

Abstract
Cholera toxin (CT) is an archetypal bacterial toxin that binds with a high affinity to the receptor ganglioside GM1 on the intestinal epithelial surface and that causes the severe watery diarrhea characteristic of the disease cholera. Blockage of the interaction of CT with the GM1 receptor is an attractive approach for therapeutic intervention. We report here that crude bile prevents the interaction of CT with GM1 and reduces CT-mediated fluid accumulation in the rabbit intestine. The unsaturated fatty acids detected in crude bile, arachidonic, linoleic, and oleic acids, were found to be the most effective. Crude bile and the unsaturated fatty acids interacted with CT but not GM1 to prevent CT-GM1 binding. Neither crude bile nor the unsaturated fatty acids had any effect on the subunit structure of CT. The binding of CT to unsaturated fatty acids resulted in a shift of the apparent pI of CT from 6.8 to 8.2 and a marked decrease in intrinsic fluorescence. The Kd was calculated from fluorescence quenching assays. It was demonstrated by the rabbit ileal loop model that practically no fluid accumulated in the intestinal loops when CT was administered together with inhibitory concentrations of linoleic acid. The bile present in the intestine was sufficient to inhibit the activity of up to 300 ng CT. Bile and unsaturated fatty acids also inhibited the binding of Escherichia coli heat-labile enterotoxin (LT) to GM1, and no fluid accumulation was observed in rabbit ileal loops when LT was administered together with linoleic acid.
Infectious diarrheal diseases are a major cause of human mortality, especially in developing countries, where conditions of inadequate sanitation, a lack of safe drinking water, malnourishment, war, and famine contribute to regular episodes of cholera, dysentery, traveler's diarrhea, and other forms of enteric disease, which claim nearly 2 million lives a year (10). Many enteric pathogens, including Vibrio cholerae, Escherichia coli, and Shigella spp., release toxins that are the primary cause of disease. The archetypal bacterial toxins are the cholera toxin (CT) and heat-labile enterotoxin (LT), derived from V. cholerae and enterotoxigenic E. coli (ETEC), respectively. Both CT and LT exploit binding to the cell surface glycolipid ganglioside GM1 as a means of entering intestinal epithelial cells (8). These toxins have a common heterohexameric structure consisting of a single A subunit attached to a pentameric core of five B subunits. The A subunit possesses ADP ribosyltransferase activity, and the B subunits selectively bind to the oligosaccharide portion of the ganglioside GM1. The biological action of CT and LT is initiated by the binding of the B subunits to the receptor, ganglioside GM1, on the intestinal epithelial cell membrane, followed by internalization of the A subunit into the cell (6, 7). The consequence of this activity is the impaired absorption of sodium ions and the rapid loss of water from the cells, resulting in the copious “rice water” diarrhea characteristic of the disease. The CT B subunit (CTB) and the LT B subunit (LTB) share 80% sequence identity (4) and are closely related immunologically.
Oral rehydration salt solution in combination with antibiotics is generally recommended for the treatment of diarrheal diseases of bacterial origin (2). However, in view of the rapid emergence of multiple-antibiotic-resistant strains of both V. cholerae and E. coli all over the world (1, 16), it is believed that the development of new pharmacological agents that inactivate the toxins and suppress the diarrhea would be advantageous. Since the binding of CT and LT to the GM1 receptor is the critical step in translocating the toxin into epithelial cells and the consequent fluid loss from the cells (8), the blockage of the GM1 binding of CT and LT with other ligands is an attractive approach for a therapeutic intervention that would prevent the action of the toxins. We report here that crude bile inhibits the GM1 binding of CT and LT. The active components present in crude bile were identified as unsaturated fatty acids, which can bind to CT and LT with a high affinity. The unsaturated fatty acids also prevent fluid accumulation in the intestine when they are administered with the toxins.
MATERIALS AND METHODS
Materials.
CT, CTB, GM1, crude ox bile, bile salts, and fatty acids were purchased from Sigma-Aldrich. LT-containing cell lysates were prepared as follows: E. coli strain 12566 was grown for 16 h; and the cells were centrifuged, washed and suspended in phosphate-buffered saline (PBS), and sonicated. The samples were centrifuged again to remove the debris, and the supernatant was used in a GM1 enzyme-linked immunosorbent assay (ELISA) or rabbit ligated ileal loops to examine the effect of bile or unsaturated fatty acids on LT.
Estimation of CT and LT by GM1 ELISA.
Samples containing 50 ng CT-, 50 ng CTB-, or 90 μl LT-containing cell lysates were incubated without or with different concentrations of ligand in a total volume of 100 μl for 15 min at room temperature. The samples were serially diluted and added to microtiter plates containing immobilized GM1. ELISA was performed as described previously (13) by using polyclonal rabbit serum raised against pure CT as the primary antibody, since it has been reported that anti-CT sera can effectively react with LT (9).
IC50 determination.
CT and CTB (500 ng/ml) were incubated with increasing concentrations of each ligand for 15 min at room temperature, and a GM1 ELISA was performed as described previously (13). The concentration of ligand required for 50% inhibition of CT binding to GM1 (IC50) was determined. The IC50 values were calculated from duplicate sets of data for at least 10 different concentrations of antagonists. The values reported are the averages of at least three independent determinations.
Immunoprecipitation.
CT or CTB (5 μg) was incubated without or with 3% bile or 85 mM sodium linolate in PBS (50 μl volume) at room temperature for 15 min, anti-CT immunoglobulin G (1:10 dilution) was added to each sample, and each sample was further incubated at 4°C for 3 h. Finally, 50 μl of swollen protein A-agarose beads (a 100-μl slurry in 500 μl PBS) was added, and the mixture was incubated overnight at 4°C with mild shaking. The immunoprecipitated complexes were collected by centrifugation (3,000 rpm, 5 min) at 4°C, washed, and suspended in Laemmli sample buffer (35 μl); and the suspension was analyzed by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (PAGE).
Fluorescence spectroscopy.
The emission spectrum of CT (15 μg) was recorded with a Perkin-Elmer luminescence spectrometer. Excitation was at a wavelength of 282 nm, emission was monitored at wavelengths between 300 and 400 nm, and a band pass of 5 nm was used. To estimate CT-fatty acid binding, the relative decrease in the intrinsic fluorescence of CT upon addition of increasing amounts of fatty acids was monitored at approximately 10-min intervals. The quenching of the fluorescence occurred rapidly and was stable for at least 30 min. Corrections were made for the inner filter effect for the ligand.
Isoelectric focusing.
Isoelectric focusing of CT (6 μg) and CT complexed with 100 mM linoleic acid was performed by using a Bio-Rad Protean isoelectric focusing cell and 17-cm linear (pH 3 to 10) immobilized pH gradient (IPG) strips (Bio-Rad). The samples were loaded onto the IPG strips by using the rehydration loading protocol, and isoelectric focusing was performed as recommended by the manufacturer. The IPG strips were stained with Coomassie blue to visualize the proteins.
Ligated rabbit ileal loop assay.
The in vivo effect of linoleic acid on CT and LT was examined by using the ligated rabbit ileal loop model, essentially as described by De and Chatterjee (5). Briefly, the rabbits were starved overnight but had free access to water, and they were then anesthetized. The small intestine was exteriorized through a midline incision and tied into consecutive 7-cm segments proximally to the mesoappendix. CT in 500 μl PBS or 750 μl LT containing ETEC cell lysates was injected alone or with different concentrations of linoleic acid into the rabbit ileal loops. After 16 h the animals were killed and the fluid that had accumulated in each loop was collected separately, measured, and expressed as a ratio of the amount (ml) of fluid per unit length (cm) of loop.
RESULTS
CT cannot be detected by GM1 ELISA in the presence of crude bile.
Since ganglioside GM1 is the receptor for CT, the binding of CT to GM1 immobilized on microtiter plates is widely used as an assay (the GM1 ELISA) for CT. Surprisingly, we observed that in the presence of crude bile, CT could not be detected by the GM1 ELISA. To examine whether the inhibition of CT-GM1 binding in the presence of crude bile was due to the interaction of bile with GM1 or CT, GM1 immobilized on microtiter plates was incubated with crude bile (0.2%), washed, and then incubated with CT. The results obtained clearly show that prior incubation of GM1 with crude bile had no effect on the subsequent binding of CT to GM1 (Fig. 1). However, when CT was incubated with crude bile before it was loaded on GM1, no CT was detected with a concentration of 0.05% bile or greater (Fig. 1). Taken together, these results suggest that crude bile interacts with CT and not GM1 to prevent CT-GM1 binding. The effect of bile on CT-GM1 binding was insensitive to the presence of up to 2 M NaCl and changes in pH in the range of 6.5 to 8.2 (data not shown).
FIG. 1.
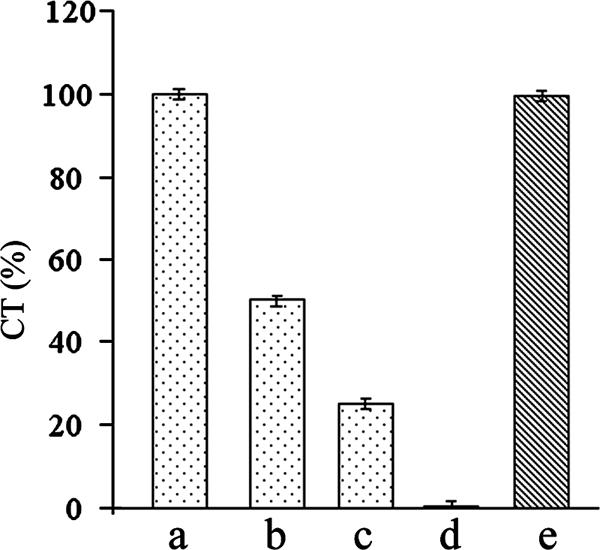
Effect of bile on CT and GM1. CT (100 ng) was incubated alone (bar a) or with 0.025% (bar b), 0.04% (bar c), or 0.05% (bar d) bile; and the amount of CT was estimated by the GM1 ELISA. GM1-coated wells were preincubated with 0.2% bile and washed, 100 ng CT was added to the wells, and the amount of CT was estimated by ELISA (bar e). Values obtained for 100 ng CT were taken as 100%. The results of three independent experiments are represented as the means ± standard deviations.
Bile does not block the binding of primary antibody to CT.
We also considered the possibility that bile blocks the anti-CT antibody binding sites on CT, so that CT was no longer detectable by GM1 ELISA in the presence of crude bile. To address this possibility, we examined if CT incubated with bile could be immunoprecipitated with anti-CT antibodies. CT (5 μg) and CT incubated with excess crude bile (3%) were immunoprecipitated with anti-CT antiserum, and the precipitate was analyzed by sodium dodecyl sulfate-PAGE. The results obtained indicated that comparable amounts of CT were precipitated by anti-CT antisera, irrespective of whether the CT was preincubated with bile or not, suggesting that the bile did not block the anti-CT antibody binding sites on CT (Fig. 2).
FIG. 2.

Immunoprecipitation of CT incubated with bile. CT (5 μg) was incubated without bile (lane a) or with 3% bile (lane b) and precipitated with anti-CT antisera, followed by precipitation with protein A-agarose. The controls were CT (5 μg; lane c), anti-CT antisera precipitated with protein A-agarose (lane d), and CT treated with protein A-agarose without anti-CT antisera (lane e). Lane M, standard molecular size marker. The numbers on the right indicate the sizes of the markers (kDa). The positions of immunoglobulin G (IgG) and the CTA (A) and CTB (B) subunits are indicated.
Bile has no effect on the subunit structure of CT.
The major components of crude bile, cholate and deoxycholate, have detergent-like properties. To examine if crude bile affects the subunit structure of CT (AB5), the holotoxin and the pentameric B subunit were incubated with bile and electrophoresed on native polyacrylamide gels. A single ∼87-kDa band, which is the expected size of the CT holotoxin, was observed when CT treated with crude bile or not treated with crude bile was analyzed by native PAGE. CTB also gave a single band of about 60 kDa, the expected size of the B-subunit pentamer, even when it was incubated with bile (Fig. 3). Thus, crude bile does not dissociate the quaternary structure of CT or CTB. When CT was heat denatured, bands corresponding to about 27 kDa (A subunit) and 12 kDa (B subunit) were observed.
FIG. 3.

Effect of bile on quaternary structure of CT. The holotoxin (CT) and the pentameric B subunit (CTB) of CT were incubated alone (lanes −) or with 0.4% of bile (lanes +) and electrophoresed on 7.5% native polyacrylamide gels. The top and bottom arrows indicate the positions of CT and CTB, respectively.
Identification of active bile components and IC50 determination.
Crude bile is a heterogeneous mixture, and we have previously reported on a fractionation method that separates the components of crude bile (3). Conjugated and unconjugated bile salts were identified as the major components of crude bile, and cholesterol and fatty acids were identified as minor components of crude bile. The GM1 ELISA with the different fractions indicated that the fraction containing the fatty acids was the most effective in preventing the binding of CT to GM1 (data not shown). This fraction contained the saturated fatty acids palmitic and stearic acids and the unsaturated fatty acids linoleic, oleic, and arachidonic acids (3). The IC50s of the fatty acids were determined, and the unsaturated fatty acids were found to be the most effective in preventing the binding of CT to GM1, with IC50 values being about 10-fold lower than those of the saturated fatty acids. The IC50s were as follows: linoleic acid, 0.41 mM; oleic acid, 0.41 mM; arachidonic acid, 0.38 mM; palmitic acid, 5.8 mM; stearic acid, 5.27 mM. Subsequent experiments were therefore performed with linoleic acid at the inhibitory concentration. As has been demonstrated for crude bile, linoleic acid did not affect the quaternary structure of CT and preincubation of CT with linoleic acid did not block the anti-CT antibody binding sites on CT (data not shown).
Isoelectric focusing.
Two methods were employed to directly evaluate the interaction of linoleic acid with CT: isoelectric focusing and quenching of intrinsic fluorescence. It has been demonstrated previously that protein-ligand binding might change the apparent pI of the protein, probably as a result of a surface charge redistribution upon ligand binding (17). As has been reported previously (11), the pI of CT was determined to be 6.8 by isoelectric focusing on IPG strips (pH 3 to 10). A remarkable shift in the apparent pI of CT to 8.2 was observed when CT was incubated with linoleic acid prior to isoelectric focusing (Fig. 4).
FIG. 4.

Alteration of pI of CT by fatty acids. Isoelectric focusing was performed with IPG strips (pH 3 to 10) with CT (6 μg; strip a) and CT incubated with sodium linolate (strip b), and their positions and pIs are indicated by arrows. The positions and pIs of the isoelectric focusing marker proteins are also indicated.
Fluorescence spectroscopy and determination of Kd.
To analyze the binding of linoleic acid to CT in greater detail, the emission spectrum of CT was recorded in the presence and absence of linoleic acid. Linoleic acid produced a marked decrease in the fluorescence intensity of CT, with a moderate blue shift in the emission spectrum (Fig. 5A). In order to estimate the dissociation constant of CT-linoleic acid binding, the quenching of the intrinsic fluorescence of pure CT upon addition of increasing amounts of sodium linolate was measured. The decrease in intrinsic fluorescence of CT following linolate binding was used to quantitate the CT-fatty acid binding affinity. After correction for the inner filter effect, the apparent Kd was determined to be 0.13 mM (Fig. 5B). Similarly, the Kds of oleic acid and arachidonic acid binding to CT were determined to be 0.16 mM and 0.18 mM, respectively. Thus, the affinities of binding of all three unsaturated fatty acids to CT were comparable.
FIG. 5.

Quenching of intrinsic fluorescence of CT by fatty acid. (A) Typical fluorescence scan of CT titrated with increasing concentrations of sodium linolate. The concentration of linolate (mM) added to 15 μg CT is indicated. RFU, relative fluorescence units. (B) A simple two-state binding model.
In vivo assay.
Since the major biological function of CT is to promote the loss of fluid from epithelial cells, we used the ligated rabbit ileal loop model to examine intestinal fluid accumulation when CT was administered in the ileal loops with or without linoleic acid. Fluid accumulation was inhibited in a dose-dependent manner when sodium linolate was administered with CT (Fig. 6). Although about 1.6 ± 0.2 ml/cm fluid accumulated in ileal loops inoculated with 500 ng CT, the amount was reduced to 0.76 ml/cm when CT was administered together with 1.65 mM linoleic acid. Practically no fluid accumulation was observed in loops containing 3.3 mM linoleic acid. Interestingly, it was noted that no fluid accumulated in loops inoculated with up to 300 ng CT, suggesting that the crude bile present in the intestine might be sufficient to completely inhibit the activity of up to 300 ng CT.
FIG. 6.

Effect of linoleic acid on CT-mediated fluid accumulation in rabbit ileal loops (A). Rabbit ileal loops were inoculated with saline (loop 1), 100 ng CT (loop 2), 300 ng CT (loop 3), 500 ng CT (loop 4), 500 ng CT with 1.65 mM sodium linolate (loop 5), and 500 ng CT with 3.3 mM sodium linolate (loop 6). The loop lengths and the amount of fluid accumulated in each loop were measured, and the amount of fluid (ml) per unit length (cm) of loop was determined (B). Values represent the means ± standard deviations from three independent experiments.
Unsaturated fatty acids inhibit LT-GM1 binding.
Since, like CT, the closely related E. coli LT also utilizes GM1 as the natural cell surface receptor, the effect of crude bile on the interaction of LT with GM1 was examined. Sonicated cell lysates of ETEC strain 12566 were used as a source of LT. The LT in the cell lysates could be detected by the GM1 ELISA; however, upon preincubation of the lysates with crude bile (0.2%), LT could no longer be detected by the GM1 ELISA (data not shown). Of the bile components examined, unsaturated fatty acids were found to be the most effective in preventing the LT-GM1 interaction. The effect of linoleic acid on LT in vivo was examined by using the rabbit ligated ileal loop model. About 5.5 ± 1 ml fluid accumulated in the ileal loops when 750 μl of crude E. coli cell lysate was administered into 7-cm segments of ileum. Practically no fluid accumulation was observed when the same amount of E. coli lysate was preincubated with 21 mM sodium linolate, while 1 ml of fluid accumulation was observed when the lysate was incubated with 10.5 mM sodium linolate (Fig. 7). Upon comparison with the results obtained with CT, the results of these experiments indicated that the effect of linoleic acid on LT was less pronounced than that on CT.
FIG. 7.

Effect of linoleic acid on LT-mediated fluid accumulation in rabbit ileal loops (A). Rabbit ileal loops were inoculated with saline (loop 1); cell lysates containing LT (loop 2); or cell lysates with 4.2 mM (loop 3), 10.5 mM (loop 4), or 21 mM (loop 5) sodium linolate. The loop lengths and the amount of fluid accumulated in each loop were measured, and the amount of fluid (ml) per unit length (cm) of loop was determined (B). Values represent the means ± standard deviations from three independent experiments.
DISCUSSION
Bile is a major constituent of the small intestine which is secreted into the lumen of the duodenum from the gallbladder through the bile duct and which is inevitably encountered by all enteric bacteria during infection. We have recently shown that in addition to the major components, which were conjugated and unconjugated bile salts and bile pigments, crude bile also contains fatty acids and cholesterol as minor components (3). In this paper we report that the unsaturated fatty acids present in crude bile can bind to CT and the closely related LT and can prevent the interaction of these toxins with the intestinal receptor GM1, a critical step in the translocation of the toxins into the intestinal epithelial cells and the resultant severe fluid loss that is characteristic of the diseases caused by these toxins. It seems paradoxical that bile should inhibit the interaction of CT and LT with GM1, since bile is certainly present in the intestine, the physiological site of action of these toxins. A possible explanation is as follows. The growth of V. cholerae in rabbit ileal loops (7 cm long) produced about 10 μg CT in the loops (unpublished observation); the amount of bile present in similar intestinal loops is sufficient to inactivate only about 300 ng CT (Fig. 6). Although this may be an underestimation, since starved rabbits were used in the experiments and starvation is known to reduce the secretion of bile into the duodenum, it still seems reasonable to assume that the amount of bile present in the intestine may be insufficient to prevent GM1 binding of the massive amounts of CT and LT produced by V. cholerae and ETEC during infection. Supplementation with bile or unsaturated fatty acids drastically reduced the level of fluid accumulation due to CT and also LT (Fig. 6 and 7).
Several compounds that inhibit GM1 binding of CT and LT have been identified earlier. Since galactose, the terminal sugar of GM1, is of primary importance in CT and LT binding (12), several galactose derivatives were screened to develop receptor antagonists, and m-nitrophenyl-α-galactoside (MNPG) was found to be the most effective; MNPG was about 100-fold more effective than galactose itself (14). Next-generation leads based on MNPG, 3,5-substituted phenylgalactosides, displayed efficacy two- to sixfold greater than that of MNPG (15). However, linoleic acid is 200-fold more effective than MNPG as a GM1-CT binding antagonist.
The data presented in this paper suggest the possibility that the oral administration of unsaturated fatty acids, preferably together with oral rehydration salt, could be used as a precautionary and therapeutic measure for cholera and, possibly, traveler's diarrhea. Unsaturated fatty acids are abundantly present in our daily diets: oils like walnut oil, soybean oil, corn oil, and sunflower seed oil contain more than 50% linoleic acid, while evening primrose oil and safflower seed oil contain about 75% linoleic acid. Thus, small amounts of these oils might be sufficient to inactivate the CT and LT produced during infection. They are not toxic to humans; indeed, linoleic acid is an essential fatty acid. Thus, they might be used as drugs without the fear of toxic side effects. Moreover, since they act on secreted toxins and not on the bacteria, the possibility of bacterial resistance does not arise. Further studies with patients with cholera are necessary to confirm the efficacy of unsaturated fatty acids for the treatment of cholera.
Acknowledgments
We thank all members of the Biophysics Division for cooperation, encouragement, and helpful discussions during the study and I. Guha Thakurta, K. Paul, and P. Majumdar for excellent technical support.
This work was supported by a research grant from the Network Program (grant SMM 003), Council of Scientific and Industrial Research (CSIR), Government of India. A.C. is grateful to CSIR for a research fellowship.
Footnotes
Published ahead of print on 22 October 2007.
REFERENCES
- 1.Ash, R. J., B. Mauck, and M. Morgan. 2002. Antibiotic resistance of gram-negative bacteria in rivers, United States. Emerg. Infect. Dis. 8:713-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhan, M. K., E. Chea Woo, O. Fontaine, I. Maulen-Radovan, N. F. Pierce, and H. Ribeiro, Jr. 1995. Multicentre evaluation of reduced-osmolarity oral rehydration salt solution. Lancet 345:282-285. [PubMed] [Google Scholar]
- 3.Chatterjee, A., P. K. Dutta, and R. Chowdhury. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect. Immun. 75:1946-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dallas, W. S., and S. Falkow. 1980. Amino acid sequence homology between cholera toxin and Escherichia coli heat-labile toxin. Nature 288:499-501. [DOI] [PubMed] [Google Scholar]
- 5.De, S. N., and S. N. Chatterjee. 1953. An experimental study of the mechanisms of action of Vibrio cholerae on the intestinal mucous membrane. J. Pathol. Bacteriol. 46:559-562. [DOI] [PubMed] [Google Scholar]
- 6.Finkelstein, R. A., M. Boesman, S. H. Neoh, M. K. LaRue, and R. Delaney. 1974. Dissociation and recombination of the subunits of the cholera enterotoxin (choleragen). J. Immunol. 113:145-150. [PubMed] [Google Scholar]
- 7.Fishman, P. H. 1990. Mechanism of action of choleratoxin, p. 127-140. In J. Moss and M. Vaughan (ed.), ADP-ribosylating toxins and G proteins. American Society for Microbiology, Washington, DC.
- 8.Ganguly, N. K., and T. Kaur. 1996. Mechanism of action of cholera toxin & other toxins. Indian J. Med. Res. 104:28-37. [PubMed] [Google Scholar]
- 9.Gilligan, P. H., J. C. Brown, and D. C. Robertson. 1983. Immunological relationships between cholera toxin and Escherichia coli heat-labile enterotoxin. Infect. Immun. 42:683-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hénock Blaise, N. Y., and D. B. Dovie. 2007. Diarrheal diseases in the history of public health. Arch. Med. Res. 38:159-163. [DOI] [PubMed] [Google Scholar]
- 11.Kabir, S. 1986. Charge heterogeneity of cholera toxin and its subunits. FEMS Microbiol. Lett. 37:155-162. [Google Scholar]
- 12.King, C. A., and W. E. Van Heyningen. 1973. Deactivation of cholera toxin by a sialidase-resistant monosialosylganglioside. J. Infect. Dis. 127:639-647. [DOI] [PubMed] [Google Scholar]
- 13.Krishnan, H. H., A. Ghosh., K. Paul, and R. Chowdhury. 2004. Effect of anaerobiosis on expression of virulence factors in Vibrio cholerae. Infect. Immun. 72:3961-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minke, W. E., C. Roach, W. J. Hol, and C. M. J. Verlinde. 1999. Structure based exploration of the ganglioside GM1 binding sites of Escherichia coli heat-labile enterotoxin and cholera toxin for the discovery of receptor antagonists. Biochemistry 38:5684-5692. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell, D. D., J. C. Pickens, K. Korotkov, E. Fan, and W. J. Hol. 2004. 3,5-Substituted phenyl galactosides as leads in designing effective cholera toxin antagonists; synthesis and crystallographic studies. Bioorg. Med. Chem. 12:907-920. [DOI] [PubMed] [Google Scholar]
- 16.Mwansa, J. C., J. Mwaba, C. Lukwesa, N. A. Bhuiyan, M. Ansaruzamman, T. Ramamurthy, M. Alam, and G. B. Nair. 2006. Multiply antibiotic-resistant Vibrio cholerae O1 biotype El Tor strains emerge during cholera outbreaks in Zambia. Epidemiol. Infect. 23:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudnick, D. A., C. A. McWherter, S. P. Adams, I. J. Ropson, R. J. Duronio, and J. I. Gordon. 1990. Structural and functional studies of Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase produced in Escherichia coli. Evidence for an acyl-enzyme intermediate. J. Biol. Chem. 265:13370-13378. [PubMed] [Google Scholar]