

Abstract
Methylene blue (MB) has experienced a renaissance mainly as a component of drug combinations against Plasmodium falciparum malaria. Here, we report biochemically relevant pharmacological data on MB such as rate constants for the uncatalyzed reaction of MB at pH 7.4 with cellular reductants like NAD(P)H (k = 4 M−1 s−1), thioredoxins (k = 8.5 to 26 M−1 s−1), dihydrolipoamide (k = 53 M−1 s−1), and slowly reacting glutathione. As the disulfide reductases are prominent targets of MB, optical tests for enzymes reducing MB at the expense of NAD(P)H under aerobic conditions were developed. The product leucomethylene blue (leucoMB) is auto-oxidized back to MB at pH 7 but can be stabilized by enzymes at pH 5.0, which makes this colorless compound an interesting drug candidate. MB was found to be an inhibitor and/or a redox-cycling substrate of mammalian and P. falciparum disulfide reductases, with the kcat values ranging from 0.03 s−1 to 10 s−1 at 25°C. Kinetic spectroscopy of mutagenized glutathione reductase indicates that MB reduction is conducted by enzyme-bound reduced flavin rather than by the active-site dithiol Cys58/Cys63. The enzyme-catalyzed reduction of MB and subsequent auto-oxidation of the product leucoMB mean that MB is a redox-cycling agent which produces H2O2 at the expense of O2 and of NAD(P)H in each cycle, turning the antioxidant disulfide reductases into pro-oxidant enzymes. This explains the terms subversive substrate or turncoat inhibitor for MB. The results are discussed in cell-pathological and clinical contexts.
Methylene blue (MB or MB+) [also known as methylthionine hydrochloride or 3,7-bis(dimethylamino)phenothiazin-5-ium chloride] was the very first synthetic compound to be used as a drug. Paul Ehrlich, who introduced the concept of modern target-based chemotherapy using MB as an example, and Paul Guttmann described the compound as being an effective antimalarial agent (28). Despite its beneficial antimalarial activity, the drug disappeared from the scene because up-and-coming compounds such as chloroquine were more effective; in addition, soldiers resented taking MB because of inevitable but harmless side effects: green or blue urine and bluish sclerae (47, 55).
After MB was revisited as an antimalarial agent (6, 53) and found to be an inhibitor of Plasmodium falciparum glutathione (GSH) reductase (GR) (23), it was studied as a partner drug in antimalarial drug combinations (2, 39, 48, 51).
Compared with other antiparasitic agents, MB is affordable and registered in most countries (as the treatment of choice for acute and chronic methemoglobinemia [16, 17, 36]), and it can be made internationally available in sufficient dosages (51). The price for treating a malaria episode in a child with an MB-containing drug combination would be less than €0.50 (40). Drug resistance has not been reported for MB and could not be provoked in rodent malaria models (52, 53).
Because of its favorable properties, including the differential staining of cell biological structures and protein crystals, medicinal utility, and unique physicochemical and photochemical characteristics, MB has been studied in practically all scientific and technical disciplines (6, 41, 54). The classical review of Clark et al. (20) quoted more than 400 papers on MB written by Bernthsen, Brönsted, Clark, Ehrlich, Feulgen, Guillemont, Hopkins, Koch, Laveran, Marshall, Michaelis, Meyerhof, Neisser, Phelps, Schardinger, Thunberg, Warburg, and Wieland, among others.
However, very few systematic studies have been conducted on the interaction of MB with enzymes and other proteins under quasiphysiological conditions. Accordingly, we studied properties of MB that are relevant for biochemical and cell pharmacological investigations such as UV/Vis absorption and spontaneous reactions of MB with cell physiological reductants. The product of these reactions is leucomethylene blue (leucoMB), the two-electron-reduced form of MB (20, 41).
Our main focus is the interaction of MB with the homodimeric flavoenzymes of the GR family that are present both in the malarial parasite and in the mammalian host cell (4, 9, 34). The physiological reactions catalyzed by these enzymes are as follows (with equation 1 in the case of GR and equation 2 in the case of thioredoxin reductase [TrxR]):
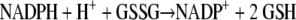 |
(1) |
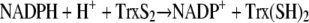 |
(2) |
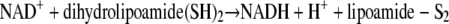 |
(3) |
The reaction in equation 3, which is catalyzed by dihydrolipoamide dehydrogenase (LipDH), is readily reversible. A major cellular function of GR and TrxR is to keep GSH and redoxins in the reduced state and thus to maintain the reducing milieu of cytosolic spaces (10, 34, 43, 50). Mitochondrial LipDH is also of importance for the aerobic energy metabolism (13, 38).
It is worth mentioning that LipDH is still called diaphorase because its diaphoretic activity, the reduction of naphthoquinones and other xenobiotics, was known before its physiological function was discovered (37). Studies on redox-cycling naphthoquinone derivatives where MB was used as a control indicated that MB is not only an inhibitor but also a substrate of P. falciparum GR (12). As reported here, we tested this hypothesis and extended it to the other disulfide reductases of P. falciparum and the human host cells.
Accordingly, the disulfide reductases would catalyze the following reaction:
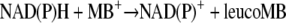 |
(4) |
The uncolored product leucoMB is readily auto-oxidized:
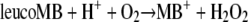 |
(5) |
Thus, the overall reaction would be as follows:
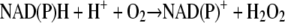 |
(6) |
In equation 6, MB serves as a redox-cycling catalyst whose biochemical and pharmacological activities depend on the presence of disulfide reductase and molecular oxygen.
MATERIALS AND METHODS
Reagents, stock solutions, and enzyme assay buffers.
NAD(P)H, glutathione disulfide (GSSG), and reduced GSH were obtained from Biomol, and lipoamide disulfide was obtained from Sigma. Other chemicals and biologicals, purchased from Roth, Serva, Sigma, and Qiagen, were of the highest available purity.
A 75 mM stock solution of oxidized lipoamide was prepared in ethanol. When 5 to 10 μl of this solution was added to a 1-ml sample, the resulting ethanol concentration was 100 to 200 mM. NADH (10 mM) or NADPH (4 mM) was prepared fresh every day by adding the respective buffers to weighed-out samples and kept at 0°C; when buffers with a pH of <5.2 were used, the NAD(P)H solutions were made up every hour. Enzyme stock solutions (0.2 mM [10 mg/ml]) (see below) were dialyzed against the respective buffers in 50-μl quantities.
GR assay buffer contained 47 mM potassium phosphate, 1 mM EDTA, and 200 mM KCl (adjusted to pH 6.9); TrxR assay buffer contained 100 mM potassium phosphate and 2 mM EDTA (adjusted to pH 7.4), and LipDH assay buffer contained 50 mM potassium phosphate and 1 mM EDTA (adjusted to pH 7.4).
Disulfide reductase assay buffer contained 100 mM potassium acetate and 200 mM KCl (adjusted to pH 5.0).
MB.
MB (CAS 61-73-4; EC 200-515-2) is 3,7-bis(dimethyl-amino)-phenol-thiazin-5-ium chloride. MB as a hydrochloride (methylthioninium chloride from Roth) has an Mr of 320; Sigma sells MB trihydrate (Mr 373.9). MB (1%) (10 mg/ml MB hydrochloride trihydrate) is a 26.7 mM aqueous solution. Uncharged leucoMB has an Mr of 285. Stock solutions of 1 mM MB were kept at room temperature in dark bottles for up to 1 week.
MB was quantitated using a method described previously by Clark et al. (20). Alternatively, for routine determinations of MB+ in stock solutions, we diluted aliquots with 100 mM potassium acetate-200 mM KCl adjusted to pH 5 and measured the absorption at 613 nm. An absorption coefficient of 40.0 mM−1 cm−1 was found to apply for an MB concentration of 2 to 50 μM in the pH range of 2 to 7.5. Occasionally, we observed light-dependent bleaching of MB (20, 41) and ascribed this to the presence of EDTA in our buffers. Although the effect was not reproducible, we repeated the respective experiments in EDTA-free buffer. Mills and Wang previously discussed whether MB itself can act as sacrificial electron donor when exposed to light (41).
Disulfide reductases and other proteins.
The enzymes GR (23), TrxR (29, 42), and mitochondrial LipDH of P. falciparum (38) were expressed in recombinant forms, purified, and assayed at 25°C as described previously. For the mammalian counterparts, we used recombinant human GR (44), human TrxR (from placenta as well as the recombinant enzyme) (24, 27), and pig heart mitochondrial LipDH (obtained from Sigma), the best-studied mammalian LipDH (4, 56).
Expression plasmids in GR-free Escherichia coli SG5 cells of human GR mutants lacking one or both active-site cysteine residues were kindly provided by Rimma Iozef, Heidelberg University. The recombinant GR species including the yellow Cys63Ala mutant, the yellow double mutant Cys58Ala/Cys63Ala, and the relatively unstable orange-colored Cys58Ala mutant were purified as described previously for recombinant wild-type GR (44). As the mutants were found to have no detectable GSSG reduction activity, they were identified by their color during the purification procedure. An ɛ value of 11.3 mM−1 cm−1 at a λmax,vis of 455 nm was assumed for these mutants.
P. falciparum thioredoxin 1, which is a substrate of both TrxRs, was prepared as previously described (29). Human thioredoxin 1 is not suitable for routine measurements; consequently, we used the His-tagged Cys73Ser mutant of this protein (H. Merkle and S. Gromer, unpublished data). Drosophila melanogaster thioredoxin 2 (8) served as a general eukaryotic thioredoxin.
Determination of second-order rate constants at 25°C.
The rate for the reaction NAD(P)H + MB+→NAD(P)+ + leucoMB was measured in 1-ml cuvettes. Starting out with 200 μM NADPH or NADH (ɛ = 6.22 mM−1 cm−1 at 340 nm) in phosphate buffer at pH 7, MB (10 to 50 μM) was added, and the rate of disappearance of NADPH or NADH was measured. As the absorption at 613 nm did not change, we assumed that leucoMB was auto-oxidized so rapidly that the MB concentration remained constant and that the contribution of leucoMB to the absorption at 340 nm can be neglected.
When the reduction of MB by GSH, thioredoxin, or dihydrolipoamide was studied, the thiols were kept in the reduced form using only 10 mU ml−1 disulfide reductase and 100 to 200 μM NADPH or, in the case of LipDH, 100 to 200 μM NADH. Otherwise, the procedure corresponded to that used for the GHOST assay (25), using MB instead of GSSG as the final oxidant of NADPH or NADH. The spontaneous oxidation of NAD(P)H by MB and the enzyme-catalyzed reduction of MB (see below) were accounted for.
In an alternative procedure, the rate of oxidation of SH groups by MB was measured by determining the residual thiol concentration at given time points using a method described by Ellman (22). Dithioerythritol (10 μM) (20 μM thiol), 40 μM MB, and incubation times of up to 120 min at 25°C represent optimal conditions.
NAD(P)H auto-oxidase activities of disulfide reductases.
At 25°C in the presence of 100 μM NAD(P)H and atmospheric O2, the turnover of NAD(P)H was recorded at 340 nm first in the absence and consecutively in the presence of the enzyme studied, and the inherent NADPH auto-oxidase activity, corrected for the spontaneous NAD(P)H oxidation rate, was calculated (13). At pH 5.0, the apparent NADPH auto-oxidase activity of P. falciparum GR was found to be 40 times higher than that at pH 7.0.
MB reduction activity of disulfide reductases.
In a standard experiment, 25 μl 4 mM NADPH was added to 940 μl assay buffer, leading to an absorption of 0.62 at 340 nm. This was followed by 3 to 30 μl 1 mM MB in water. After 5 min, the rate ΔAspont/min, representing the spontaneous reaction between NADPH and MB, was measured. Subsequently, we added 5 μl of 0.2 mM (11 mg/ml) GR. This led to ΔAtotal/min, the rate of the maximal decrease in absorption. As MB is regenerated by the auto-oxidation of leucoMB, the oxidation of NADPH proceeded until it was completely consumed. In a separate experiment, a sample containing 970 μl buffer, 25 μl 4 mM NADPH, and 5 μl concentrated enzyme solution was mixed. The rate of absorbance decrease (ΔANOX/min) represents the intrinsic NADPH auto-oxidase activity of the enzyme. The oxidation rate of NADPH due to the reduction of MB is given by the following equation:
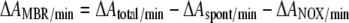 |
(7) |
The reaction of H2O2, resulting from the auto-oxidation of leucoMB, with enzyme-bound NADPH (57) and the auto-oxidation of NADPH must not be considered at pH 7 but certainly should be considered at acidic pH values.
Enzymatic bleaching of MB under aerobic conditions at pH 5.0.
MB can be reduced to leucoMB using P. falciparum TrxR or GR at pH 5.0. The absorption at 613 nm was zeroed for 950 μl buffer at pH 5.0. Subsequently, we added 20 μl 1.00 mM MB in H2O and measured an absorbance of 0.830. Twenty-five microliters of 4 mM NADPH in buffer at pH 5 and 5 μl 10 mg/ml TrxR or GR were then added. After 1 min, the absorbance had fallen to a minimum value of 0.005 and stayed there for approximately 3 min. After NADPH had been consumed, the absorbance at 613 nm rose again, but this could be reversed by adding NADPH (final concentration, 50 μM).
RESULTS
Physicochemical properties of MB at pH 7.0.
Most physical and chemical properties of MB have been studied under nonphysiological conditions (20, 41). In contrast, our data here refer to an MB solution in 50 to 100 mM phosphate buffers at pH 7 at 25°C or in acetate buffer at pH 5.0 (Table 1). The latter buffer imitates the milieu of the parasite's digestive vesicles.
TABLE 1.
Biochemically and pharmacologically relevant characteristics of MB and leucoMBa
| Parameter | Value(s) | Reference(s) and/or source |
|---|---|---|
| Redox potential (mV) | ||
| MB+/leucoMB | +11 | 20, 41 |
| MB+/MB radicalb | −230 | |
| pKa | ||
| MB | ∼0 | 41, 51 |
| leucoMB | 4.5, 5.8 | |
| MB radical | 9 | |
| Solubility | ||
| MB trihydrate at pH 7.0 [mg/ml (mM)] | 20 (53.4) | 20 and this report |
| LeucoMB (μM) | <50 | |
| ε value (mM−1 cm−1)c | ||
| MB | ||
| 340 nm | 3.90 | This report |
| 455 nm | 1.1 | 45, 46 |
| 613 nm | 40.0 | 45, 46 |
| leucoMB | ||
| 258 nm | 17.2 (peak) | This report |
| 320 nm | 4.0 (peak) | |
| 340 nm | 3.30 | |
| Between 380 and 800 nm | <0.1 | |
| Fluorescence (nm) | 664 (excitation), 682 (emission) | 21 |
| Monomer-dimer equilibrium of MB (μM) | 170 < Kdiss < 252 | 3, 41, 51 |
| Binding of MB to bovine serum albumin (Kdiss) (μM) | 2.90 | 58 |
| Adsorption to surfaces | Langmuir data | 20, 41, 47 |
| EC50 of MB against P. falciparum in vitro (nM) ± SDd | 6.5 ± 1.8 | 2 |
| Therapy of malaria in children using MB (mg/kg of body wt) orally over 3 days | 36-72 | 39 |
| Treatment of hereditary methemoglobinemia using MB (mg) orally per day | 250 | 17 |
Unless stated otherwise, the in vitro data refer to 100 mM phosphate buffer at pH 7.0.
Two MB radicals disproportionate to give MB and leucoMB. This explains why erroneous midpoint potentials of less than −200 mV have also been reported for the MB+/leucoMB pair, e.g., by Atamna et al. and Schirmer et al. (6, 51).
MB exhibits absorption peaks at 250, 292, and 663 nm with ε values of 18, 38, and ∼75 mM−1 cm−1, respectively. leucoMB showed peaks at 210 nm, 258 nm, and 320 nm, with the ε values being >60, 17.4, and 4.0 mM−1 cm−1, respectively.
EC50, 50% effective concentration.
For biological and pharmacological measurements, the absorption coefficients of MB at 340 nm but also at 613 nm and 663 nm are relevant (Table 1). Absorbance was found to be proportional to concentration up to 50 μM at 340 nm and at 613 nm but only up to 3 μM at 663 nm. The major reason for this is that the MB monomer has a higher absorbance at 663 nm (ɛ = 75 mM−1 cm−1) than the dimer (45). As the Kdiss of the dimer is approximately 200 μM in phosphate buffers at pH 7.0, one expects low ɛ values when measurements are done at total MB concentrations above 50 μM. This explains, e.g., the low ɛ value of 45 mM−1 cm−1 reported previously (51). Under aerobic conditions at physiological pH, leucoMB is auto-oxidized rapidly so that the absorptions at 340 nm or at 613 nm do not change while MB is transiently reduced.
Reactions of MB with cellular reductants.
MB reacts spontaneously with NADPH according to equation 4. The second-order rate constant for the reaction was found to be 3.6 ± 0.2 M−1 s−1 in TrxR assay buffer at pH 7.4 and 6.6 ± 0.3 M−1 s−1 in GR assay buffer at pH 6.9. With 100 μM NADPH and 25 μM MB, we observed a decrease in absorbance of 0.0062 min−1, corresponding to a turnover of 1 μM NADPH per min. Using NADH instead of NADPH, the reaction rates were very similar (Table 2). This is also true when the rates were measured in anaerobic cuvettes.
TABLE 2.
Second-order rate constants for the reaction of MB with biologically relevant reductants under aerobic conditions
| Reductant | k (M−1 s−1) | Buffer system used (pH) |
|---|---|---|
| NADH | 3.8 | LipDH assay buffer (7.4) |
| 2.5 | 0.3 M ethanol and 1 mM LipS2 in LipDH assay buffer | |
| NADPH | 6.6 | GR assay buffer (6.9) |
| 8.0 | GR assay buffer (6.5) | |
| 3.6 | TrxR assay buffer (7.4) | |
| GSH | <0.5a | GR assay buffer (6.9) |
| P. falciparum thioredoxin | 8.5 | TrxR assay buffer (7.4) |
| D. melanogaster thioredoxin | 27.0 | TrxR assay buffer (7.4) |
| d,l-Dihydrolipoamide | 53.1 | LipDH assay buffer (7.4) |
| 1,4-Dithioerythritol | 12.4 | TrxR assay buffer (7.4) |
For the slow reaction 2 GSH + MB+→leucoMB + GSSG + H+, no rate constant could be determined. The half-life of GSH was approximately 6 h when 100 μM GSH was incubated with 25 μM MB (31).
According to previous reports, MB can be reduced by the monothiol GSH (31); an addition-displacement mechanism for the reaction of thiols with MB was suggested previously by Kosower (32). While we did not observe any significant oxidation of GSH at pH 7, the following reaction could be measured, with the second-order rate constants being 8.5 M−1 s−1 for P. falciparum thioredoxin 1, 27.0 M−1 s−1 for D. melanogaster thioredoxin 2, and 53 M−1 s−1 for dihydrolipoamide:
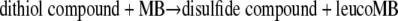 |
(8) |
Using dithiol compounds, it is possible to produce leucoMB under quasiphysiological conditions; in contrast, the famous blue bottle experiment (1) employs strongly alkaline solutions. After varying a number of parameters (pH, ionic strength, and the nature and concentration of reductants), we found the following experimental conditions to be optimal. An anaerobic cuvette (1 ml) containing 7 mM dithioerythritol and 30 μM MB in 100 mM potassium phosphate adjusted to pH 8.0 was flushed with N2 for 30 min. The absorptions at 613 nm dropped from 1.2 to less than 0.01, which indicated that more than 99% of MB had been reduced by dithioerythritol.
Enzyme-catalyzed conversion of MB to leucoMB under aerobic conditions.
The problem of keeping leucoMB in the reduced state by the NADPH/disulfide reductase system in the presence of O2 remained to be solved. The production of leucoMB under aerobic conditions is of interest for the study of further properties of this compound, particularly with the goal of using leucoMB (methylene white) as an uncolored drug formulation.
At neutral pH, the auto-oxidation rate for leucoMB is so fast that very high disulfide reductase activities must be present to reinforce MB reduction and to stabilize the product leucoMB. This is probably the case in mitochondrion-containing cells where the activities of the disulfide reductases are in the range of 1 to 10 units/ml cytosolic space and the oxygen concentration is below 10 μM (34, 43). For practical purposes, we chose pH 5.0 under aerobic conditions for our in vitro experiments. At this pH, MB was quantitatively bleached, that is, reduced, by NADPH in a P. falciparum GR- or TrxR-catalyzed reaction; this was shown by the disappearance of the absorption at 613 nm. The resulting leucoMB was found to be kinetically stable at pH 5.0, but it was visibly reoxidized after the NADPH had been consumed. The addition of NADPH (final concentration, 50 μM) led to bleaching again, and this cycle could be repeated. Thus, the enzymatic reduction of MB can be considered to be a quasiphysiological analogue of the chemical blue bottle experiment (1).
On the basis of these observations, one may approach enzyme-stabilized pharmaceutical formulations of leucoMB in colorless antimalarial syrups, which are preferred to blue ones by some patients (2, 39). The nature of the formulation will of course not change the systemic redox equilibria between MB and leucoMB. As measured in anaerobic cuvettes, leucoMB has no absorption in the visible range; the absorption coefficient at 258 nm is 17.4 mM−1 cm−1, and it is 3.30 mM−1 cm−1 at 340 nm. leucoMB is kinetically unstable in the presence of micromolar concentrations of O2 and auto-oxidizes readily. The midpoint potential of the pair MB/leucoMB is +10 mV (Table 1). In contrast to MB, leucoMB has two biochemically relevant pKa values between pH 4 and pH 6 (Table 1), which implies that it is partially charged at pH 7. Nevertheless, leucoMB is poorly soluble at neutral pH (<40 μM at 25°C) and tends to precipitate. In many redox reactions, leucoMB results from the two-electron reduction of MB; alternatively, MB can be reduced by one electron, which results in the uncharged MB radical. Two molecules of this species readily disproportionate to give MB and leucoMB (41). In mitochondrion-containing cells, the O2 concentration is probably 100-fold lower than that in erythrocytes, and the ratio of NADPH to NADP+ is higher than 10. This means that the predominant species in cytosolic spaces is uncolored leucoMB. In urine that is stained blue or green by MB, a mixture of MB and leucoMB is excreted (47).
MB as an inhibitor of disulfide reductases.
The inhibitory effects of MB on the physiological reactions of the enzymes studied (equations 1 to 3) are summarized in Table 3. As a case in point, MB is an inhibitor of recombinant Plasmodium GR with a 50% inhibitory concentration (IC50) value in the low micromolar range (23). The type of inhibition could not be unambiguously determined, but it was not competitive. Assuming that it is noncompetitive (23) rather than uncompetitive (12), the IC50 values in Table 3 correspond to the Ki values. When MB acts as an inhibitor of human GR, it is probably bound at the inhibitory site in the central cavity between the two subunits (49, 59).
TABLE 3.
Activities of the disulfide reductases studied as MB reductasesa
| Enzyme | Subunit Mr | KM of MB (μM) | kcat for MB (s−1) | kcat/KM (M−1 s−1) | IC50 for MB (μM) | KM for cogn. substrate (μM) | kcat for cogn. substrate (s−1) | NADPH auto-oxidase (s−1) |
|---|---|---|---|---|---|---|---|---|
| Human GR | 52.4 | 6.3 | 0.03 | 4,760 | 16.4 | 65 | 200 | 0.003 |
| Human GR C58A | 52.4 | 17 | 0.13 | 7,650 | NA | NA | NA | 0.005 |
| Human GR C63A | 52.4 | 56 | 0.62 | 11,100 | NA | NA | NA | 0.070 |
| Human GR C58A/C63A | 52.4 | 67 | 0.60 | 8,900 | NA | NA | NA | 0.065 |
| Human TrxR1 | 55.2 | 95 | 10.00 | 105,000 | 30 | 10 | 25 | 0.001 |
| Porcine LipDH | 55.0 | 28 | 3.40 | 121,000 | >1,000 | >1,000 | 290 | 0.220 |
| P. falciparum GR | 57.2 | 50 | 2.50 | 50,000 | 5.4 | 83 | 150 | 0.030 |
| P. falciparum TrxR | 60.3 | 68 | 6.30 | 88,200 | 59 | 2-10 | 50 | 0.006 |
| P. falciparum mtLipDH | 57.2 | 49 | 7.40 | 151,000 | >1,000 | >1,000 | 320 | ND |
GR was assayed in phosphate buffer at pH 6.9, TrxR was assayed at pH 7.4, and LipDH at pH 7.3. cogn., cognate; NA, for not applicable since the human GR mutants had no measurable activity when assayed with GSSG; ND, not determined.
In contrast to GR and TrxR, the LipDH orthologues from P. falciparum and mammals were not inhibited by MB, with the IC50 values being above 1 mM. This is in keeping with reports that dihydrolipoamide dehydrogenases are not inhibited by their diaphorase substrates (13, 18, 19).
MB as a substrate of disulfide reductases.
The potential of MB as a substrate for the enzymes investigated is shown in Table 3; for this purpose, enzyme activities were measured with the physiological disulfide substrate and the artificial substrate MB in parallel. Assays with MB as a substrate were performed with 1 U enzyme per ml, whereas 10 mU/ml was employed for assays with the cognate substrate. It should be noted that in situ the enzyme concentrations (>1 U ml−1) (34, 43) are indeed >100-fold higher than those normally used in enzyme kinetic studies in vitro (10 mU/ml).
MB reduction by NAD(P)H was measured using the enzymatic optical test at 340 nm for NAD(P)H oxidation. At this wavelength, ɛ values were determined to be 3.90 mM−1 cm−1 for MB and 3.30 mM−1 cm−1 for leucoMB (Table 1). The antidromic contribution of the product leucoMB to the overall absorbance decrease was neglected, as under aerobic conditions leucoMB is auto-oxidized at a high rate so that the concentration of MB remains constant and the concentration of leucoMB is very low. The reaction proceeded until all NAD(P)H was consumed; the steepest part of the slope was taken for determining the reaction rate.
Corrections for the spontaneous reaction of MB with NAD(P)H (Table 2) and for the NAD(P)H auto-oxidase activities of the respective enzymes were accounted for as described in Materials and Methods.
Dihydrolipoamide dehydrogenase, reaching a catalytic efficiency of more than 105 M−1 s−1, was found to be an excellent catalyst for MB reduction (Table 3). The catalytic efficiency of an enzyme, kcat/KM, often represents the second-order rate constant for the rate-limiting step. This illustrates that the disulfide reductases act as efficient catalysts for the reduction of MB by NAD(P)H, with the spontaneous reaction rate (k = 3.6 M−1 s−1) being enhanced 103- to 105-fold in the presence of the enzymes.
In conclusion, MB not only inhibits the natural reactions of the enzymes but also serves as a subversive substrate since the product leucoMB is auto-oxidized back to MB with the concomitant production of reactive oxygen species (Fig. 1). The possible cell biochemical and cell-pathological consequences are discussed below.
FIG. 1.

MB as a redox-cycling substrate of P. falciparum GR. The disulfide reductase catalyzes the reduction of MB by NADPH. The resulting leucoMB, a most efficient auto-oxidator, is then oxidized by O2. From a cell pharmacologic perspective, each reaction cycle, catalyzed by the MB-enzyme ensemble, leads to the consumption of NADPH and O2 and to the production of parasitotoxic reactive oxygen species, predominantly to H2O2.
Which active-site groups of the enzymes are involved?
As dithiols are able to reduce MB (Table 2), the question of which redox active group(s) in disulfide reductases reduces MB arises; both the flavin of the prosthetic group flavin adenine dinucleotide (FAD) and the active-site dithiol pair, e.g., Cys58 and Cys63 in human GR, appear to be good candidates.
To study the role of the thiols, we prepared mutants lacking either cysteine residue (Cys58Ala and Cys63Ala) or both cysteines (Cys58Ala/Cys63Ala). Compared to the wild type, with these mutants the kcat values for MB reduction increased by a factor of more than 20 (Table 3). This supports the notion that it is the flavin rather than the thiols that mediates MB reduction by disulfide reductases and other flavoenzymes. The double mutant Cys58Ala/Cys63Ala can be visibly reduced by NADPH, thereby losing the absorption of oxidized flavin at around 455 nm, and it can be reoxidized by adding MB (Fig. 2A and B). Assuming that this reoxidation represents the following reaction, we estimated the second-order rate constant k, correcting for instantaneous auto-oxidation of the product leucoMB and the intrinsic auto-oxidation activity of protein-bound FADH−:
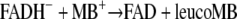 |
(9) |
The estimated value of 7,500 ± 1,000 M−1 s−1 compares reasonably well to the kcat/KM value of the flavoprotein-catalyzed reaction NADPH + H+ + MB→NADP+ + leucoMB, with a value of 8,900 M−1 s−1 (Table 3), indicating that the oxidation of FADH− by MB is probably the rate-limiting step in the overall enzyme-catalyzed reaction.
FIG. 2.

Reduction of a flavoprotein (human GR mutant A58/A63) with NADPH and reoxidation by O2 or by MB in assay buffer at pH 6.9. (A) The black curve shows the original spectrum of the oxidized flavoprotein (8.2 μM) with an absorption maximum at 455 nm. The protein-bound FAD was then reduced with 200 μM NADPH under aerobic conditions to give FADH−. This resulted in a decrease in the absorption at 455 nm by 78% (not shown). Subsequent auto-oxidation led to partially reoxidized protein-bound flavin and indirectly to the production of NADP+ (gray curve). Here, the characteristic broad peak at around 680 nm represents the complex between protein-bound FADH− and NADP+ (33). The original spectrum of oxidized protein-bound flavin (black curve) was recovered after 20 min by auto-oxidation and was 10 times faster by oxidation with 3 μM MB (Fig. 2B). (B). Traces representing the kinetics of FADH− reoxidation by MB. Mutant flavoprotein (9.2 μM) had been incubated under aerobic conditions in GR assay buffer containing 0 to 3 μM MB and monitored at 455 nm in a total volume of 700 μl. The addition of NADPH at 100 μM led to a decrease in the absorption at 455 nm. Reoxidation took place according to the equation protein-bound FADH− + MB+→protein-bound FAD + leucoMB, with leucoMB being auto-oxidized instantaneously. The highest rate of absorbance increase was 0.120 min−1. Using a Δɛ of 9.0 mM−1 cm−1 for the difference between reduced flavin and oxidized flavin and correcting for the auto-oxidation of reduced flavin, the second-order rate constant for the reaction of equation 9 was estimated to be 7,500 ± 1,200 M−1 s−1 (15, 33). Solid curve, 3 μM MB; dashed-dotted curve, 0.6 μM MB; dotted curve, 0 μM MB.
In contrast to the double mutant, wild-type human GR can be reduced only with extremely low yield to the so-called EH4 form, where not only the catalytic-site cysteines but also the flavin is present in the two-electron reduced form. According to the literature, it is an EH4 form of a given disulfide reductase that reduces nonphysiological substrates such as quinones and O2 as a “diaphorase” (4, 7). This EH4 form is readily accessible in LipDH and in P. falciparum GR but not in human GR (5, 15, 56).
These observations for the EH4 forms, together with our results for Cys-free mutants, support the notion that the reduced flavin ring is responsible for MB reduction and diaphorase activity in general by disulfide reductases. The site of contact between reduced flavin and MB remains to be established. Crystallographic studies on human GR soaked with MB described by Zappe (59) revealed an MB binding site close to the contact zone between the nicotinamide of NADPH and the flavin (Fig. 3). Those crystallographic experiments must be extended to other disulfide reductases.
FIG. 3.

Scheme of dimeric human GR representing the active-site geometry of disulfide reductases. There are two identical active sites per homodimeric enzyme. The dimer interface is shown as a diagonal line, with a central filled circle representing the twofold symmetry axis as viewed from above. The reducing equivalents flow from the nicotinamide of NADPH via the flavin to the active-site disulfide, which is reduced to give the catalytic dithiol. Subsequent dithiol-disulfide exchanges lead to the reduction of the substrate GSSG (R1-S-S-R2, with R1 equaling R2). Two binding sites for MB have been identified by crystallography at low resolution, an intersubunit cavity at the twofold axis and a site close to the nicotinamide-binding site (30, 59). In the case of LipDH, the disulfide of lipoamide binds as R1-S-S-R2 at the disulfide site; in the case of TrxR, it is the peripheral disulfide of the other subunit which binds here.
MB reduction activity versus other enzyme activities of disulfide reductases.
As shown in Table 3, there are no obvious correlations when MB and the cognate disulfide are compared as substrates of disulfide reductases. However, with respect to the NADPH auto-oxidase activity of different GR species, there is a tendency that the higher the NADPH auto-oxidase activity, the higher is the MB reductase activity of a GR species. This is most obvious when the kcat values of wild-type human GR and the three mutants were measured or when the human enzyme and the orthologue from P. falciparum were compared.
DISCUSSION
MB as a redox-cycling agent.
Our data suggest that besides its inhibitory potential, MB has interesting characteristics as a subversive substrate of different well-established drug target proteins. The term subversive substrate (or turncoat inhibitor) is defined from a pharmacological perspective: it indicates that this compound changes the physiological function of the enzyme to the opposite (13, 34). The antioxidant disulfide reductases reported here are thiol-producing enzymes guarding the reducing milieu of cytosolic spaces. In the presence of MB, they turn into pro-oxidant H2O2-producing enzymes and challenge the reducing milieu that they are meant to protect (9, 34, 50). As shown in Fig. 1, the pharmacologic mechanism of MB is that of a redox-cycling agent. In each cycle, MB is reduced by NADPH or NADH in an enzyme-dependent manner. The resulting leucoMB undergoes rapid auto-oxidation, with the products being MB and H2O2. In balance, each catalytic cycle leads to the loss of NAD(P)H and O2, while H2O2, a reactive oxygen species, is produced. (There were no indications that radicals like MB· and superoxide play a role in the redox cycling of MB under the conditions described here, but this will be studied further for each disulfide reductase [13]).
Furthermore, NAD(P)H and O2, which are needed for the pathogen's metabolism, are consumed in the pathological reaction cycles, and the NADPH-to-NADP+ ratio is likely to be affected. GSSG, the physiological substrate of GR, is expected to be more slowly reduced, which leads to toxic effects of GSSG. In addition, there is less GSH available in the parasite as a substrate of GSH S-transferase for the detoxification of heme and other lipophilic compounds (10).
When extrapolating these findings to in vivo conditions, we assume that the GR activity in P. falciparum is 3 to 10 U/ml under Vmax conditions (2, 33); the kcat for MB is 1.6% of that for GSSG, which is approximately 100 mU/ml. With a concentration of MB of 30 μM and a KM value for MB of 50 μM (Table 3), the turnover rate can be estimated to be 40 mU/ml or 40 μM/min at 25°C and 100 μM/min at 40°C, the temperature of a malaria attack. It should also be noted that the concentrations of the cognate substrates in the disulfide form are likely to be low because a high dithiol-to-disulfide ratio is maintained by the disulfide reductases. Consequently, when assuming a concentration of 0.5 μM for P. falciparum TrxR in situ, this enzyme is probably as important for turning over MB as P. falciparum GR. In contrast, human GR has a kcat value of only 0.03 s−1, which indicates that under MB therapy, less than 0.5% of glucose consumption of healthy and parasitized erythrocytes will be used for maintaining the MB-driven redox cycle in erythrocytes. With respect to human TrxR, this enzyme is not present in erythrocytes (26, 35).
Binding properties of MB.
Quantitative biochemical and pharmacological studies can be complicated by the binding properties of MB (Table 1). MB dimerizes, with the dissociation constant being 170 μM (3). This means that at 10 μM total MB, 90% is monomeric, but at 100 μM, only 59% is present as a monomer. MB is reversibly bound to proteins in an unspecific or a specific manner. Indeed, this property is used in protein crystallization experiments. If the emerging microcrystals are stainable with MB, they are most likely to consist of protein and not of buffer components. Crystals of colorless proteins turn blue, but the crystals of the yellow FAD-containing disulfide reductases turn green when incubated with MB (Fig. 4).
FIG. 4.

Protein crystals of human GR with (A) and without (B) bound MB. In the oxidized form, GR is a yellow enzyme; thus, the color of the enzyme and the blue of MB yield green crystals.
MB binds to bovine serum albumin with a stoichiometry of 1:1, with the dissociation constant being 2.90 μM (58). If the value for human serum albumin is similar, the plasma concentration of free MB is only 60 nM when the total MB concentration is 10 μM and the total albumin concentration is 500 μM and when competing ligands of albumin are absent. The binding of MB to solid surfaces is notorious (20, 41, 47). We recommend soaking glassware and quartz cuvettes after experiments with MB extensively in the buffer used, preferably overnight; otherwise, the glassware will give off MB in visible amounts in subsequent experiments.
When parasitized red blood cells and normal erythrocytes are incubated together in MB-containing solution, the drug becomes concentrated selectively in the parasitized erythrocytes (2). The mechanism that leads to this sequestration is not clear but may be due to the binding of MB to structures in the parasite. Another hypothesis is that MB is reduced to uncharged leucoMB, which easily permeates the membrane of the digestive vesicles and is auto-oxidized to the MB cation and is thus trapped in the vesicles. It should be noted that this is not a weak base effect but a redox mechanism.
One major reason for the renewed interest in MB as an antiparasitic compound is that it fulfils the criteria for a BONARIA drug (11, 34). In the acronym BONARIA, BON stands for safe and efficacious, A stands for affordable, R stands for registered, and IA stands for internationally accessible. MB is a registered drug that has been used in clinical work for many decades, mainly in pediatric clinics as an antidote against methemoglobinemia-inducing toxic compounds (16, 51). This means that the extreme costs of drug development, a major obstacle when new drug programs against diseases of the poor are considered, can be reduced. Due to the fact that parasite resistance develops faster than drug development, new approaches for the treatment of parasitic diseases are urgently needed. To counteract drug resistance development, a general recommendation is to search for drug combinations rather than to use a single drug. Along these lines, our group found synergistic effects of MB in combination with artemisinin derivatives when drug combinations were tested against P. falciparum in culture (2). Additionally, a combination of MB and chloroquine (BlueCQ) has been tested in vitro and in clinical trials in Burkina Faso (39, 40). Due to the rapid spread of chloroquine-resistant strains, also in West Africa, chloroquine is not a suitable partner drug anymore. Consequently, other MB-containing antimalarial drug combinations are being tested in clinical pilot studies (A. Zoungrana, O. Müller, R. H. Schirmer, et al., unpublished data).
Flavoenzymes of trypanosomes and leishmania are also of interest as targets of MB since this compound has been shown to be effective against African trypanosomes in vitro (14). When tested against Trypanosoma cruzi enzymes, MB was found to be a subversive substrate of LipDH and a strong inhibitor of the trypanosomatid-specific disulfide reductase trypanothione reductase (R. L. Krauth-Siegel, S. Gromer, et al., unpublished data).
Acknowledgments
We thank Irene König and Uschi Göbel for their excellent technical assistance.
The support provided by the Deutsche Forschungsgemeinschaft (Be 1540/4-4 and subproject B2 of the SFB 544 Control of Tropical Infectious Diseases) is gratefully acknowledged.
Footnotes
Published ahead of print on 29 October 2007.
REFERENCES
- 1.Adamcikova, L., and P. Sevcik. 1998. The blue bottle experiment—simple demonstration of self-organisation. J. Chem. Educ. 75:1580. [Google Scholar]
- 2.Akoachere, M., K. Buchholz, E. Fischer, J. Burhenne, W. E. Haefeli, R. H. Schirmer, and K. Becker. 2005. In vitro assessment of methylene blue on chloroquine-sensitive and -resistant Plasmodium falciparum strains reveals synergistic action with artemisinins. Antimicrob. Agents Chemother. 49:4592-4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonov, L., G. Gergov, V. Petrov, M. Kubista, and J. Nygren. 1999. UV-Vis spectroscopic and chemometric study on the aggregation of ionic dyes in water. Talanta 49:99-106. [DOI] [PubMed] [Google Scholar]
- 4.Argyrou, A., and J. S. Blanchard. 2004. Flavoprotein disulfide reductases: advances in chemistry and function. Prog. Nucleic Acid Res. Mol. Biol. 78:89-142. [DOI] [PubMed] [Google Scholar]
- 5.Argyrou, A., G. Sun, B. A. Palfey, and J. S. Blanchard. 2003. Catalysis of diaphorase reactions by Mycobacterium tuberculosis lipoamide dehydrogenase occurs at the EH4 level. Biochemistry 42:2218-2228. [DOI] [PubMed] [Google Scholar]
- 6.Atamna, H., M. Krugliak, G. Shalmiev, E. Deharo, G. Pescarmona, and H. Ginsburg. 1996. Mode of antimalarial effect of methylene blue and some of its analogues on Plasmodium falciparum in culture and their inhibition of P. vinckei petteri and P. yoelii nigeriensis in vivo. Biochem. Pharmacol. 51:693-700. [DOI] [PubMed] [Google Scholar]
- 7.Bauer, H., K. Fritz-Wolf, A. Winzer, S. Kühner, S. Little, V. Yardley, H. Vezin, B. Palfey, R. H. Schirmer, and E. Davioud-Charvet. 2006. A fluoro analogue of the menadione derivative 6-[2′-(3′-methyl)-1′,4′-naphthoquinolyl]hexanoic acid is a suicide substrate of glutathione reductase. Crystal structure of the alkylated human enzyme. J. Am. Chem. Soc. 128:10784-10794. [DOI] [PubMed] [Google Scholar]
- 8.Bauer, H., S. M. Kanzok, and R. H. Schirmer. 2002. Thioredoxin-2 but not thioredoxin-1 is a substrate of thioredoxin peroxidase-1 from Drosophila melanogaster: isolation and characterization of a second thioredoxin in D. melanogaster and evidence for distinct biological functions of Trx-1 and Trx-2. J. Biol. Chem. 277:17457-17463. [DOI] [PubMed] [Google Scholar]
- 9.Becker, K., S. Koncarevic, and N. H. Hunt. 2005. Oxidative stress and antioxidant defense in malarial parasites, p. 365-383. In I. Sherman (ed.), Molecular approaches to malaria. ASM Press, Washington, DC.
- 10.Becker, K., S. Rahlfs, C. Nickel, and R. H. Schirmer. 2003. Glutathione—functions and metabolism in the malarial parasite Plasmodium falciparum. Biol. Chem. 384:551-566. [DOI] [PubMed] [Google Scholar]
- 11.Becker, K., and R. H. Schirmer. 2007. Antioxidative Enzyme des Malariaerregers als Drug-Targets. BIOspektrum 13:138-141. [Google Scholar]
- 12.Biot, C., H. Bauer, R. H. Schirmer, and E. Davioud-Charvet. 2004. 5-substituted tetrazoles as bioisosteres of carboxylic acids. Bioisosterism and mechanistic studies on glutathione reductase inhibitors as antimalarials. J. Med. Chem. 47:5972-5983. [DOI] [PubMed] [Google Scholar]
- 13.Blumenstiel, K., R. Schöneck, V. Yardley, S. L. Croft, and R. L. Krauth-Siegel. 1999. Nitrofuran drugs as common subversive substrates of Trypanosoma cruzi lipoamide dehydrogenase and trypanothione reductase. Biochem. Pharmacol. 58:1791-1799. [DOI] [PubMed] [Google Scholar]
- 14.Boda, C., B. Enanga, B. Courtioux, J. C. Breton, and B. Bouteille. 2006. Trypanocidal activity of methylene blue. Evidence for in vitro efficacy and in vivo failure. Chemotherapy 52:16-19. [DOI] [PubMed] [Google Scholar]
- 15.Böhme, C. C., L. D. Arscott, K. Becker, R. H. Schirmer, and C. H. Williams, Jr. 2000. Kinetic characterization of glutathione reductase from the malarial parasite Plasmodium falciparum. Comparison with the human enzyme. J. Biol. Chem. 275:37317-37323. [DOI] [PubMed] [Google Scholar]
- 16.Bradberry, S. M. 2003. Occupational methaemoglobinaemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicol. Rev. 22:13-27. [DOI] [PubMed] [Google Scholar]
- 17.Cawein, M., C. H. Behlen II, E. J. Lappat, and J. E. Cohn. 1964. Hereditary diaphorase deficiency and methemoglobinemia. Arch. Intern. Med. 113:578-585. [DOI] [PubMed] [Google Scholar]
- 18.Cenas, N. K., L. D. Arscott, C. H. Williams, Jr., and J. S. Blanchard. 1994. Mechanism of reduction of quinones by Trypanosoma congolense trypanothione reductase. Biochemistry 33:2509-2515. [DOI] [PubMed] [Google Scholar]
- 19.Cenas, N. K., G. A. Rakauskiene, and J. J. Kulys. 1989. One- and two-electron reduction of quinones by glutathione reductase. Biochim. Biophys. Acta 973:399-404. [DOI] [PubMed] [Google Scholar]
- 20.Clark, W. M., B. Cohen, and H. D. Gibbs. 1925. Studies on oxidation-reduction VIII—methylene blue. U.S. Public Health Rep. 40:1131-1201. [Google Scholar]
- 21.Dilgin, Y., and G. Nisli. 2005. Fluorimetric determination of ascorbic acid in vitamin C tablets using methylene blue. Chem. Pharm. Bull. (Tokyo) 53:1251-1254. [DOI] [PubMed] [Google Scholar]
- 22.Ellman, G. L. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70-77. [DOI] [PubMed] [Google Scholar]
- 23.Färber, P. M., L. D. Arscott, C. H. Williams, Jr., K. Becker, and R. H. Schirmer. 1998. Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett. 422:311-314. [DOI] [PubMed] [Google Scholar]
- 24.Gromer, S., L. D. Arscott, C. H. Williams, Jr., R. H. Schirmer, and K. Becker. 1998. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 273:20096-20101. [DOI] [PubMed] [Google Scholar]
- 25.Gromer, S., H. Merkle, R. H. Schirmer, and K. Becker. 2002. Human placenta thioredoxin reductase: preparation and inhibitor studies. Methods Enzymol. 347:382-394. [DOI] [PubMed] [Google Scholar]
- 26.Gromer, S., S. Urig, and K. Becker. 2004. The thioredoxin system—from science to clinic. Med. Res. Rev. 24:40-89. [DOI] [PubMed] [Google Scholar]
- 27.Gromer, S., L. A. Wessjohann, J. Eubel, and W. Brandt. 2006. Mutational studies confirm the catalytic triad in the human selenoenzyme thioredoxin reductase predicted by molecular modeling. Chembiochem 7:1649-1652. [DOI] [PubMed] [Google Scholar]
- 28.Guttmann, P., and P. Ehrlich. 1891. Über die Wirkung des Methylenblau bei Malaria. Berl. Klin. Wochenschr. 28:953-956. [Google Scholar]
- 29.Kanzok, S. M., R. H. Schirmer, I. Türbachova, R. Iozef, and K. Becker. 2000. The thioredoxin system of the malaria parasite Plasmodium falciparum. Glutathione reduction revisited. J. Biol. Chem. 275:40180-40186. [DOI] [PubMed] [Google Scholar]
- 30.Karplus, P. A., E. F. Pai, and G. E. Schulz. 1989. A crystallographic study of the glutathione binding site of glutathione reductase at 0.3-nm resolution. Eur. J. Biochem. 178:693-703. [DOI] [PubMed] [Google Scholar]
- 31.Kelner, M. J., and N. M. Alexander. 1985. Methylene blue directly oxidizes glutathione without the intermediate formation of hydrogen peroxide. J. Biol. Chem. 260:15168-15171. [PubMed] [Google Scholar]
- 32.Kosower, E. M. 1987. Structure and reaction of thiols with special emphasis on glutathione, p. 103-146. In D. Dolphin, R. Poulson, and O. Avramovic (ed.), Coenzymes and cofactors, vol. IIIA. John Wiley & Sons, New York, NY. [Google Scholar]
- 33.Krauth-Siegel, R. L., L. D. Arscott, A. Schönleben-Janas, R. H. Schirmer, and C. H. Williams, Jr. 1998. Role of active site tyrosine residues in catalysis by human glutathione reductase. Biochemistry 37:13968-13977. [DOI] [PubMed] [Google Scholar]
- 34.Krauth-Siegel, R. L., H. Bauer, and R. H. Schirmer. 2005. Dithiol proteins as guardians of the intracellular redox milieu in parasites: old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew Chem. 44:690-715. [DOI] [PubMed] [Google Scholar]
- 35.Low, F. M., M. B. Hampton, A. V. Peskin, and C. C. Winterbourn. 2007. Peroxiredoxin-2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 109:2611-2617. [DOI] [PubMed] [Google Scholar]
- 36.Mansouri, A., and A. A. Lurie. 1993. Concise review: methemoglobinemia. Am. J. Hematol. 42:7-12. [DOI] [PubMed] [Google Scholar]
- 37.Massey, V. 1960. The identity of diaphorase and lipoyl dehydrogenase. Biochim. Biophys. Acta 37:314-322. [DOI] [PubMed] [Google Scholar]
- 38.McMillan, P. J., L. M. Stimmler, B. J. Foth, G. I. McFadden, and S. Müller. 2005. The human malaria parasite Plasmodium falciparum possesses two distinct dihydrolipoamide dehydrogenases. Mol. Microbiol. 55:27-38. [DOI] [PubMed] [Google Scholar]
- 39.Meissner, P. E., G. Mandi, B. Coulibaly, S. Witte, T. Tapsoba, U. Mansmann, J. Rengelshausen, W. Schiek, A. Jahn, I. Walter-Sack, G. Mikus, J. Burhenne, K.-D. Riedel, R. H. Schirmer, B. Kouyaté, and O. Müller. 2006. Methylene blue for malaria in Africa: results from a dose-finding study in combination with chloroquine. Malar. J. 5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meissner, P. E., G. Mandi, S. Witte, B. Coulibaly, U. Mansmann, J. Rengelshausen, W. Schiek, A. Jahn, M. Sanon, T. Tapsoba, I. Walter-Sack, G. Mikus, J. Burhenne, K.-D. Riedel, H. Schirmer, B. Kouyaté, and O. Müller. 2005. Safety of the methylene blue plus chloroquine combination in the treatment of uncomplicated falciparum malaria in young children of Burkina Faso. Malar. J. 4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mills, A., and J. Wang. 1999. Photobleaching of methylene blue sensitised by TiO2: an ambiguous system? J. Photochem. Photobiol. A Chem. 127:123-134. [Google Scholar]
- 42.Müller, S., T. W. Gilberger, P. M. Färber, K. Becker, R. H. Schirmer, and R. D. Walter. 1996. Recombinant putative glutathione reductase of Plasmodium falciparum exhibits thioredoxin reductase activity. Mol. Biochem. Parasitol. 80:215-219. [DOI] [PubMed] [Google Scholar]
- 43.Müller, S., T. W. Gilberger, Z. Krnajski, K. Luersen, S. Meierjohann, and R. D. Walter. 2001. Thioredoxin and glutathione system of the malaria parasite Plasmodium falciparum. Protoplasma 217:43-49. [DOI] [PubMed] [Google Scholar]
- 44.Nordhoff, A., U. S. Bücheler, D. Werner, and R. H. Schirmer. 1993. Folding of the four domains and dimerization are impaired by the Gly446→Glu exchange in human glutathione reductase. Implications for the design of antiparasitic drugs. Biochemistry 32:4060-4066. [DOI] [PubMed] [Google Scholar]
- 45.Patil, K., P. Rajesh, and P. Talap. 2000. Self-aggregation of methylene blue in aqueous medium and aqueous solutions of Bu4NBr and urea. Phys. Chem. Chem. Phys. 2:4313-4317. [Google Scholar]
- 46.Prahl, S. 4 March 1999, posting date. Optical absorption of methylene blue. Oregon Medical Laser Center, Beaverton, OR. http://omlc.ogi.edu/spectra/mb/index.html.
- 47.Rengelshausen, J., J. Burhenne, M. Fröhlich, Y. Tayrouz, S. K. Singh, K.-D. Riedel, O. Müller, T. Hoppe-Tichy, W. E. Haefeli, G. Mikus, and I. Walter-Sack. 2004. Pharmacokinetic interaction of chloroquine and methylene blue combination against malaria. Eur. J. Clin. Pharmacol. 60:709-715. [DOI] [PubMed] [Google Scholar]
- 48.Rosin, D. 1893. Quinine and methylene blue in malaria. Dtsche. Med. Wochenschr. 44:Editorial.
- 49.Sarma, G. N., S. N. Savvides, K. Becker, M. Schirmer, R. H. Schirmer, and P. A. Karplus. 2003. Glutathione reductase of the malarial parasite Plasmodium falciparum: crystal structure and inhibitor development. J. Mol. Biol. 328:893-907. [DOI] [PubMed] [Google Scholar]
- 50.Schafer, F. Q., and G. R. Buettner. 2001. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30:1191-1212. [DOI] [PubMed] [Google Scholar]
- 51.Schirmer, R. H., B. Coulibaly, A. Stich, M. Scheiwein, H. Merkle, J. Eubel, K. Becker, H. Becher, O. Müller, T. Zich, W. Schiek, and B. Kouyaté. 2003. Methylene blue as an antimalarial agent. Redox Rep. 8:272-275. [DOI] [PubMed] [Google Scholar]
- 52.Thurston, J. P. 1953. The chemotherapy of Plasmodium berghei. I. Resistance to drugs. Parasitology 43:246-252. [DOI] [PubMed] [Google Scholar]
- 53.Vennerstrom, J. L., M. T. Makler, C. K. Angerhofer, and J. A. Williams. 1995. Antimalarial dyes revisited: xanthenes, azines, oxazines, and thiazines. Antimicrob. Agents Chemother. 39:2671-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wainwright, M., and L. Amaral. 2005. The phenothiazinium chromophore and the evolution of antimalarial drugs. Trop. Med. Int. Health 10:501-511. [DOI] [PubMed] [Google Scholar]
- 55.Weber, F. P. 1901. The occurrence of green or blue urine and its most frequent cause. Lancet 2:774. [Google Scholar]
- 56.Williams, C. H., Jr. 1992. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase—a family of flavoenzyme transhydrogenases, p. 121-211. In F. Müller (ed.), Chemistry and biochemistry of flavoenzymes, vol. 3. CRC Press, Boca Raton, FL. [Google Scholar]
- 57.Wolff, S. P., and M. J. Crabbe. 1985. Low apparent aldose reductase activity produced by monosaccharide autoxidation. Biochem. J. 226:625-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yi, P. G., J. F. Liu, Z. C. Shang, and Q. S. Yu. 2001. Study on the interaction between methylene blue and bovine serum albumin by fluorescence spectroscopy. Guang Pu Xue Yu Guang Pu Fen Xi 21:826-828. (In Chinese.) [PubMed] [Google Scholar]
- 59.Zappe, H. A. 1980. Die Bindungsstellen Hämolyse-induzierender Pharmaka an der Glutathionreduktase aus menschlichen Erythrocyten. Röntgenstrukturanalyse von Enzym-Pharmakon-Komplexen. Heidelberg University, Heidelberg, Germany.