. Sallie, H. Conjeevaram, A. M. Di Bisceglie, Y. Park, B. Savarese, D. Kleiner, M. Tsokos, C. Luciano, et al. 1995. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N. Engl. J. Med. 333:1099-1105. [DOI] [PubMed] [Google Scholar]
- 20.Schinazi, R. F., C. K. Chu, A. Peck, A. McMillan, R. Mathis, D. Cannon, L. S. Jeong, J. W. Beach, W. B. Choi, and S. Yeola. 1992. Activities of the four optical isomers of 2′,3′-dideoxy-3′-thiacytidine (BCH-189) against human immunodeficiency virus type 1 in human lymphocytes. Antimicrob. Agents Chemother. 36:672-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skalski, V., C. N. Chang, G. Dutschman, and Y. C. Cheng. 1993. The biochemical basis for the differential anti-human immunodeficiency virus activity of two cis enantiomers of 2′,3′-dideoxy-3′-thiacytidine. J. Biol. Chem. 268:23234-23238. [PubMed] [Google Scholar]
- 22.Whittington, R., and R. N. Brogden. 1992. Zalcitabine. A review of its pharmacology and clinical potential in acquired immunodeficiency syndrome (AIDS). Drugs 44:656-683. [DOI] [PubMed] [Google Scholar]