

Abstract
Recombinant vesicular stomatitis virus (rVSV) has shown great potential as a new viral vector for vaccination. However, the prototypic rVSV vector described previously was found to be insufficiently attenuated for clinical evaluation when assessed for neurovirulence in nonhuman primates. Here, we describe the attenuation, neurovirulence, and immunogenicity of rVSV vectors expressing human immunodeficiency virus type 1 Gag. These rVSV vectors were attenuated by combinations of the following manipulations: N gene translocations (N4), G gene truncations (CT1 or CT9), noncytopathic M gene mutations (Mncp), and positioning of the gag gene into the first position of the viral genome (gag1). The resulting N4CT1-gag1, N4CT9-gag1, and MncpCT1-gag1 vectors demonstrated dramatically reduced neurovirulence in mice following direct intracranial inoculation. Surprisingly, in spite of a very high level of attenuation, the N4CT1-gag1 and N4CT9-gag1 vectors generated robust Gag-specific immune responses following intramuscular immunization that were equivalent to or greater than immune responses generated by the more virulent prototypic vectors. MncpCT1-gag1 also induced Gag-specific immune responses following intramuscular immunization that were equivalent to immune responses generated by the prototypic rVSV vector. Placement of the gag gene in the first position of the VSV genome was associated with increased in vitro expression of Gag protein, in vivo expression of Gag mRNA, and enhanced immunogenicity of the vector. These findings demonstrate that through directed manipulation of the rVSV genome, vectors that have reduced neurovirulence and enhanced immunogenicity can be made.
The dire need for a human immunodeficiency virus (HIV) vaccine has led to an increase in the development of novel approaches for vaccination. Early attempts at designing HIV vaccines that could induce a neutralizing antibody response against the envelope of HIV have not been clinically successful; this lack of success has led to the examination of whether cell-mediated immunity against HIV antigens can protect individuals from infection with HIV or at least prevent the progression to AIDS following infection (9, 40).
Several groups have examined the use of recombinant viruses expressing HIV antigens as vaccines that can induce a potent cell-mediated immune response (23). A successful virally vectored vaccine will require the ability to carefully balance its immunogenicity and safety. In general, as viruses are attenuated to increase safety, their immunogenicities can be diminished. An ideal viral vaccine vector should replicate sufficiently and express high levels of foreign antigen, inducing potent immune responses without causing pathology in the recipient.
Vesicular stomatitis virus (VSV) is an enveloped, negative-stranded RNA virus of the family Rhabdoviridae (34). In nature, VSV is transmitted by insects and infects livestock, causing a self-limiting disease that is marked by vesicular lesions of the mouth and teats. VSV rarely infects humans, but when infection does occur, it can result in disease ranging from asymptomatic infection to mild flu-like illness (7, 16). VSV has many characteristics that render it an ideal candidate for development as a vaccine vector, including the absence of preexisting immunity to VSV in human populations, ease of genetic manipulation, robust gene expression, and vigorous virus propagation in vitro. Since the development of a system for the recovery of recombinant VSV (rVSV) from plasmid DNA (22, 43, 44), rVSV vectors have been assessed in animal models as vaccine vectors for numerous pathogens including HIV-1, herpes simplex virus type 2, influenza virus, respiratory syncytial virus, hepatitis C virus, cottontail rabbit papillomavirus, measles virus, Ebola virus, Lassa fever virus, Marburg virus, and severe acute respiratory syndrome coronavirus (1, 5, 6, 8, 17-20, 24, 29, 31-33, 35, 38). The early development of rVSV for use in humans as an HIV-1 vaccine vector has been recently reviewed (2).
The prototypic rVSV vectors originally developed as HIV-1 vaccines were based on an rVSV genome that was generated from two laboratory-adapted strains of the VSV Indiana serotype (VSVIN) (10, 12, 33, 35). This rVSV vector backbone was slightly attenuated compared to wild-type VSV (wtVSV) when examined in mice (33). The prototypic rVSV-HIV-1 vectors were shown to induce strong cellular immunity to HIV Gag and HIV Env in small animals (11, 12), and this led to the demonstration that rVSV vectors expressing HIV Env and simian immunodeficiency virus Gag were highly protective in the rhesus macaque simian-human immunodeficiency virus (SHIV) 89.6P challenge model (35), supporting future clinical assessments of these vectors in humans. While VSV does not cause neurological disease in livestock following natural infection, it does cause a well-described viral encephalitis in rodents following intranasal (i.n.) or intracranial (i.c.) inoculation of young mice (37, 42). Since regulatory agencies typically require nonhuman primate (NHP) neurovirulence testing of replication-competent viral vaccines prior to clinical assessment in humans, we evaluated the neurotropism and neurotoxicity of the prototypic HIV-1 rVSV vectors in an exploratory NHP neurovirulence study. While this study demonstrated no dissemination of the rVSV vectors to the brain following i.n. administration, there was significant neuropathology following intrathalamic inoculation of cynomolgus macaques (15). From that study, it was concluded that the prototypic rVSV vectors were insufficiently attenuated for assessment in humans, and further vector attenuation would be required to reduce the potential for neurovirulence.
Recently, we described strategies for rVSV attenuation, including the combination of G gene truncations with either N gene translocations or noncytopathic M gene mutations (3). Here, we extend those studies and describe the construction of genetically modified rVSV vectors expressing HIV Gag and their assessment in mice for neurovirulence and immunogenicity. Through these studies, we have identified rVSV vectors that are dramatically less neurovirulent than the prototypic rVSV vector. Moreover, and somewhat surprisingly, these highly attenuated vectors exhibited enhanced Gag-specific immunogenicity compared to the more virulent prototypic rVSV gag vectors. These results were achieved through vector design that maximized foreign gene expression while reducing the expression and function of some key viral proteins, thereby reducing virus replication efficiency while enhancing immunogenicity.
MATERIALS AND METHODS
Cells and virus.
Vero and Baby hamster kidney (BHK) cell lines were obtained from the American Type Culture Collection (ATCC) and propagated at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS), sodium pyruvate (20 mM), and gentamicin (50 μg/ml). The tissue culture-adapted San Juan strain of VSVIN, a recombinant form of VSVIN (rVSVIN) (22), and rVSVIN expressing HIV-1 Gag protein (rVSVIN-gag5) were kindly provided by Jack Rose (Yale University, CN). A modified form of vaccinia virus Ankara that expressed the bacteriophage T7 RNA polymerase (45) was obtained from Bernard Moss (NIH, Bethesda, MD) and further modified to express T7 RNA polymerase under the control of an early transcription promoter (21).
rVSV vector preparation.
The construction of rVSV vectors containing combinations of G gene truncations, N gene translocations, and noncytopathic M gene mutations, including rVSVINN2CT1, rVSVINN3CT1, rVSVINN2CT9, rVSVINN3CT9, and rVSVINMncpCT1, was described in detail previously (3). The genomic organization and nomenclature for the vectors described in this study are depicted in Fig. 1A. For the purposes of this study, the rVSVINN3CT1 vector was modified to express HIV-1 p55 Gag protein from a transcription unit inserted into the first position of the genome, adjacent to the viral 3′ transcription promoter (Fig. 1B). To carry out this modification, three PCR products were generated: (i) a DNA fragment extending from the unique BsaAI site in the plasmid vector to the P gene 5′ untranslated region of rVSVINN3CT1, including the P gene transcription start signal followed by eight additional noncoding nucleotides and a flanking XhoI site; (ii) a fragment spanning from the P gene transcriptional start signal to the unique XbaI site in the P gene, including an NheI restriction site followed by a transcription termination signal, followed by a CT intergenic dinucleotide, all added upstream of the P gene transcriptional start signal; and (iii) a DNA fragment containing the HIV-1 HXB2 strain p55 gag gene open reading frame (ORF) flanked by XhoI and NheI sites. The three DNA fragments were digested with XhoI and NheI and then ligated in vitro. The resulting ligation product was gel purified, BsaAI/XbaI digested, and cloned into the BsaAI/XbaI sites of rVSVINN3CT1 to generate rVSVINN4CT1-gag1. The rVSVINN4CT9-gag1 vector was similarly generated by cloning the gag gene into BsaAI/XbaI-digested rVSVINN3CT9.
FIG. 1.

Design and protein expression of rVSV-gag vectors. (A) The genomes of the rVSV vectors created for this study are diagramed in the 3′-to-5′ orientation of the negative-stranded viral RNA next to the nomenclature used for each vector. (B) Generation of rVSV vector genomic cDNAs. Endonuclease sites used for the assembly of cDNA constructs are indicated. T7-Prom represents the T7 RNA polymerase promoter sequence. Le and Tr represent the virus untranslated leader and trailer sequences, respectively. Shaded bars represent viral transcription start signals, and each viral transcription unit is separated by the nontranscribed intergenic dinucleotides GT and CT, as indicated. A T7 RNA polymerase transcription-termination signal (T7 Term) and hepatitis delta virus (HDV) ribozyme lead to the generation of a precise viral 5′ end during the virus rescue process. (C) Western blots showing in vitro HIV-1 Gag expression of rVSV vectors 24 h postinfection of replicate BHK cell monolayers. Lanes 1 and 2 contain proteins from uninfected and “empty” rVSV-infected cells, respectively. Lanes 3 to 7 contain proteins from cells infected with the different Gag-expressing rVSV vectors. Protein size markers (24 kDa to 50 kDa) were run alongside test samples.
The analogous rVSV New Jersey serotype (rVSVNJ) glycoprotein-exchange vectors rVSVNJN4CT1-gag1 and rVSVNJN4CT9-gag1 were generated by replacing the Indiana serotype G (GIN) gene with truncated forms of the New Jersey serotype G (GNJ) gene that retained the CT1 and CT9 of the Indiana serotype. The modified GNJ gene cDNAs were generated by PCR from a plasmid DNA template containing the GNJ gene (Jack Rose, Yale University, CT). Briefly, the forward primer contained the GIN MluI site and four adjacent downstream noncoding nucleotides, followed by the first 25 nucleotides of the GNJ ORF. Reverse primers contained an NheI site flanking a translation stop codon, an adjacent sequence encoding either the single arginine residue of GIN CT1 or the 9 amino acids of GIN CT9, and 36 nucleotides encoding the GNJ trans-membrane region. Both PCR products were digested with MluI and NheI and cloned into the NheI/MluI sites of rVSVINN3CT1 cDNA to produce rVSVNJN3CT1 and rVSVNJN3CT9. The PCR products could not be directly cloned into rVSVINN4CT1-gag1 and rVSVINN4CT9-gag1 because of the presence of a second NheI site in the Gag cloning cassette. The rVSVNJN4CT1-gag1 and rVSVNJN4CT9-gag1 cDNAs were then generated by swapping the BsaAI/XbaI fragments of rVSVNJN3CT1 and rVSVNJN3CT9 with that from rVSVINN4CT1-gag1.
The rVSVIN-gag1 vector was generated by cloning the Gag cDNA into pVSV1XN (Jack Rose, Yale University, CT) (28), which contained an XhoI/NheI expression cassette in the first position of the genome. To make the pVSVNJ-gag1 vector, we first used PCR to remove a natural HpaI site in the GNJ ORF (175 nucleotides downstream of the ATG codon). The GNJ gene was amplified in two pieces using primers that changed the HpaI site in GNJ into an HincII site without changing the amino acid sequence and that extended the PCR products into the 5′ portion of the VSV L ORF (to include a unique HpaI site 232 nucleotides downstream of the VSV L ATG codon). The PCR product encoding the 5′ portion of GNJ was first inserted into a shuttle vector using a unique MluI site (upstream of the NJ G start codon) and the original GNJ HpaI site. The second PCR product encoding the 3′ part of the GNJ gene through a unique HpaI site in VSV L was then inserted using HincII in the GNJ gene and an NdeI site added to the 3′ end of the PCR product to facilitate cloning. Upon ligation of these two fragments, the HpaI site in the GNJ ORF was destroyed, leaving only a unique HpaI site in VSV L (232 nucleotides downstream of the VSV L ATG codon) to facilitate cloning. Thus, to generate pVSVNJ-gag1, the complete GNJ ORF and a portion of the VSV L gene were removed from the shuttle vector and inserted into pVSVIN-gag1 via MluI and HpaI.
The rVSVINMncpCT1-gag1 vector was generated by swapping the BsaAI/XbaI DNA fragment from rVSVINMncpCT1 (3) with the analogous fragment from rVSVIN-gag1 genome cDNA. The rVSVNJMncpCT1-gag1 vector was then generated by swapping the GIN gene (CT1) with that from rVSVNJN4CT1-gag1.
Virus propagation, purification, and titration.
Virus was amplified on BHK cell monolayers and titrated on Vero cell monolayers as previously described (3). Virus used for neurovirulence and immunogenicity studies was purified by centrifugation through a 10% (wt/vol) sucrose cushion and resuspended in phosphate-buffered saline (PBS) as previously described (3).
Western blot analysis of HIV Gag expression.
Briefly, replicate confluent BHK cell monolayers in six-well plates were infected at a multiplicity of infection of 5 PFU per cell. Virus inoculum was adsorbed for 15 min at room temperature (RT) followed by 30 min at 37°C. Additional growth medium was then added, and cells were incubated at 37°C in 5% CO2 for 24 h. At 24 h postinfection, cells were scraped into suspension and collected by centrifugation for 10 min at 3,000 × g. Supernatant was removed, and cell pellets were treated with 0.5 ml of lysis buffer (0.05 M Tris-HCl [pH 7.5], 0.01 M NaCl, 1× Triton X-100). Cell lysates were then diluted 1:1 in Laemmli sample buffer (catalog number 161-0737; Bio-Rad) and heated at 90°C for 5 min to denature proteins. Samples were electrophoresed on 4 to 12% Bis-Tris-polyacrylamide gel electrophoresis gels (NuPAGE, catalog number NP0321) with a Precision Plus protein standard (catalog number 1610375; Bio-Rad), and proteins were then transferred onto a nitrocellulose membrane using the iBlot system (catalog numbers IB1001 and IB3010-02; Invitrogen). The nitrocellulose membrane was then blocked in 5% milk in TTBS (0.02% Tween 20, 0.9% NaCl, 100 mM Tris-HCl [pH 7.5]) overnight, followed by three 5-min washes in TTBS. The blot was incubated with HIV-1 p24 Gag-specific monoclonal antibody (catalog no. 1103; ImmunoDiagnostics, Inc.) diluted 1:2,000 in 5% milk-TTBS for 1 h at RT, followed by three 5-min washes in TTBS. The blot was then incubated with biotinylated goat anti-mouse immunoglobulin G (IgG) (catalog number BA-9200; Vector Labs) diluted 1:2,000 in 5% milk-TTBS for 1 h, followed by three 5-min washes in TTBS. Protein-antibody complexes were then visualized using Vectastain ABC and TMB substrate kits (catalog numbers PK-6100 and SK4400; Vector Labs).
Animals.
All mice were obtained from Taconic Farms (Germantown, NY). Outbred female Swiss Webster mice were used for neurovirulence studies and were approximately 5 weeks old at the time of inoculation. Inbred female BALB/c mice were used for immunogenicity studies and were approximately 7 weeks old at the time of inoculation. All animal care and procedures conformed to Institutional Animal Care and Use Committee guidelines. The facilities are fully accredited by the American Association for Accreditation of Laboratory Animal Care. Five mice were housed per cage and were acclimated for 1 week in the vivarium prior to inoculation. Mice were euthanized if any severe signs of disease were observed.
Neurovirulence studies.
Neurovirulence was assessed by monitoring morbidity and mortality of Swiss Webster mice following i.c. inoculation of rVSV vectors as previously described (3). Following i.c. inoculation, mice were weighed daily and assessed for signs of neurological disease over 3 weeks. The relative level of neurovirulence was assessed by measuring morbidity and mortality in the mice as an endpoint. Any mice showing severe signs of disease or any mice that were clearly moribund were promptly euthanized. The 50% lethal dose (LD50) was calculated based on the cumulative mortality at each dose level (30). Likewise, the 50% paralyzing dose (PD50) was calculated based on the cumulative induced paralysis at each dose level.
Vaccination protocol for immunogenicity studies.
Prior to intramuscular (i.m.) or i.n. inoculation, mice were anesthetized with an intraperitoneal injection of ketamine (105 mg/kg) and xylazine (10 mg/kg). BALB/c mice were primed either i.m. or i.n. with the indicated dose of rVSVIN. For prime-boost studies, mice were boosted 8 weeks postprime by the same route with the corresponding rVSVNJ glycoprotein-exchange vector. Negative controls consisted of naïve mice or mice immunized with an “empty” prototypic rVSV vector. i.m. immunizations were given by injection into the calf muscle of anesthetized mice in a 50-μl volume of PBS. Intranasal immunizations were given by dropwise instillation into the nares of anesthetized mice by a micropipettor in a 10-μl volume of PBS (5 μl per naris). Serum samples and spleen cells were harvested from mice (n = 5) 7 days after priming, 5 days after boosting, and 4 weeks after boosting to test for humoral and cellular immune responses.
IFN-γ ELISPOT.
Enzyme-linked immunospot (ELISPOT) assays were performed in 96-well MultiScreen-IP plates (Millipore) coated with 1 μg/well of anti-murine gamma interferon (IFN-γ) capture antibody (BD Pharmingen) in PBS overnight at 4°C. The plates were then washed with PBS and blocked with T-cell medium (RPMI-1640 [Cellgro-Mediatech], 10% fetal bovine serum [HyClone], 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 100 μM nonessential amino acids, 1 mM sodium pyruvate, 55 μM 2-mercaptoethanol, and 10 mM HEPES [Invitrogen]) for at least 2 h at 37°C in 5% CO2. Cell suspensions from individual spleens were processed, and red blood cells were lysed with ACK lysis buffer (BioWhittaker). The cell suspensions were filtered using a sterile, 100-μm mesh cell strainer (BD Labware), and the cells were counted using a Guava personal cytometer (Guava Technologies). The cells were added to the ELISPOT plates in triplicate at 3 × 105 cells per well in T-cell medium and cocultured with either 10 μM Gag H-2Kd immunodominant peptide (AMQMLKETI purchased at 85% purity; New England Peptide), 10 μM VSV N H-2Ld immunodominant peptide (MPYLIDFGL purchased at 85% purity; New England Peptide), 1 μg/ml concanavalin A (Sigma), or T-cell medium. The cultures were incubated for 16 to 20 h at 37°C in 5% CO2, and the plates were then decanted and washed twice with double-distilled H2O. The plates were then washed six times with PBS-0.01% Tween 20, and biotinylated anti-murine IFN-γ detection antibody (1:250 dilution in PBS-10% FBS; BD Pharmingen) was added. The plates were incubated for 2 h at RT and washed five times, and streptavidin-horseradish peroxidase (1:100 dilution in PBS-10% FBS; BD Pharmingen) was added. The plates were incubated for 1 h at RT, washed three times with PBS-0.01% Tween 20, and then washed three times with PBS. AEC substrate solution (BD Pharmingen) was then added, and the spots were allowed to develop for 5 to 10 min. Spot development was stopped by decanting the substrate and gently rinsing the plate with tap water. After the membranes were dry, the plates were scanned using an ImmunoSpot reader, and the spots were counted using ImmunoSpot software (Cellular Technology Ltd.).
Gag tetramer staining.
Spleens from immunized mice were individually harvested and processed as described above. T cells specific for the H2-Kd-restricted immunodominant CD8 epitope of HIV-1 Gag in BALB/c mice (AMQMLKETI) were detected by staining with phycoerythrin-labeled H2-Kd tetramers (Beckman Coulter). All antibody-staining reagents were from BD Pharmingen. Spleen cells (1 × 106 cells) were first blocked with 50 μl of a 1/50 dilution of anti-Fc antibody (2G12, “Fc block”) in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline with 5% fetal bovine serum and 0.1% sodium azide; BD Pharmingen) for 15 min at RT in the well of a V-bottom 96-well plate. Next, 50 μl of a 1/10 dilution of the phycoerythrin-labeled tetramer in FACS buffer was added and incubated in the dark and on ice for 30 min. Forty microliters of an antibody-costaining cocktail (CD8-fluorescein isothiocyanate, CD3-allophycocyanin, CD4-peridinin-chlorophyll protein (PerCP), and CD19-PerCP; 1:40 dilution of each antibody) was then added and incubated in the dark for 15 min at RT. Cells were washed twice with 200 μl of FACS buffer and resuspended in 400 μl of CytoFix buffer (BD Pharmingen). Samples were analyzed within 24 h on a FACSCalibur flow cytometer (BD Pharmingen) by gating on live CD3/CD8+ and CD4/CD19− (dump channel) cells. Later studies utilized H2-Kd pentamers (ProImmune, Inc.) specific for the same H2-Kd-restricted immunodominant CD8 epitope of HIV-1 Gag in BALB/c mice using the same protocol conditions as those described above for tetramers. Bridging studies confirmed that both Gag tetramers and Gag pentamers yielded similar results.
Ex vivo cytotoxic T-lymphocyte (CTL) assay.
Spleens from immunized mice were harvested and processed individually as described above. Spleen (effector) cells were resuspended at 2 × 107 cells/ml in T-cell medium and added in triplicate (200 μl/well) to the first row of each test plate. Cells were diluted in twofold serial dilutions from rows A to F. Spontaneous and maximum release controls were included on each plate (rows G and H) whereby either 100 μl medium alone or 2% Triton X-100 was added, respectively. Target cells in log phase (p815) (107 cells/ml) were washed twice in HEPES buffer, electroporated (25 μf, 0.43 V) in europium (Eu) labeling buffer, washed twice, and resuspended in T-cell medium. Cells were incubated with either 10 μM Gag H-2Kd immunodominant peptide or without peptide (1 h at 37°C in 5% CO2). Following incubation, cells were washed three times in T-cell medium, resuspended at 105 cells/ml, and added to test plates (100 μl/well). Test plates were incubated for 3 h at 37°C in 5% CO2 and then centrifuged (300 × g for 3 min at RT). Supernatants (20 μl) were transferred into 200 μl enhancement solution (Perkin-Elmer), and Eu release was detected by time-resolved fluorescence on a Wallac 1420 Victor2 multilabel counter (Perkin-Elmer). Mean percent lysis was calculated from the average of triplicates based on the following formula:
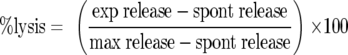 |
“Percent specific lysis” was determined by subtracting the “percent lysis” of targets not loaded with peptide from the “percent lysis” of peptide-loaded targets for each group.
In vivo CTL assay.
Spleen cells to be used as targets were isolated from naïve BALB/c mice and processed as described above. Isolated spleen cells were divided equally into two populations and differentially labeled with a “high” or “low” concentration of carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE) (5.0 or 0.5 μM, respectively; Molecular Probes) in Dulbecco's PBS for 15 min at 37°C. The cells were pelleted following labeling, and the carboxyfluorescein succinimidyl ester (CFSE)-high population was loaded with 10 μM Gag H-2Kd immunodominant peptide. Both populations were incubated 1 h at 37°C in 5% CO2. After incubation, cells were washed three times, and the two populations were mixed 1:1 in Dulbecco's PBS. Approximately 3 × 107 cells in 200 μl were injected intravenously into the tail vein of immunized mice and naïve control mice. Spleens were harvested from recipient mice 18 h after injection. The presence of transferred cells was determined by the detection of CFSE-positive cells by flow cytometry. Percent specific lysis was determined by the following formula:
 |
Gag (p24)-specific immunoglobulin ELISA.
Enzyme-linked immunosorbent assays (ELISAs) were performed in 96-well Dynatech Immulon II plates coated with 20 ng/well of recombinant HIV-I IIIB Gag p24 (Immunodiagnostics) diluted in carbonate-bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. The plates were washed five times with PBS-0.1% Tween and then blocked with 3% bovine serum albumin (BSA)-0.1% Tween-PBS for at least 2 h. The plates were washed five times, and mouse sera were serially diluted threefold in 1% BSA-0.1% Tween-PBS. The plates were incubated overnight at 4°C. After washing the plates, bound Gag-specific antibody was detected by adding biotin-SP-conjugated Affinipure goat anti-mouse IgG (Jackson ImmunoResearch Laboratories) diluted 1:15,000 in 1% BSA-0.1% Tween-PBS for 1 h at RT. The plates were then washed, and Streptavidin-POD conjugate (Roche Diagnostics) diluted 1:10,000 in 1% BSA-0.1% Tween-PBS was added and incubated at RT for 1 h. The plates were washed, and SureBlue TMB peroxidase substrate (Kirkegaard & Perry Laboratories) was then added and incubated at RT for 10 min. The substrate reaction mixture was stopped by the addition of 2 N sulfuric acid. The intensity of the resulting color was measured at 450 nm, and the endpoint titer was defined as the reciprocal of the serum dilution resulting in an optical density at 450 nm that was equal to the mean plus 2 standard deviations of the control naïve sera. The geometric mean and standard errors of the geometric means of titers for each group were calculated.
Detection of in vivo expression of Gag mRNA by real-time quantitative PCR.
For sample preparation, BALB/c mice (n = 5) were primed by i.m. inoculation of rVSVIN vectors (1 × 107 PFU) and boosted 8 weeks later by i.m. inoculation of the corresponding rVSVNJ vectors as described above. At the indicated times, fresh muscle tissues were weighed, and 10% (wt/vol) homogenates were prepared in PBS-sucrose phosphate buffer with glutamine. Fresh draining lymph nodes (pooled popliteal, iliac, and inguinal) were suspended in 2 ml PBS-sucrose phosphate buffer with glutamine to make 10% (wt/vol) homogenates. Homogenates were frozen in 140-μl aliquots at −70°C for later RNA extraction. Viral nucleic acid was extracted from homogenized tissues using the RNeasy Mini kit (Qiagen, Valencia, CA) and was reverse transcribed into cDNA using methods previously described (4). Briefly, detection of mRNA was done in a 20-μl reaction mix containing total RNA from tissue samples (5.0 μl or 0.5 μg), 10 μM of the anchored oligo(dT) primer, and reagents from the Sensiscript reverse transcriptase kit (Qiagen).
A primer-and-probe set for detecting RNA-encoding HIV gag and the primer nucleotide sequence for the anchored oligo(dT) primer were described previously (4). The primer-probe set for the ribosomal protein L15 (RPLO) housekeeping gene was selected using Primer Express software, version 2.0 (Applied Biosystems, Foster City, CA).
The duplex real-time quantitative PCR to detect Gag mRNA was carried out using the QuantiTect multiplex PCR kit (Qiagen). A 30-μl PCR mixture containing 5 μl of each 10-fold dilution of the HIV gag and RPLO cDNA used to generate standard curves or 5 μl of cDNA from tissue samples, 15 μl of 2× QuantiTect Multiplex PCR Master mix, 400 nM each of the forward and reverse primers for HIV gag and RPLO, and 200 nM of each probe. In order to distinguish the amplicons, the probes for HIV gag were labeled with VIC (6-carboxyrhodamine 6G), and those for RPLO were labeled with NED. Amplification and detection were performed with an ABI Prism 7500 Fast real-time PCR system using the following conditions: 15 min at 95°C to activate the HotStar Taq DNA polymerase, followed by 40 cycles of 60 s at 94°C and 90 s at 60°C. All samples were tested in duplicate.
Statistical analyses.
Data are expressed as means ± standard errors of the means of five individually assayed mice per test group. Where appropriate, statistical comparisons were conducted on log-transformed data using one-way analysis of variance followed by Dunnett's posttest. A P value of <0.05 was considered to be statistically significant (represented in figures by * [P < 0.05] and ** [P < 0.001]).
RESULTS
Generation of attenuated rVSV vectors expressing HIV gag.
The goal of these studies was to identify highly attenuated rVSV gag vectors that exhibit low neurotoxicity but high levels of Gag-specific immunogenicity. A prototypic rVSV-gag5 vector, similar in design to those of vectors which had shown efficacy in a rhesus macaque SHIV 89.6P challenge model (35), served as a reference standard for the studies described here in the hope that the further-attenuated vectors would exhibit reduced neurovirulence but equivalent immunogenicity. An “empty” rVSV vector not expressing a foreign antigen served as a negative control for the immunogenicity studies. Previously, we showed that the neurovirulence of VSV could be dramatically reduced through the combination of specific N gene translocations and G gene truncations (3). We chose two of these double mutant vectors, N3CT9 and N3CT1, for the expression of HIV-1 Gag. The N3CT9 and N3CT1 vectors have the N gene translocated to the third position of the viral genome and have the G protein CT truncated from 29 amino acids to 9 and 1 amino acids, respectively. Since we knew that these viruses were highly attenuated, we chose to place the HIV gag gene into the first position of the genome in order to maximize Gag expression. Likewise, the placement of the gag gene into the first position in the genome should also further attenuate the vectors by translocation of the viral genes one position further away from the 3′ transcription promoter. Therefore, these new vectors were designated N4CT9-gag1 and N4CT1-gag1, reflecting the new position of the N gene in the fourth position of the viral genome, the number of amino acids remaining in the CT of the G protein, and the position of gag in the genome (Fig. 1A).
Additionally, we previously showed that the combination of the CT1 truncation and a noncytopathic mutation of the M gene (14), MncpCT1, resulted in reduced neurovirulence (3). For these studies, the gene for Gag was placed into the first position of this vector genome to generate an MncpCT1-gag1 construct. The genetic organization and nomenclature of all the vectors used in these studies are shown in Fig. 1A. Gag expression was confirmed by Western blot analysis of infected BHK cell lysates (Fig. 1C). Vectors that had the gag gene in the first position of the genome expressed more Gag protein than was observed with rVSV-gag5.
Neurovirulence of attenuated rVSV vectors expressing HIV gag.
Mice are exquisitely sensitive to the i.c. instillation of wtVSV (37, 42). Young mice are also sensitive to the i.n. instillation of wtVSV, where it can infect and migrate through the olfactory nerve and into the olfactory lobe (13, 41). Infection by either route can lead to lethal viral encephalitis. Previous work demonstrated that prototypic rVSV vectors were less virulent than wtVSV, as they were less lethal following i.n. inoculation, although they still caused significant weight loss (11, 12, 25, 32). In contrast, the introduction of either the CT1 or CT9 mutation generated virus that no longer caused weight loss following i.n. inoculation (25, 32, 33, 39). Therefore, to assess the neurovirulence of the further-attenuated rVSV vectors described here, we utilized the heightened sensitivity of mice to VSV infection following direct i.c. inoculation. Five-week-old outbred Swiss Webster mice were inoculated i.c. with attenuated rVSV vectors expressing HIV-Gag in a dose-ranging study with log10 PFU increments such that an LD50 and a PD50 could be calculated. While the prototypic rVSV-gag5 vector was less virulent than wtVSV following i.n. inoculation, rVSV-gag5 exhibited lethality comparable to that of wtVSV following i.c. inoculation, and both had LD50s of less than 5 PFU (Fig. 2A). We previously showed that rVSV vectors containing a combination of two mutations (N3CT1 and N3CT9) were markedly less neurovirulent than rVSV vectors containing a single mutation (3). Assessed here, the N4CT1-gag1 and N4CT9-gag1 double mutants containing gag in the first position were even less virulent than their respective N3CT1 and N3CT9 progenitor vectors, demonstrating the attenuating effects of placing gag in the first position of the genome (Fig. 2). Remarkably, N4CT1-gag1 caused only mild weight loss (data not shown) and occasional illness at the highest dose tested, so neither an LD50 nor PD50 could be calculated for this vector (>108 PFU). Similarly, the MncpCT1-gag1 vector did not induce significant illness or death up to an i.c. dose of 108 PFU. N4CT9-gag1 was markedly less neurovirulent than the N3CT9 backbone but still induced paralysis and mortality at doses ranging from 104 to 107 PFU, although the time to death at the higher doses was longer than what was observed with N3CT9 (Fig. 2B).
FIG. 2.

Neurovirulence properties of rVSV-gag vectors in mice following i.c. inoculation. Groups of 5-week-old Swiss Webster mice (n = 10) were inoculated i.c. with log10-fold dilutions of rVSVIN vectors. Mice were monitored for 3 weeks for mortality and morbidity (paralysis). (A) The LD50 and PD50 were determined by the method described previously by Reed and Muench (30). Arrowheads indicate where the LD50 and PD50 were not achieved at the highest dose tested of 108 PFU. (B) Time to death was recorded for mice in the group receiving the dose immediately above the determined LD50.
Immunogenicity of attenuated rVSV vectors expressing HIV gag.
Previous studies with prototypic rVSV vectors in mice showed these vectors to be immunogenic following either i.n. or intraperitoneal inoculation (11, 12, 25). Our further studies with the prototypic rVSV-gag5 vector demonstrated that it was also immunogenic when administered by other routes (subcutaneous and intradermal) and that relatively robust immune responses were seen following i.m. inoculation (data not shown). As both the i.n. and i.m. routes can be readily tested in NHP and clinical studies, both routes of immunization were used to evaluate the attenuated rVSV vectors in mice. Mice were primed with rVSVIN vectors and boosted 2 months later with the corresponding GNJ switch vector (rVSVNJ) (36). The G protein serotype switch allows the boosting virus to circumvent the neutralizing antibody response that is generated after the priming inoculation. For all studies, sets of mice were immunized such that immune responses could be monitored at the peak of the effector phase 7 days following the prime, at the peak of the effector phase 5 days following the boost, and at 4 weeks following the boost as an assessment of the early memory level generated following boosting.
Following i.n. immunization, the rVSV-gag5 vector induced moderate T-cell responses to HIV Gag that increased upon boosting 2 months later (Fig. 3). While all three attenuated vectors were immunogenic, the T-cell responses trended lower than those observed in rVSV-gag5-vaccinated mice when either Gag tetramer staining (Fig. 3A) or IFN-γ ELISPOT responses to a Gag epitope (Fig. 3B) were assessed. There were detectable CTL responses in mice immunized with either N4CT1-gag1 or N4CT9-gag1, but they were slightly weaker than the CTL responses in mice immunized with the rVSV-gag5 vector (Fig. 3C and D). Serum antibody responses to the Gag p24 subunit were assessed 4 weeks after the boost. Surprisingly, the serum Gag-specific IgG titers in mice immunized with either N4CT1-gag1 or N4CT9-gag1 trended somewhat higher than titers induced by rVSV-gag5, while MncpCT1-gag1 did not induce detectable antibodies to Gag. Overall, MncpCT1-gag1 was the least immunogenic among the attenuated constructs following i.n. inoculation.
FIG. 3.

Immunogenicity of rVSV-gag vectors following i.n. inoculation. Mice (n = 5) were primed by i.n. inoculation of rVSVIN vectors (1 × 107 PFU) and boosted 8 weeks later by i.n. inoculation with the corresponding rVSVNJ vectors. T-cell responses were assessed in splenocytes at 7 days postprime and both 5 days and 4 weeks postboost. Humoral responses were assessed in serum at 4 weeks postboost. (A) HIV-1 Gag tetramer responses. Isolated splenocytes were stained with Gag tetramer (H-2Kd) (AMQMLKETI) and assessed by flow cytometry for the percentage of CD3+ CD8+ T cells that were Gag tetramer positive. (B) HIV-1 Gag IFN-γ ELISPOT responses. Isolated splenocytes were cultured overnight with the H-2Kd Gag immunodominant peptide (AMQMLKETI), and IFN-γ secretion was determined by ELISPOT analysis. The data are normalized to 106 spleen cells. (C) HIV-1 Gag ex vivo CTL responses at 5 days postboost. Isolated splenocytes were used in 3-h ex vivo CTL assays to lyse Eu-labeled P815 target cells loaded with the H-2Kd Gag immunodominant peptide. (D) HIV-1 Gag in vivo CTL responses at 4 weeks postboost. Gag-specific in vivo cytolytic activity was assessed by 18-h in vivo CTL assays in immunized mice. (E) HIV-1 Gag serum antibody responses at 4 weeks postboost. Serum was collected from immunized mice and assessed for Gag p24-specific total IgG by ELISA. Data are presented as the geometric mean titers. (F) VSV N IFN-γ ELISPOT responses. Vector-specific responses were assessed by measuring the T-cell response to VSV nucleoprotein (VSV N). Isolated splenocytes were cultured overnight with the H-2Ld VSV N immunodominant peptide (MPYLIDFGL), and IFN-γ secretion was determined by ELISPOT analysis. Immune responses induced by attenuated rVSVs were compared to immune responses induced by rVSV-gag5. *, P < 0.05; **, P < 0.001.
Following i.m. immunization, the rVSV-gag5 vector induced moderate and equivalent postprime and postboost peak T-cell responses to HIV-Gag (Fig. 4A and B). After priming, the T-cell responses induced by the three attenuated vectors were similar to that generated by rVSV-gag5. However, after boosting, there was a marked increase in the T-cell responses in mice immunized with either N4CT1-gag1 or N4CT9-gag1 that resulted in strong responses remaining 4 weeks after the boost (Fig. 4A and B). CTL responses in mice immunized with either N4CT1-gag1 or N4CT9-gag1 were notably more robust than the moderate CTL responses induced by the prototypic rVSV-gag5 vector (Fig. 4C and D). Moreover, the serum titers of Gag-specific IgG in mice immunized i.m. with either N4CT1-gag1 or N4CT9-gag1 were substantially higher than the low titers induced in mice immunized with rVSV-gag5. The humoral and cellular responses induced by MncpCT1-gag1 were generally similar to those induced by the rVSV-gag5 vector (Fig. 4).
FIG. 4.

Immunogenicity of rVSV-gag vectors following i.m. inoculation. Mice (n = 5) were primed by i.m. inoculation of rVSVIN vectors (1 × 107 PFU) and boosted 8 weeks later by i.m. inoculation of the corresponding rVSVNJ vectors. T-cell responses were assessed in splenocytes at 7 days postprime and both 5 days and 4 weeks postboost. Humoral responses were assessed in serum at 4 weeks postboost. Assays were performed as described in the legend of Fig. 3. (A) HIV-1 Gag tetramer responses. (B) HIV-1 Gag IFN-γ ELISPOT responses. (C) HIV-1 Gag ex vivo CTL responses 5 days postboost. (D) HIV-1 Gag in vivo CTL responses 4 weeks postboost. (E) HIV-1 Gag serum antibody responses 4 weeks postboost. (F) VSV N IFN-γ ELISPOT responses. Immune responses induced by attenuated rVSVs were compared to immune responses induced by rVSV-gag5. *, P < 0.05; **, P < 0.001.
Antivector immune responses.
The antivector immune responses were assessed by measuring T-cell responses to VSV N protein, which is the immunodominant antigen for T-cell responses in BALB/c mice (27, 46). While VSV-neutralizing antibody generated in mice following priming should not inhibit the boosting virus, due to serotype switching, T-cellular responses to the internal proteins of VSV might affect the boosting viruses, as both priming and boosting viruses have N, P, M, and L proteins from the Indiana serotype. The T-cell epitope from VSV N protein in BALB/c mice was mapped to a 9-amino-acid peptide (VSV-N275-283) (MPYLIDFGL) using a series of overlapping peptides (data not shown). This epitope was used in IFN-γ ELISPOT assays to assess the T-cell response to the VSV vectors in BALB/c mice.
Following i.n. immunization, the prototypic rVSV-gag5 vector induced a moderate T-cell response to the N protein that increased upon boosting 2 months later, yielding an IFN-γ ELISPOT response that peaked at over 2,000 spot-forming cells/million spleen cells (Fig. 3F). As seen with responses to HIV Gag, responses to the N protein in mice immunized i.n. with either N4CT1-gag1 or N4CT9-gag1 were less than the response induced by the prototype. This most likely reflects the reduced ability of these attenuated vectors to replicate and express antigens in murine nasal tissue. Following i.m. immunization, rVSV-gag5 induced strong T-cell responses to the N protein that increased upon boosting to levels averaging above 2,300 spot-forming cells/million spleen cells.
Effect of dose on immunogenicity.
We previously showed that prototypic rVSV vectors do not follow a dose response when inoculated by the i.n. route because the robust replication of prototypic vectors in nasal tissues allows lower doses of virus to be as immunogenic as higher doses (24). On the other hand, the attenuated vectors with reduced replication capacity described here compared more favorably to the prototypic vectors with regard to immunogenicity when they were inoculated into muscle, suggesting that this route of immunization is less dependent on the replicative capacity of the rVSV vector. To evaluate this phenomenon further, a dose-ranging study was performed with N4CT9-gag1 inoculated i.m. N4CT9-gag1 was inoculated at doses ranging from 102 to 106 PFU by the i.m. route, and cellular and humoral responses were assessed in splenocytes and serum 5 days postboost. A clear dose response was seen in both the cellular and humoral responses as depicted by Gag-specific CD8+ T-cell expansion (Fig. 5A), IFN-γ secretion (Fig. 5B), in vivo cytolytic activity (Fig. 5C), and the serum p24 Gag-specific IgG titers (Fig. 5D). The strongest cellular and humoral responses were detected at 106 PFU, the highest dose tested. Gag-specific cellular responses above background were seen at doses as low as 103 PFU, and Gag-specific IgG was detected at doses as low as 105 PFU. The presence of a clear dose response is indicative of the limited replication of this attenuated rVSV vector in vivo following i.m. inoculation, as immunogenicity was proportional to the inoculating dose size. However, robust immunogenicity was achieved by maximizing the inoculating dose, overcoming the need for robust replication.
FIG. 5.

Dose response of N4CT9-gag1 following i.m. inoculation. Mice (n = 5) were primed by i.m. inoculation with ascending doses of N4CT9IN-gag1 and boosted 8 weeks later by i.m. inoculation with the corresponding dose of N4CT9NJ-gag1. T-cell responses in splenocytes and humoral responses in serum were assessed 5 days postboost. Assays were performed as described in the legend of Fig. 3. (A) HIV-1 Gag tetramer responses. (B) HIV-1 Gag IFN-γ ELISPOT responses. (C) HIV-1 Gag in vivo CTL responses. (D) HIV-1 Gag serum antibody responses.
In vivo expression of HIV gag mRNA following i.m. immunization.
Both cellular and humoral immune responses to Gag were substantially higher postboost than was observed for rVSV-gag5 when the highly attenuated N4CT1-gag1 and N4CT9-gag1 vectors were administered i.m. (Fig. 4). The enhanced immunogenicity of attenuated vectors could have resulted from reduced antivector immunity following priming (allowing multiple infectious cycles of the boosting virus) or from enhanced expression of Gag due to a 3′-proximal position in the genome. To address the role of antivector immunity, we assessed VSV N protein-specific cellular immune responses in mice 2 months following priming (the time point at which boosting had occurred in the previous studies) with rVSV-gag5 and attenuated vectors. No significant differences in cellular responses to the N protein were found, suggesting that differences in anti-VSV immune responses were not playing a significant role in the enhanced Gag-specific immunogenicity of the attenuated vectors (data not shown).
To further understand how these attenuated viruses perform at levels similar to those of the prototype, we assessed the in vivo levels of Gag mRNA expression following i.m. priming and boosting with either rVSV-gag5, expressing Gag from the fifth gene in the genome, or N4CT9-gag1, which expresses Gag from the first gene in the genome. Gag mRNA levels were assessed both at the site of inoculation (muscle) and in the draining lymph nodes over the course of 8 days. For both viruses, Gag mRNA was detected in both muscle and draining lymph nodes as early as 4 h postinoculation and persisted for 2 to 4 days (Fig. 6). After boosting with N4CT9-gag1, Gag mRNA levels dropped more rapidly than was seen after priming, most likely due to the effect of T-cell-based antivector immunity limiting virus propagation. However, overall Gag mRNA levels were notably higher in muscles of mice inoculated with N4CT9-gag1 than in those inoculated with rVSV-gag5, consistent with genome position of the gag gene influencing in vivo expression. Importantly, there was no evidence of Gag mRNA persistence in mice inoculated with N4CT9-gag1.
FIG. 6.

Detection of in vivo gag mRNA transcription following i.m. priming and boosting with rVSV-gag vectors. Mice (n = 5 per time point) were primed by i.m. inoculation of rVSVIN vectors (1 × 107 PFU) and boosted 8 weeks later by i.m. inoculation of the corresponding GNJ protein exchange vectors. At 4 h and 1, 2, 4, and 8 days postpriming and postboosting, muscle (A) and draining lymph nodes (B) were excised from mice and homogenized in sucrose-phosphate-glutamic acid buffer. Tissue homogenates were frozen and then assessed later for Gag mRNA by quantitative real-time PCR. The limit of detection for the quantitative real-time PCR assay is 5 × 103 Gag mRNA copies per 10 mg tissue.
Effects of gag gene position in the VSV genome on immunogenicity.
The attenuated rVSV vectors were designed with the gag gene in the first position of the genome in order to maximize antigen expression and immunogenicity. The contribution of gag gene location to the increased immunogenicity that was observed with these vectors was of interest. Initially, we chose rVSV-gag5 as our benchmark for comparisons of immunogenicity, as this vector was similar to the rVSV-HIV/SHIV vectors that had shown efficacy in primate challenge studies (35). To evaluate the effect of gag gene placement on immunogenicity, we constructed a prototypic rVSV vector with gag in the first position of the genome (rVSV-gag1) (Fig. 1A). It should be noted that this vector was slightly less virulent than rVSV-gag5, as all the VSV genes are translocated further away from the 3′ transcription promoter, down-regulating viral gene expression (Fig. 2). Consequently, the LD50 for rVSV-gag1 was 125 PFU, compared to less than 5 PFU for rVSV-gag5 (Fig. 2A). We then compared the immunogenicity of rVSV-gag1 with that of the original prototypic rVSV-gag5 vector and with N4CT9-gag1 and N4CT1-gag1. At all three times points and with all assays, rVSV-gag1 induced more robust immune responses to Gag than did rVSV-gag5 (Fig. 7 and data not shown). This was most striking with the Gag-specific serum IgG titers, as rVSV-gag5 induced very low titers, averaging 1,241, while rVSV-gag1 induced an average titer of 79,838 (Fig. 7D). With this increased immunogenicity, the immune responses induced by rVSV-gag1 were similar in magnitude to those observed for N4CT1-gag1 and N4CT9-gag1, reaffirming the immunological benefit of placing the gag gene in the first position of the genome.
FIG. 7.

Effect of gag gene position on immunogenicity. Mice (n = 5) were primed by i.m. inoculation of rVSVIN vectors (1 × 107 PFU) and boosted 8 weeks later by i.m. inoculation of rVSVNJ vectors. T-cell responses were assessed in splenocytes at 7 days postprime and at 5 days and 4 weeks postboost. Humoral responses were assessed in serum at 4 weeks postboost. Assays were performed as described in the legend of Fig. 3. (A) HIV-1 Gag IFN-γ ELISPOT responses. (B) HIV-1 Gag serum antibody responses 4 weeks postboost. Immune responses induced by rVSV-gag5 and the attenuated rVSVs were compared to immune responses induced by rVSV-gag1. *, P < 0.05; **, P < 0.001.
DISCUSSION
The use of viral vectors based on VSV is a promising vaccine platform that is under development (reviewed in reference 2). While prototypic rVSV vectors are highly immunogenic in NHPs and small-animal models, there are concerns regarding vector safety, as an exploratory NHP neurovirulence study demonstrated that these prototypic rVSV vectors could cause significant neurological injury after intrathalamic inoculation (15). The results of that study prompted an attempt to generate attenuated forms of the prototypic rVSV vector that would exhibit reduced neurovirulence while retaining immunogenicity. Here, we describe the generation of three attenuated rVSV vectors expressing HIV-1 Gag and their assessment for neurovirulence and immunogenicity in mice. The attenuated vectors were dramatically less neurovirulent in mice and induced immune responses to Gag following i.m. immunization that were equivalent to, if not stronger than, responses induced by the prototypic vector.
The design of the attenuated vectors presented here utilized unique combinations of previously known attenuation phenotypes. We previously showed that the combination of N gene translocations with G protein CT tail truncations dramatically reduced vector neurovirulence in mice (3). Here, we found that the placement of the gag gene in the first position of the genome of these viruses resulted in further attenuation. Specifically, N4CT1-gag1 only rarely induced signs of disease following i.c. inoculation at the highest feasible dose tested, 108 PFU. This is a tremendous reduction in neurovirulence from what was observed previously with the prototypic rVSV-gag5 vector, which had an i.c. LD50 of less than 5 PFU. Placing the gag gene in the first position of the genome is presumed to further attenuate the virus by decreasing the transcription of all five VSV genes due to their increased distance from the 3′ transcription promoter. These attenuated VSV vector designs are therefore based on the combination of gene translocations and G protein CT truncation. Unlike viruses that are attenuated by point mutations, the chance of these attenuated VSV vectors reverting to a wild-type phenotype is highly unlikely, as large sections of the genome would need to be recombined or regenerated in a very specific manner. Viable VSV recombinants have not been previously isolated, and therefore, a recombination event producing viable virus is presumed to be an extremely-low-probability event. Ultimately, an assessment of these attenuated rVSV vectors in an NHP intrathalamic neurovirulence model will determine if they are safe for clinical evaluation.
While the attenuated rVSV vectors demonstrated dramatically reduced neurovirulence, they retained robust immunogenicity following i.m. inoculation. In contrast, the immunogenicity of attenuated rVSV vectors following i.n. inoculation was reduced compared to that observed with rVSV-gag5. Our data suggest that the two routes of immunization have different requirements for viral replication in order to support the induction of an immune response. More replication capacity is required for i.n. administration of rVSV vectors, and less replication capacity is required for i.m administration. This may reflect that virus, virally infected cells, and/or antigen can readily drain to lymph nodes following i.m. injection, while virus must spread across the nasal epithelium in order to have access to the nasal-associated lymphoid tissue (NALT). These findings with our attenuated rVSV vectors correlate with findings from a previous report demonstrating that G-protein-deleted, propagation-defective rVSV vectors are immunogenic following i.m. inoculation but not i.n. inoculation (26). Interestingly, in our studies, equivalent levels of Gag-specific antibodies were present in the sera of mice immunized i.n. with either prototypic or attenuated vectors, suggesting that there are different constraints on the vectors for the induction of humoral and cellular immunity. It is possible that high levels of antigen expression at the nasal epithelium produced sufficient quantities of Gag protein that could drain to NALT and initiate humoral immunity, while the level of virally infected cells in NALT required for optimal cellular responses was not achieved. Furthermore, there was evidence that the attenuated vectors replicated and expressed recombinant antigen to some extent when inoculated i.n., as evidenced by the presence of Gag-specific cellular immune responses following i.n. inoculation that were low but still well above the background levels of the various assays. This suggests that with further optimization, these, or similar, attenuated rVSV vectors might be sufficiently immunogenic by the i.n. route.
Our rVSV vector designs clearly benefited from placing the gene for the foreign antigen into the first position of the viral genome. Placement of the gag gene in front of all of the VSV genes enhanced Gag expression while reducing VSV gene expression, thereby both improving immunogenicity and reducing neurovirulence. When the prototypic rVSV-gag1 vector was compared to N4CT9-gag1, we found them to have equivalent immunogenicities following i.m. inoculation but dramatically different neurovirulences following i.c. inoculation. This suggests that immunogenicity is closely related to antigen expression levels and not replication capacity, while neurovirulence is associated with replication capacity. Overall, for rVSV vectors, this suggests that optimal immunogenicity may be achieved by maximizing antigen expression within an infected cell rather than by maximizing the replication or propagation capacity of the virus. Our data therefore highlight the importance of foreign gene placement within VSV for the ultimate success of these vectors.
In conclusion, we have generated three highly attenuated rVSV vectors expressing HIV-1 Gag that exhibited minimal neurotoxicity in a very stringent murine i.c. LD50 model yet elicited robust Gag-specific immune responses that were equivalent to, or stronger than, those elicited by the more virulent prototypic rVSV-gag5 vector. Further assessment of these attenuated rVSV vectors in NHP immunogenicity and neurovirulence models will help define a candidate rVSV HIV-1 vaccine vector for clinical evaluation.
Acknowledgments
This work was sponsored in part by an HIV-1 Vaccine Design and Development Team contract from the National Institutes of Health and the National Institute of Allergy and Infectious Diseases (HVDDT NO1-A1-25458).
We thank Deborah Branch, Ayuko Ota-Setlik, and Julia Li for assistance with animal studies; Dawn DeThomas for assistance with graphical design; and Christopher Parks for critical review of the manuscript. We are very grateful to John Rose for provision of pVSV1XN, and we reiterate our thanks to Michael Whitt for the plasmid containing the Mncp gene.
Footnotes
Published ahead of print on 17 October 2007.
REFERENCES
- 1.Brandsma, J. L., M. Shlyankevich, L. Buonocore, A. Roberts, S. M. Becker, and J. K. Rose. 2007. Therapeutic efficacy of vesicular stomatitis virus-based E6 vaccination in rabbits. Vaccine 25751-762. [DOI] [PubMed] [Google Scholar]
- 2.Clarke, D. K., D. Cooper, M. A. Egan, R. M. Hendry, C. L. Parks, and S. A. Udem. 2006. Recombinant vesicular stomatitis virus as an HIV-1 vaccine vector. Springer Semin. Immunopathol. 28239-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke, D. K., F. Nasar, M. Lee, J. E. Johnson, K. Wright, P. Calderon, M. Guo, R. Natuk, D. Cooper, R. M. Hendry, and S. A. Udem. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J. Virol. 812056-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coleman, J. W., E. Ogin-Wilson, J. E. Johnson, F. Nasar, T. P. Zamb, D. K. Clarke, R. M. Hendry, and S. A. Udem. 2007. Quantitative multiplex assay for simultaneous detection of the Indiana serotype of vesicular stomatitis virus and HIV gag. J. Virol. Methods 14355-64. [DOI] [PubMed] [Google Scholar]
- 5.Egan, M. A., S. Y. Chong, S. Megati, D. C. Montefiori, N. F. Rose, J. D. Boyer, M. K. Sidhu, J. Quiroz, M. Rosati, E. B. Schadeck, G. N. Pavlakis, D. B. Weiner, J. K. Rose, Z. R. Israel, S. A. Udem, and J. H. Eldridge. 2005. Priming with plasmid DNAs expressing interleukin-12 and simian immunodeficiency virus gag enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. AIDS Res. Hum. Retrovir. 21629-643. [DOI] [PubMed] [Google Scholar]
- 6.Egan, M. A., S. Y. Chong, N. F. Rose, S. Megati, K. J. Lopez, E. B. Schadeck, J. E. Johnson, A. Masood, P. Piacente, R. E. Druilhet, P. W. Barras, D. L. Hasselschwert, P. Reilly, E. M. Mishkin, D. C. Montefiori, M. G. Lewis, D. K. Clarke, R. M. Hendry, P. A. Marx, J. H. Eldridge, S. A. Udem, Z. R. Israel, and J. K. Rose. 2004. Immunogenicity of attenuated vesicular stomatitis virus vectors expressing HIV type 1 Env and SIV Gag proteins: comparison of intranasal and intramuscular vaccination routes. AIDS Res. Hum. Retrovir. 20989-1004. [DOI] [PubMed] [Google Scholar]
- 7.Fields, B. N., and K. Hawkins. 1967. Human infection with the virus of vesicular stomatitis during an epizootic. N. Engl. J. Med. 277989-994. [DOI] [PubMed] [Google Scholar]
- 8.Geisbert, T. W., S. Jones, E. A. Fritz, A. C. Shurtleff, J. B. Geisbert, R. Liebscher, A. Grolla, U. Stroher, L. Fernando, K. M. Daddario, M. C. Guttieri, B. R. Mothe, T. Larsen, L. E. Hensley, P. B. Jahrling, and H. Feldmann. 2005. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girard, M. P., S. K. Osmanov, and M. P. Kieny. 2006. A review of vaccine research and development: the human immunodeficiency virus (HIV). Vaccine 244062-4081. [DOI] [PubMed] [Google Scholar]
- 10.Haglund, K., J. Forman, H. G. Krausslich, and J. K. Rose. 2000. Expression of human immunodeficiency virus type 1 Gag protein precursor and envelope proteins from a vesicular stomatitis virus recombinant: high-level production of virus-like particles containing HIV envelope. Virology 268112-121. [DOI] [PubMed] [Google Scholar]
- 11.Haglund, K., I. Leiner, K. Kerksiek, L. Buonocore, E. Pamer, and J. K. Rose. 2002. High-level primary CD8+ T-cell response to human immunodeficiency virus type 1 gag and env generated by vaccination with recombinant vesicular stomatitis viruses. J. Virol. 762730-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haglund, K., I. Leiner, K. Kerksiek, L. Buonocore, E. Pamer, and J. K. Rose. 2002. Robust recall and long-term memory T-cell responses induced by prime-boost regimens with heterologous live viral vectors expressing human immunodeficiency virus type 1 Gag and Env proteins. J. Virol. 767506-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huneycutt, B. S., I. V. Plakhov, Z. Shusterman, S. M. Bartido, A. Huang, C. S. Reiss, and C. Aoki. 1994. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 63581-95. [DOI] [PubMed] [Google Scholar]
- 14.Jayakar, H. R., and M. A. Whitt. 2002. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J. Virol. 768011-8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson, J. E., F. Nasar, J. W. Coleman, R. E. Price, A. Javadian, K. Draper, M. Lee, P. A. Reilly, D. K. Clarke, R. M. Hendry, and S. A. Udem. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 36036-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson, K. M., J. E. Vogel, and P. H. Peralta. 1966. Clinical and serological response to laboratory-acquired human infection by Indiana type vesicular stomatitis virus (VSV). Am. J. Trop. Med. Hyg. 15244-246. [DOI] [PubMed] [Google Scholar]
- 17.Jones, S. M., H. Feldmann, U. Stroher, J. B. Geisbert, L. Fernando, A. Grolla, H. D. Klenk, N. J. Sullivan, V. E. Volchkov, E. A. Fritz, K. M. Daddario, L. E. Hensley, P. B. Jahrling, and T. W. Geisbert. 2005. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 11786-790. [DOI] [PubMed] [Google Scholar]
- 18.Kahn, J. S., A. Roberts, C. Weibel, L. Buonocore, and J. K. Rose. 2001. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J. Virol. 7511079-11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahn, J. S., M. J. Schnell, L. Buonocore, and J. K. Rose. 1999. Recombinant vesicular stomatitis virus expressing respiratory syncytial virus (RSV) glycoproteins: RSV fusion protein can mediate infection and cell fusion. Virology 25481-91. [DOI] [PubMed] [Google Scholar]
- 20.Kapadia, S. U., J. K. Rose, E. Lamirande, L. Vogel, K. Subbarao, and A. Roberts. 2005. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology 340174-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovacs, G. R., C. L. Parks, N. Vasilakis, and S. A. Udem. 2003. Enhanced genetic rescue of negative-strand RNA viruses: use of an MVA-T7 RNA polymerase vector and DNA replication inhibitors. J. Virol. Methods 11129-36. [DOI] [PubMed] [Google Scholar]
- 22.Lawson, N. D., E. A. Stillman, M. A. Whitt, and J. K. Rose. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. USA 924477-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liniger, M., A. Zuniga, and H. Y. Naim. 2007. Use of viral vectors for the development of vaccines. Expert Rev. Vaccines 6255-266. [DOI] [PubMed] [Google Scholar]
- 24.Natuk, R. J., D. Cooper, M. Guo, P. Calderon, K. J. Wright, F. Nasar, S. Witko, D. Pawlyk, M. Lee, J. DeStefano, D. Tummolo, A. S. Abramovitz, S. Gangolli, N. Kalyan, D. K. Clarke, R. M. Hendry, J. H. Eldridge, S. A. Udem, and J. Kowalski. 2006. Recombinant vesicular stomatitis virus vectors expressing herpes simplex virus type 2 gD elicit robust CD4+ Th1 immune responses and are protective in mouse and guinea pig models of vaginal challenge. J. Virol. 804447-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Publicover, J., E. Ramsburg, and J. K. Rose. 2004. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J. Virol. 789317-9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Publicover, J., E. Ramsburg, and J. K. Rose. 2005. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J. Virol. 7913231-13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puddington, L., M. J. Bevan, J. K. Rose, and L. Lefrancois. 1986. N protein is the predominant antigen recognized by vesicular stomatitis virus-specific cytotoxic T cells. J. Virol. 60708-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsburg, E., J. Publicover, L. Buonocore, A. Poholek, M. Robek, A. Palin, and J. K. Rose. 2005. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J. Virol. 7915043-15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramsburg, E., N. F. Rose, P. A. Marx, M. Mefford, D. F. Nixon, W. J. Moretto, D. Montefiori, P. Earl, B. Moss, and J. K. Rose. 2004. Highly effective control of an AIDS virus challenge in macaques by using vesicular stomatitis virus and modified vaccinia virus Ankara vaccine vectors in a single-boost protocol. J. Virol. 783930-3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reed, E., and H. Muench. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27493-497. [Google Scholar]
- 31.Reuter, J. D., B. E. Vivas-Gonzalez, D. Gomez, J. H. Wilson, J. L. Brandsma, H. L. Greenstone, J. K. Rose, and A. Roberts. 2002. Intranasal vaccination with a recombinant vesicular stomatitis virus expressing cottontail rabbit papillomavirus L1 protein provides complete protection against papillomavirus-induced disease. J. Virol. 768900-8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberts, A., L. Buonocore, R. Price, J. Forman, and J. K. Rose. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J. Virol. 733723-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts, A., E. Kretzschmar, A. S. Perkins, J. Forman, R. Price, L. Buonocore, Y. Kawaoka, and J. K. Rose. 1998. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J. Virol. 724704-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose, J. K., and M. A. Whitt. 2001. Rhabdoviridae: the viruses and their replication, p. 1223. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 35.Rose, N. F., P. A. Marx, A. Luckay, D. F. Nixon, W. J. Moretto, S. M. Donahoe, D. Montefiori, A. Roberts, L. Buonocore, and J. K. Rose. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106539-549. [DOI] [PubMed] [Google Scholar]
- 36.Rose, N. F., A. Roberts, L. Buonocore, and J. K. Rose. 2000. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J. Virol. 7410903-10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabin, A., and P. Olitsky. 1938. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus. I. Effect of age on the invasion of the brain by virus instilled in the nose. J. Exp. Med. 6615-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlereth, B., J. K. Rose, L. Buonocore, V. ter Meulen, and S. Niewiesk. 2000. Successful vaccine-induced seroconversion by single-dose immunization in the presence of measles virus-specific maternal antibodies. J. Virol. 744652-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schnell, M. J., L. Buonocore, E. Boritz, H. P. Ghosh, R. Chernish, and J. K. Rose. 1998. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 171289-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorner, A. R., and D. H. Barouch. 2007. HIV-1 vaccine development: progress and prospects. Curr. Infect. Dis. Rep. 971-75. [DOI] [PubMed] [Google Scholar]
- 41.van den Pol, A. N., K. P. Dalton, and J. K. Rose. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 761309-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wertz, G. W., V. P. Perepelitsa, and L. A. Ball. 1998. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc. Natl. Acad. Sci. USA 953501-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whelan, S. P., L. A. Ball, J. N. Barr, and G. T. Wertz. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. USA 928388-8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witko, S. E., C. S. Kotash, R. M. Nowak, J. E. Johnson, L. A. Boutilier, K. J. Melville, S. G. Heron, D. K. Clarke, A. S. Abramovitz, R. M. Hendry, M. S. Sidhu, S. A. Udem, and C. L. Parks. 2006. An efficient helper-virus-free method for rescue of recombinant paramyxoviruses and rhadoviruses from a cell line suitable for vaccine development. J. Virol. Methods 13591-101. [DOI] [PubMed] [Google Scholar]
- 45.Wyatt, L. S., B. Moss, and S. Rozenblatt. 1995. Replication-deficient vaccinia virus encoding bacteriophage T7 RNA polymerase for transient gene expression in mammalian cells. Virology 210202-205. [DOI] [PubMed] [Google Scholar]
- 46.Yewdell, J. W., J. R. Bennink, M. Mackett, L. Lefrancois, D. S. Lyles, and B. Moss. 1986. Recognition of cloned vesicular stomatitis virus internal and external gene products by cytotoxic T lymphocytes. J. Exp. Med. 1631529-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]