

Abstract
We previously reported that defined components of the host transcription machinery are recruited to human cytomegalovirus immediate-early (IE) transcription sites, including cdk9 and cdk7 (S. Tamrakar, A. J. Kapasi, and D. H. Spector, J. Virol. 79:15477-15493, 2005). In this report, we further document the complexity of this site, referred to as the transcriptosome, through identification of additional resident proteins, including viral UL69 and cellular cyclin T1, Brd4, histone deacetylase 1 (HDAC1), and HDAC2. To examine the role of cyclin-dependent kinases (cdks) in the establishment of this site, we used roscovitine, a specific inhibitor of cdk1, cdk2, cdk7, and cdk9, that alters processing of viral IE transcripts and inhibits expression of viral early genes. In the presence of roscovitine, IE2, cyclin T1, Brd4, HDAC1, and HDAC2 accumulate at the transcriptosome. However, accumulation of cdk9 and cdk7 was specifically inhibited. Roscovitine treatment also resulted in decreased levels of cdk9 and cdk7 RNA. There was a corresponding reduction in cdk9 protein but only a modest decrease in cdk7 protein. However, overexpression of cdk9 does not compensate for the effects of roscovitine on cdk9 localization or viral gene expression. Delaying the addition of roscovitine until 8 h postinfection prevented all of the observed effects of the cdk inhibitor. These data suggest that IE2 and multiple cellular factors needed for viral RNA synthesis accumulate within the first 8 h at the viral transcriptosome and that functional cdk activity is required for the specific recruitment of cdk7 and cdk9 during this time interval.
Human cytomegalovirus (HCMV), a herpesvirus, is a prevalent pathogen infecting between 50 to 80% of adults in the United States. HCMV causes disease largely in immunocompromised patients, organ transplant recipients, and the developing fetus. Congenital HCMV is the major viral cause of birth defects, leading to permanent disabilities such as hearing and vision loss, mental disabilities, and even death. As yet, there is no cure or effective preventative treatment for HCMV.
Immediately after the viral particles contact the cellular plasma membrane, normal host cellular functions are disrupted. The combination of the interactions between the virus and the host that are established and the altered cellular functions result in an environment that is optimal for viral replication (for a review, see reference 16). Viral gene expression is temporally regulated, beginning with the immediate-early (IE) genes, which do not require de novo host or viral protein synthesis for expression. The IE gene products activate the expression of viral early genes, which in turn initiate and regulate viral DNA synthesis. After the onset of viral DNA synthesis, the late viral genes, which encode primarily structural proteins, are expressed and lead to the eventual release of virus from the cell.
In order for a productive infection to ensue, HCMV must commandeer the host transcriptional machinery to establish efficient viral IE RNA synthesis. Understanding how the host transcriptional machinery is redirected for viral gene transcription is key to unveiling how a productive HCMV infection develops in the host cell. Initially, this is achieved by the virus taking advantage of the existing nuclear architecture as well as utilizing available cellular factors in the host cell. Upon entry, a subset of the viral genomes are deposited inside the nucleus, adjacent to dynamic promyelocytic leukemia (PML) oncogenic domains (PODs) (also known as ND10 structures) (17). It is these viral genomes that serve as the template for viral IE transcription. The major IE proteins, IE1-72 and IE2-86, also localize to the sites of viral IE transcription. Between 3 and 6 h postinfection (p.i.), IE1 protein and several POD-associated proteins (including PML) become dispersed throughout the nucleus due to IE1 activity (2, 3, 26, 44). IE2 protein, however, persists at the punctate viral IE transcription sites, which we will refer to as the transcriptosome.
HCMV transcription is directed by the cellular RNA polymerase II (RNAP II). In humans, the C-terminal domain (CTD) of the largest subunit (Rpb1) of RNAP II is composed of the consensus heptapeptide sequence Try-Ser-Pro-Thr-Ser-Pro-Ser and is susceptible to high levels of phosphorylation during the transcription cycle (for a review, see references 22, 25, and 29). In the uninfected cell, the primary kinases responsible for RNAP II CTD phosphorylation are cdk7 and cdk9. A hypophosphorylated form of RNAP II (IIa) is recruited to the preinitiation complex at the gene promoter by the general transcription factors. Initiation proceeds when the cdk7 complex phosphorylates the CTD at the serine 5 residues, resulting in a hyperphosphorylated RNAP II (IIo) that recruits the RNA-capping enzymes. Eventually, further phosphorylation on serine 2 residues of the CTD by the cdk9 complex recruits factors required for RNA cleavage/polyadenylation and splicing and commits RNAP II to productive elongation. Recently, the bromodomain-containing protein Brd4 has been shown to interact with the active cdk9 complex via cyclin T1 and has been implicated in enhancing the recruitment of the cdk9 complex to the stalled RNAP II at acetylated promoter regions, stimulating cdk9 phosphorylation of RNAP II (18, 49). We previously reported that active CTD kinase complexes, CAK (cdk7/cyclin H/MAT1) and positive transcription elongation factor b (P-TEFb) (cdk9/cyclin T1), are upregulated during the HCMV infection (39). In addition, by 8 h p.i., we and others have shown that the transcriptionally active form of RNAP II, TBP, TFIIB, cdk9, and cdk7 all colocalize with IE1/2 protein at the viral transcriptosome (2, 17, 39). HCMV also encodes a kinase, UL97, which can phosphorylate the RNAP II CTD in vitro, although a study by Baek et al. suggests that UL97 may not be responsible for CTD phosphorylation in vivo (4).
The cyclin-dependent kinase (cdk) inhibitor roscovitine has been utilized to show that there is an IE requirement for cdk activity during the HCMV infection (7, 31). Roscovitine is a purine derivative that specifically inhibits cdk1, cdk2, and cdk5 (in neural cells) in vitro with an approximate 50% inhibitory concentration (IC50) of 0.7 μM and inhibits cdk7 and cdk9 with an approximate IC50 of 0.6 μM (5, 12, 15, 24). However, in addition to its use in manipulating cdk activity during infection in experimental studies, roscovitine is being closely looked at as a possible antiviral drug. Since most antiviral drugs are targeted against viral proteins, often resulting in resistant strains, roscovitine is a promising option because of its inhibition of cellular targets (for a review, see reference 37). Roscovitine has also been shown to inhibit viral replication of herpes simplex virus type 1, vesicular stomatitis virus, and Epstein-Barr virus and transcription of human immunodeficiency virus type 1 (10, 11, 14, 19, 27, 33-36, 40, 43).
Our lab has published a series of studies that initially revealed that treatment with roscovitine during an HCMV infection significantly alters viral gene expression, resulting in differential processing of several IE RNAs, inhibition of viral early and late gene expression and DNA replication, and lowered viral titers (31, 32, 39). We further showed that some of the effects of roscovitine might be attributed to alterations in the phosphorylation of RNAP II. Since the CTD acts as a platform for recruiting various transcription factors and components of the RNA-processing machinery, changes in the phosphorylation of the CTD alter the affinity of proteins for the transcription complex. During HCMV infection there is an increase in the level of hyperphosphorylated RNAP IIo, but in the presence of roscovitine, this virus-specific hyperphosphorylation is prevented. Since it seems that hyperphosphorylation of RNAP II is required for redirecting the cellular transcription machinery and initiating efficient viral IE transcription, the impairment in viral gene expression in the presence of roscovitine may be a result of an inability to recruit essential viral and cellular proteins to the sites of IE transcription. Surprisingly, the effects of roscovitine on the hyperphosphorylation of RNAP II and viral gene expression are not observed if addition of the drug is delayed until 8 h p.i. (31, 39).
The goal of this study was to further define the viral transcriptosome and determine the basis of the cdk requirement for its function during the first 8 h of the infection. We identify additional viral and cellular proteins that accumulate at the transcriptosome, including several proteins that are involved in modifying chromatin structure. Infection in the presence of roscovitine specifically affected cdk9 and cdk7 localization to the viral transcriptosome, while the accumulation of IE2 and the other identified proteins at this site was not altered by the presence of the drug. We also observed that roscovitine treatment has a significant inhibitory effect on the steady-state levels of cdk9 protein and cdk9 and cdk7 RNAs in mock- and HCMV-infected cells. However, overexpression of cdk9 does not compensate for the effects of roscovitine treatment at the beginning of HCMV infection, suggesting that the reduced levels of cdk9 are not responsible for its lack of accumulation at the transcriptosome. In accord with our previous studies, when the addition of roscovitine was delayed until 8 h p.i., the expression of cdk9 protein and its localization to the transcriptosome were no longer inhibited. This lack of effect on cdk9 expression was virus specific, as delaying the addition of drug in mock-infected cells still resulted in inhibition of cdk9 expression. These data support previous findings that many of the cellular factors needed for viral IE transcription are recruited within the first hours of infection. By 8 h p.i., the nuclear environment is sufficiently modified in infected cells, as can be seen by differences in the sensitivity to inhibitors such as roscovitine, to allow for a productive infection.
MATERIALS AND METHODS
Cell culture and virus.
Human foreskin fibroblasts (HFF) were obtained from the University of California, San Diego, Medical Center and cultured in Earle's minimal essential medium supplemented with 10% heat inactivated fetal bovine serum, 1.5 μg/ml amphotericin B, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. All reagents were from Invitrogen (Carlsbad, CA). Cells were kept in incubators maintained at 37°C with 7% CO2. The Towne strain of HCMV was obtained from the American Type Culture Collection (VR 977) and propagated as previously described (38).
Cell synchronization and infections.
HFF (passage numbers 15 to 20) were synchronized in G0 phase by allowing them to grow to confluence as previously described (30). At 3 days after confluence, the cells were trypsinized, replated at a lower density to allow progression into the cell cycle, and infected at a multiplicity of infection (MOI) of 5 with HCMV Towne or mock infected with tissue culture supernatants. Viral and mock supernatants were replaced with fresh media after 8 h p.i. For infections in the presence of roscovitine (Sigma Aldrich, St. Louis, MO), roscovitine (16 μM) or dimethyl sulfoxide (DMSO) as a control was added at the time of infection or at 8 h p.i. Medium with roscovitine or DMSO was replaced every 24 h. At various times p.i., cells were harvested and stored at −80°C for Western blot analysis or fixed in 2% formaldehyde solution for immunofluorescence assay (IFA).
Antibodies.
The antibodies used were cdk7 monoclonal antibody (MAb) MO-1 and DRB sensitivity-inducing factor (DSIF) MAb (BD Pharmingen, La Jolla, CA); CH16.0 MAb (Virusys Corporation, Sykesville, MD); cdk9 polyclonal antibody sc-484, cdk2 polyclonal antibody sc-163, cyclin T1 polyclonal antibody sc-10750, β-catenin MAb sc-7963, histone deacetylase 2 (HDAC2) polyclonal antibody sc-7899, and hemagglutinin (HA) MAb sc-7392 (Santa Cruz Biotechnology, Santa Cruz, CA); β-actin MAb (Sigma-Aldrich, St. Louis, MO); ARNA3 MAb and IE2 MAb (Chemicon, Temecula, CA); H5 MAb (Covance, Berkeley, CA); HDAC1 polyclonal antibody (Abcam, Inc., Cambridge, MA); Brd4 polyclonal antibody (a gift of K. Ozato); UL69 MAb (a gift of W. Britt); goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase (HRP) and goat anti-rabbit IgG-HRP (Calbiochem, San Diego, CA); goat anti-mouse IgM-HRP, goat anti-rabbit IgG-Cy3, fluorescein-conjugated isotype-specific secondary antibodies, ChromPure human IgG Fc fragment (HuFc), and normal rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA); and mouse isotype-specific Ig controls (Zymed, San Francisco, CA).
Western blot analysis.
Cells were lysed in Laemmli reducing sample buffer (50 mM Tris [pH 6.8], 0.2% sodium dodecyl sulfate [SDS], 10% glycerol, 5% 2-β-mercaptoethanol, 50 mM NaF, 0.5 mM Na3VO4, 5 mM β-glycerophosphate). The lysates were then sonicated briefly and boiled for 5 min. Proteins from an equivalent number of cells were separated on a 6%, 8%, or 10% SDS-polyacrylamide gel and transferred to nitrocellulose. Membranes were stained with amido black to assess protein loading in each lane. The blots were incubated with 5% nonfat dried milk in TBST (10 mM Tris [pH 8.0], 150 mM NaCl, and 0.1% Tween 20) and incubated with primary antibodies diluted in 5% milk-TBST as follows: cdk7 MAb, 1:500; rabbit anti-cdk9, 1:250; rabbit anti-cdk2 1:500; β-actin MAb, 1:10,000; ARNA3 MAb, 1:300, H5 MAb, 1:250, β-catenin MAb, 1:10,000, and CH16.0 MAb 1:10,000. Following washes with TBST, blots were incubated with anti-mouse IgG-HRP or anti-rabbit IgG-HRP diluted 1:2,000 or with anti-mouse IgM-HRP diluted 1:8,000. After washing in TBST, proteins were detected using SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL) according to the manufacturer's instructions.
IFA.
Cells were infected as described above with HCMV Towne or mock infected and were plated onto sterile glass coverslips. At specified time points, the coverslips were washed in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and fixed with 2% formaldehyde solution in PBS for 10 min. Formaldehyde-fixed cells were permeabilized in 0.2% Triton X-100 and incubated in 10% normal goat serum. Cells were subsequently incubated with one or more of the following specific antibodies diluted in 10% normal goat serum: IE2 MAb, 1:500; rabbit anti-cdk9, 1:50; HA MAb, 1:25; cdk7 MAb, 1:50; rabbit anti-Brd4, 1:1,000; rabbit anti-cyclin T1, 1:50; UL69 MAb, 1:200; DSIF MAb, 1:25; rabbit anti-HDAC1, 1:1,000; and rabbit anti-HDAC2, 1:200. Nonspecific IgG antibodies were used as controls. After PBS washes, cells were incubated with Hoechst stain and one or more of the appropriate fluorescein-conjugated secondary antibodies diluted in blocking solution: isotype-specific mouse IgG, 1:50; and goat anti-rabbit IgG-Cy3, 1:600. After PBS washes, the slips were mounted onto glass slides with SlowFade Gold (Molecular Probes, Eugene, OR). Confocal images were captured at the UCSD Cancer Center Digital Imaging Shared Resource using a Deltavision microscope and deconvolved with SoftWorx software. All images are 0.2-μm sections with a magnification of ×1,000 under oil immersion. For cdk9 IFA at 14 h p.i., the blocking step was modified by incubation with HuFc diluted 1:10 in blocking solution in order to prevent adsorption of the rabbit polyclonal cdk9 antibody to virus-encoded Fc receptor that accumulates in the Golgi apparatus.
Quantitative real-time RT-PCR.
Cells were infected as described above. The cells were harvested at various times p.i., and total RNA was isolated from the harvested cells with the RNeasy RNA miniprep kit (QIAGEN, Valencia, CA). The concentration of each sample was determined by UV spectrophotometry. Quantitative real-time reverse transcription-PCR (RT-PCR) was performed in an Applied Biosystems ABI Prism 7000 sequence detection system with the TaqMan one-step RT-PCR master mix reagent kit (Applied Biosystems, Branchburg, NJ) and oligonucleotide primers and TaqMan dual-labeled (5′ 6-carboxyfluorescein and 3′ black hole quencher-1) probes (Integrated DNA Technologies, Coralville, IA) (Table 1). The reaction volume was 50 μl, with 50 ng of RNA for each sample. A standard curve was used to calculate the relative amount of specific RNA present in a sample, from which the induction of transcription of the gene was calculated by comparison to the value obtained for the specific RNA from the 8 h p.i. untreated mock-infected cells. As an additional control for the amount of RNA in each reaction, samples were normalized with primers and a TaqMan probe specific to the cellular housekeeping glucose-6-phosphate dehydrogenase (G6PD) gene.
TABLE 1.
Quantitative real-time PCR primers and TaqMan probes
| RNA | Sequence
|
||
|---|---|---|---|
| Forward primer | Reverse primer | TaqMan probe | |
| cdk9 | 5′-TGCTGCTTAACGGCCTCTACTA-3′ | 5′-CACATTAGCAGCCTTCATGTCC-3′ | 5′ TCCACAGAAACAAGATCCTGCATAGG-3′ |
| cdk7 | 5′-TCTTATTTAATCCATGTGCTCGAATT-3′ | 5′-CCCTGGCCGATTACTGAAATAC-3′ | 5′-CGGCCACACAGGCACTGAAAATGAA-3′ |
| G6PD | 5′-TCTACCGCATCGACCACTACC-3′ | 5′-GCGATGTTGTCCCGGTTC-3′ | 5′-ATGGTGCTGAGATTTGCCAACAGGA-3′ |
cdk9HA-overexpressing cells.
The pRc-CMV-cdk9HA plasmid was obtained from X. Graña (Temple University School of Medicine, Philadelphia, PA). The plasmid was cut with XbaI and EcoRI to release the cdk9HA insert. The cdk9HA insert ends were filled in using Klenow T4 DNA polymerase. A lentivector containing the EF1α promoter was obtained from I. Verma (Salk Institute, La Jolla, CA). A hygromycin B resistance gene expressed under control of the thymidine kinase promoter was cloned into the SmaI site downstream of the EF1α promoter. The pLV-EF1α-TKHygro was cut at a unique BamHI site, and the linearized vector ends were subsequently filled in with Klenow polymerase. The blunt cdk9HA insert was ligated into the lentivector to produce pLV-EF1α-cdk9HA-TKHygro.
293FT cells were cultured and transfected with the lentiviral vector and packaging mix as described for the ViraPower lentiviral expression system (Invitrogen, Carlsbad, CA). Briefly, 6 × 107 cells were transfected in 30 ml OptiMEM (Invitrogen), 360 μl Lipofectamine 2000 (Invitrogen), 90 μg ViraPower packaging DNA mix, and 30 μg pLV-EF1-cdk9HA-TKHygro DNA. At 72 h posttransfection, the lentivirus-containing supernatant was collected, spun down to remove cellular debris, and filtered. It was immediately used to transduce subconfluent flasks of HFF cells with a 1:1 dilution of lentivirus and HFF media in the presence of 3 μg/ml Polybrene. At confluence, the HFF cells were split 1:2, and at 24 h postsplitting they were transduced a second time with fresh lentivirus. At 48 h after the second transduction, the HFF cells were put under hygromycin B selection (25 to 40 μg/ml). After 10 days under selection, the HFF flasks were confluent, and cdk9HA expression was confirmed by Western blotting and IFA with the HA MAb. The G0-synchronized cdk9HA-overexpressing HFF and untransduced HFF (passage 16) were then infected in the presence of roscovitine or DMSO as described above. Samples for Western blot analysis were harvested at 8, 24, 48, and 72 h p.i., and samples for IFA were fixed at 8 h p.i.
CTD kinase assay.
Mock cdk9HA-expressing cells were harvested by trypsinization, and 5 × 105 cells were lysed in NETN buffer (100 mM NaCl, 20 mM Tris-HCl [pH 8], 1 mM EDTA, 0.5% IGEPAL CA-630 [Sigma-Aldrich], 1× protease inhibitor cocktail [Roche Chemicals, Indianapolis, IN], 50 mM NaF, 0.5 mM Na3VO4, and 5 mM β-glycerophosphate) by rotating at 4°C for 30 min. The exogenous cdk9HA was specifically immunoprecipitated from cells using 15 μg of rabbit anti-cdk9 and control rabbit IgG serum and 30 μg of rabbit anti-HA coupled to 30 μl of protein A Plus-agarose beads (Santa Cruz Biotechnology) with dimethyl pimelimidate dihydrocholoride (Sigma-Aldrich). Lysates and antibody-coupled beads were incubated for 4 h with rotating at 4°C. Immunocomplexes were washed twice in NETN buffer, and half of each sample was boiled in 2× Laemmli buffer. For Western blot analysis, approximately 7.5 × 104 cell equivalents for the pre- and postimmunoprecipitate samples and approximately 1.25 × 105 cell equivalents for the immunoprecipitate samples were loaded onto an 8% SDS-polyacrylamide gel.
One half of the total immunoprecipitate (5 × 105 cell equivalents) was used in the kinase assay as described previously (39). Briefly, the immunocomplexes were washed once with kinase buffer (150 mM NaCl, 10 mM MgCl2, 25 mM HEPES [pH 7.4], 0.5 mM dithiothreitol). The immunocomplexes were incubated with a 30 μl kinase reaction mixture consisting of kinase buffer, 250 ng glutathione S-transferase (GST)-CTD, 5 μM ATP, and 3 μCi of [γ-32P]ATP (MP Biomedicals, Solon, OH). Reactions were carried out at 37°C for 25 min, with mixing every 5 min. The reactions were stopped with 4× Laemmli buffer, and the mixtures were boiled for 5 min. The supernatant was loaded onto an 8% SDS-polyacrylamide gel for electrophoresis. The gel was fixed, dried, and exposed to X-ray film. The GST-CTD expression vector was a gift from W. Dynan, and bacterial production of the fusion protein was as previously described (39).
RESULTS
Identification of several proteins that localize at the sites of HCMV IE transcription.
It has been reported that there are distinct nucleation sites where viral IE transcription is initiated. Establishing efficient IE transcription is important for productive HCMV infection because these sites eventually form the replication centers for viral DNA synthesis. These sites, referred to as transcriptosomes, have previously been characterized in HCMV-infected human cells by the presence of the viral input genome, viral transactivators (IE2 and UL112-113), several components of the cellular transcription machinery (RNAP II, TBP, TFIIB, cdk7, and cdk9), and viral RNA transcripts (1, 2, 17, 39, 48). We previously showed that cellular cdk9, cdk7, and transcriptionally active RNAP II (with the CTD phosphorylated on Ser2) colocalize with IE2-86 during the first 8 h of the infection in distinct aggregates adjacent to the ND10 that are undergoing dispersal (39). Through confocal microscopy analysis, we have expanded the list of proteins identified at these sites at the beginning of the infection to include the viral protein UL69 and other cellular proteins involved in regulation of transcription (cyclin T1 and DSIF) and in chromatin modification (Brd4, HDAC1, and HDAC2) (Table 2).
TABLE 2.
Viral factors and cellular proteins identified at the sites of the viral transcriptosome
| Category and protein | Reference(s) |
|---|---|
| Viral factors | |
| Input HCMV genome | 17 |
| IE2 | 1, 3, 17 |
| UL112-113 | 1 |
| UL69 | This paper |
| Cellular components of transcription | |
| RNAP II | 39 |
| TBP | 17 |
| TFIIB | 17 |
| cdk7 | 39 |
| cdk9 | 39 |
| cyclin T1 | This paper |
| DSIF | This paper |
| Cellular regulators of chromatin modification | |
| Brd4 | This paper |
| HDAC1 | This paper |
| HDAC2 | This paper |
Figure 1 shows representative confocal microscopy sections of the newly identified proteins at the viral transcriptosomes at 8 h p.i. Antibodies specific to these proteins were used to costain with either IE2 (mouse antibody) or cdk9 (rabbit antibody) to determine the sites of colocalization. In considering which viral input tegument proteins might localize to the viral transcriptosome, UL69 was a likely candidate in view of its ability to promote nuclear export of viral transcripts (21). UL69 can be detected in a subset of cells as nuclear aggregates that colocalize with cdk9 (Fig. 1, panels 1 to 3). It is possible that the reason that UL69 is not found in all cells is due to its turnover after entry into the cell. This protein is not newly synthesized in infected cells until early-late times in the infection. The finding that cyclin T1 colocalizes with IE2 at the transcriptosome is not surprising in view of the fact that cyclin T1 is in a complex with cdk9 and forms P-TEFb (panels 5 to 7). Another regulator of RNAP II-directed transcription elongation, DSIF, can also be detected at the transcriptosome as shown by its colocalization with cdk9 (panels 9 to 11). Transcriptional pausing is in part due to the interaction of DSIF with the RNAP II complex (41, 46, 47). P-TEFb is recruited to the RNAP II complex and phosphorylates the Spt5 subunit of DSIF, resulting in the disassociation of DSIF and entry into the transcription elongation phase (8, 20, 28, 42). In addition to viral factors and cellular transcription regulators, cellular chromatin-modifying proteins are being identified at these sites as a novel class of proteins. Brd4 has been found to be associated with cellular chromatin, with the mediator complex, and with active P-TEFb complexes, where it has been shown to interact with cyclin T1 (18, 45, 49). Colocalization of Brd4 and IE2 further supports the observation that the active cdk9 complex is specifically recruited to the sites of viral IE transcription (Fig. 1, panels 13 to 15). A surprising result was that two HDACs, HDAC1 and HDAC2, were recruited to this complex, as these proteins are often associated with gene silencing (panels 17 to 19 and 21 to 23, respectively) (for a review, see reference 13). However, the model associating HDACs with transcriptional repression and histone acetylases with transcriptional activation has been challenged in recent years, as more studies with HDAC inhibitors have revealed as many genes downregulated as upregulated (9). For comparison, we show the pattern of antibody staining to the above cellular proteins in the mock-infected cells (Fig. 1, panels 4, 8, 12, 16, 20, and 24). In general, the staining was diffuse nuclear with some tiny spots, and it was distinctly different from what was observed in the infected cells. Panels 25 to 30 in Fig. 1 show controls in which the infected cells were stained with a specific antibody along with a nonspecific IgG antibody that matched the second antibody used in the dual staining. Taken together, these results suggest that the process of establishing the viral IE transcription sites is more complex than was originally described, and the nuclear environment is significantly altered during the first 8 h of infection through the recruitment of multiple proteins to the viral transcriptosome.
FIG. 1.

Localization of several proteins at the sites of viral IE transcription in HCMV-infected cells. G0-synchronized cells were released into G1, infected with HCMV Towne at an MOI of 5 or with mock supernatant, and seeded onto glass coverslips. At 8 h p.i., cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with antibodies specific for the proteins indicated in each panel. Specific antibody staining was detected with fluorescein isothiocyanate- or Cy3-conjugated isotype-specific secondary antibodies. For controls, one of the specific antibodies of the pair was replaced with an isotype-specific normal IgG. Nuclei were stained with Hoechst dye. The white arrow in each panel indicates a region of colocalization, although more are present. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 under oil immersion.
HCMV infection in the presence of roscovitine impairs the localization of cdk9 and cdk7, but not IE2-86, at viral transcriptosomes.
Our lab has shown that roscovitine added at the beginning of the infection results in differential splicing of IE transcripts and inhibition of early gene expression and viral replication (31). Moreover, the virus-specific hyperphosphorylation of the CTD of RNAP II is inhibited. Since both cdk7 and cdk9 are responsible for phosphorylation of the RNAP II CTD, we wanted to investigate how cdk inhibition affects the formation of the transcriptosome with respect to IE2, cdk9, and cdk7. Cells were infected in the presence of roscovitine (or DMSO) and then fixed at 8 h p.i. for IFA analysis with an antibody to cdk9 (Fig. 2). Consistent with our previous results, cdk9 forms nuclear aggregates that can be detected by 2 h p.i. and that grow in size and abundance as the infection proceeds (39). These cdk9 aggregates colocalize with the IE2 protein, as detected with the IE2 MAb (Fig. 2, panels 1 to 3). Surprisingly, in the presence of roscovitine, there are no changes in IE2 protein accumulation at the viral transcriptosomes, while cdk9 aggregation at these sites is greatly diminished (panels 5 to 7). Although the overall intensity of the cdk9 staining is decreased in roscovitine-treated infected cells, a limited number of small cdk9 aggregates could be detected and seemed to colocalize with the IE2 protein (panels 9 and 10). Roscovitine had no effect on cdk9 localization in mock-infected cells; in both treated and untreated cells, cdk9 appears as a nonnucleolar, diffuse nuclear stain with minimal speckling (compare panels 4 and 8).
FIG. 2.

Treatment with roscovitine impairs accumulation of cdk9 at the sites of viral IE transcription. G0-synchronized cells were released into G1, infected with HCMV Towne at an MOI of 5 or with mock supernatant, and seeded onto glass coverslips. Cells were treated with 16 μM roscovitine or DMSO at the time of infection. At 8 h p.i., cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with IE2 (IgG1) and cdk9. Specific antibody staining was detected with fluorescein isothiocyanate- or Cy3-conjugated isotype-specific secondary antibodies. For controls, one of the specific antibodies of the pair was replaced with an isotype-specific normal IgG. Nuclei were stained with Hoechst dye. The white arrow in each panel indicates a region of colocalization, although more are present. The white boxes in panels 6 and 7 are enlarged in panels 9 and 10, respectively, with the modification of a 200% magnification and a 200% Cy3 intensity using Adobe Photoshop v. 7.0. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 under oil immersion. M, mock treated; V, HCMV infected; −, DMSO treated; +, roscovitine treated at the time of infection.
In order to determine whether the cdk9 localization at the transcriptosome is merely delayed due to a slower infection in the presence of roscovitine or is impaired and does not develop further as the infection progresses, we checked a series of time points after 8 h p.i. Cells that were infected in the absence of roscovitine displayed pronounced cdk9 aggregates that increased in size as the infection progressed from 8 to 14 h p.i. (Fig. 3, compare panels 2 and 14). However, cells that were infected in the presence of roscovitine displayed little to no cdk9 aggregates even at 14 h p.i. compared to cells infected in the absence of roscovitine (compare panels 6 and 2). Interestingly, cells that were treated with roscovitine after 8 h of infection displayed cdk9 aggregation, although not to the extent of the untreated infected cells (compare panels 10 and 2). Consistent with earlier observations, IE2 was still present at the viral transcriptosomes when roscovitine was added at the beginning of the infection; however, by 14 h p.i., IE2 accumulation at those sites was slightly delayed compared to the case with no drug or delayed addition of roscovitine (compare panel 5 to panels 1 and 9).
FIG. 3.

Treatment with roscovitine impairs cdk9 aggregation in infected cells. G0-synchronized cells were released into G1, infected with HCMV Towne at an MOI of 5 or with mock supernatant, and seeded onto glass coverslips. Cells were treated with 16 μM roscovitine or DMSO at the time of infection or with roscovitine at 8 h p.i. At 14 h p.i., cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with anti-cdk9 and anti-IE2 antibodies followed by Cy3- and fluorescein isothiocyanate-conjugated secondary antibodies. For controls, isotype-specific normal IgG was used in place of the specific antibody (not shown). The white arrow in each panel indicates a cdk9 aggregate, although more are present. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 under oil immersion. M, mock treated; V, HCMV infected; −R, DMSO treated; +R0, roscovitine treated at the time of infection; +R8, roscovitine treated at 8 h p.i.
In Fig. 4, we show the effects of roscovitine on the localization of cdk7 to the transcriptosome. Analogous to what was observed for cdk9, infection in the presence of roscovitine also disrupted cdk7 recruitment to the viral transcriptosome (compare panels 1 to 3 and panels 5 to 7). These data suggest that the effect of roscovitine on the recruitment of cdk9 and cdk7 to the viral transcriptosome may account, at least in part, for the altered pattern of viral transcription.
FIG. 4.

Treatment with roscovitine inhibits recruitment of cdk7 at the sites of viral IE transcription. G0-synchronized cells were released into G1, infected with HCMV Towne at an MOI of 5 or with mock supernatant, and seeded onto glass coverslips. Cells were treated with DMSO (panels 1 to 4) or 16 μM roscovitine (panels 5 to 8) at the time of infection. At 8 h p.i., cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with cdk7 (IgG2B) and IE2 (IgG1). Specific antibody staining was detected with fluorescein isothiocyanate- or Cy3-conjugated isotype-specific secondary antibodies. For controls, one of the specific antibodies of the pair was replaced with an isotype-specific normal IgG (panels 9 and 10). Nuclei were stained with Hoechst dye. The white arrow in each panel indicates a region of colocalization, although more are present. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 under oil immersion. M, mock treated; V, HCMV infected; −, DMSO treated; +, roscovitine treated at the time of infection.
The impairment in the localization of proteins to the IE transcription sites during roscovitine treatment is specific to cdk9 and cdk7.
The differential effect of roscovitine on the localization of the cellular proteins cdk7 and cdk9 versus the viral IE2 protein to the transcriptosome prompted us to determine whether other cellular proteins at the transcriptosome were similarly affected. Since we have shown that almost all cdk9 is in a complex with cyclin T1 in infected cells (39) and since it has recently been estimated that 50% of cellular cdk9/cyclin T1 is associated in an active complex with Brd4 (49), we predicted that the latter two proteins would also not localize to the transcriptosome in the presence of roscovitine. Cells were infected in the presence of roscovitine and then fixed for IFA analysis at 8 h p.i. Surprisingly, we found that both cyclin T1 and Brd4 maintain a colocalization with IE2 (Fig. 5, panels 1 to 3 for cyclin T1 and panels 5 to 7 for Brd4). The localization of HDAC1 and HDAC2 during infection in the presence of roscovitine was also examined. Confocal microscopy analysis shows that HDAC1 and HDAC2 both maintain a colocalization with IE2 at the transcriptosome that is unaffected by the presence of roscovitine (panels 9 to 11 for HDAC1 and panels 13 to 15 for HDAC2). Combined, these data suggest that addition of roscovitine specifically impairs the localization of cdk9 and cdk7 at the IE transcription sites and not that of other cellular or viral proteins that we tested.
FIG. 5.

Cyclin T1, Brd4, HDAC1, and HDAC2 are able to localize to the sites of viral IE transcription during infection in the presence of roscovitine. G0-synchronized cells were released into G1, infected with HCMV Towne at an MOI of 5 (panels 1 to 3, 5 to 7, and 13 to 15) or with mock supernatant (panels 4, 8, 12, and 16), and seeded onto glass coverslips. Cells were treated with 16 μM roscovitine or DMSO (see Fig. 1) at the time of infection. Only the roscovitine-treated samples are shown in this figure. At 8 h p.i., cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with an antibody specific for the protein indicated in each panel. Specific antibody staining was detected with fluorescein isothiocyanate- or Cy3-conjugated isotype-specific secondary antibodies. For controls, one of the specific antibodies of the pair was replaced with an isotype-specific normal IgG (not shown). Nuclei were stained with Hoechst dye. The white arrow in each panel indicates a region of colocalization, although more are present. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 magnification under oil immersion. M, mock treated; V, HCMV infected; +, roscovitine treated at the time of infection.
Roscovitine treatment leads to a decrease in the level of cdk9 protein in HCMV-infected cells.
Since we observed a decrease in cdk9 staining intensity as well as impairment of cdk9 and cdk7 localization in cells infected in the presence of roscovitine, we felt it was important to examine the effect of roscovitine on the steady-state levels of cdk9 and cdk7 protein. Cells were synchronized in G0 by allowing them to grow to confluence, trypsinized, and then infected with HCMV at an MOI of 5 upon replating and release into G1 phase. The levels of cdk9 are low when cells are in G0 and then increase after entry into G1 phase in both mock- and virus-infected cells (Fig. 6, lanes M− and V−). At 24 h p.i., there are comparable amounts of cdk9 in the untreated mock-infected (lane M−) and virus-infected (lane V−) samples. However, when roscovitine is added at the beginning of the infection, the levels of cdk9 in both the mock-infected (lane M+) and virus-infected (lane V+) cells do not increase during the 24-h period after release into G1 and remain at the low levels seen at G0. In contrast, cdk7 protein levels show only moderate changes as the cells are released into G1 phase. At 24 h p.i., the levels of cdk7 are slightly higher in the virus-infected (lane V−) cells than in the mock-infected (lane M−) cells, and this increase is not seen in the virus-infected cells treated with roscovitine (lane V+). Another target of inhibition by roscovitine, cdk2, showed only limited changes in steady-state levels.
FIG. 6.
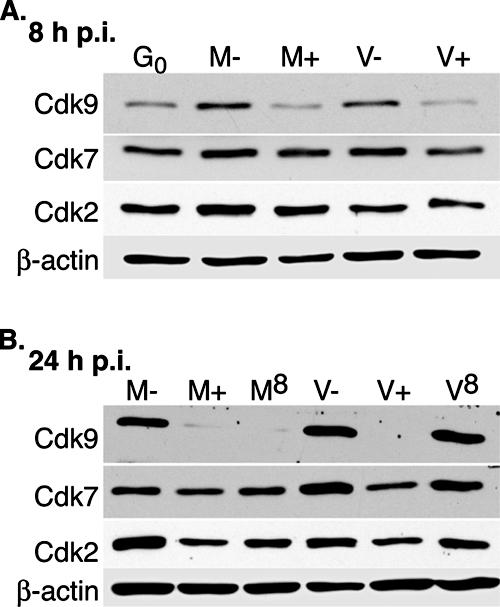
Effect of roscovitine on the steady-state levels of cdk9 and cdk7 protein. G0-synchronized cells were released into G1 and infected with HCMV Towne at an MOI of 5 or with mock supernatant. Cells were treated with 16 μM roscovitine or DMSO at the time of infection or at 8 h p.i. and were harvested at 8 h p.i. (A) or 24 h p.i. (B). For mock- and virus-infected cells that were harvested at 24 h p.i. and treated with roscovitine or DMSO at the beginning of the infection, the medium was removed at 8 h p.i., and fresh medium containing roscovitine or DMSO, respectively, was added. Cell lysates from equal numbers of cells were run on an 8% SDS-polyacrylamide gel and subjected to Western blotting using antibodies for cdk9, cdk7, cdk2, and β-actin (loading control). G0, cells released from G0; M, mock treated; V, HCMV infected; −, DMSO treated; +, roscovitine treated at the time of infection; 8, roscovitine treated at 8 h p.i.
A delay in the addition of roscovitine until 8 h p.i. does not inhibit cdk9 and cdk7 protein expression in HCMV-infected cells.
We have previously shown that many of the effects that roscovitine has on HCMV-infected cells seem to be limited to the first 8 h p.i., including alterations in viral gene expression, the virus-specific loss of RNAP II CTD phosphorylation, and, as described above, the impairment in cdk9 aggregation at the IE transcription sites (31, 39) (Fig. 2). We sought to determine if the effect of roscovitine on the steady-state levels of the RNAP II CTD kinases, particularly cdk9, was also limited to the first 8 h of the infection. Cells were infected in the presence of DMSO, followed by the addition of roscovitine at 8 h p.i. As shown in Fig. 6B, levels of both cdk9 and cdk7 in the virus-infected samples were comparable in both the delayed drug-treated (lane V8) and untreated (lane V−) cells. This difference between addition of the drug at the beginning of the infection versus at 8 h p.i. is particularly evident for cdk9. In contrast, the delay in roscovitine addition until 8 h p.i. affected cdk9 and cdk7 accumulation in mock-infected cells (lane M8) similarly to addition of roscovitine at 0 h (lane M+). Combined, these data suggest that a virus-specific event occurs within the first 8 h of the infection, after which the steady-state level of cdk9 protein in infected cells is no longer altered by the presence of roscovitine.
Inhibition of cdk9 and cdk7 expression during roscovitine treatment occurs at the RNA level.
Since infection in the presence of roscovitine affected the steady-state amounts of cdk9 protein, and to a smaller extent cdk7 protein, we proceeded to use quantitative real-time RT-PCR to determine whether the levels of the cdk9 and cdk7 transcripts were also altered. Mock- or HCMV-infected cells were treated with roscovitine (or DMSO) at the time of infection or at 8 h p.i. Total RNA was isolated at 4, 8, and 16 h p.i. and quantified with primers and probes specific for cdk9, cdk7, and G6PD (Table 1).
The quantitative real-time RT-PCR results for cdk9 are consistent with the previous Western analysis of the protein levels (Fig. 7A). In untreated samples, cdk9 RNA does not change in mock-infected cells and increases approximately 1.6-fold in infected cells by 8 h p.i. The addition of roscovitine at the time of infection results in a decrease in cdk9 RNA below the level at G0 in both mock- and virus-infected samples as early as 4 h p.i. (an approximately twofold reduction from untreated levels) and in a decrease of four- to fivefold by 8 h p.i. When the addition of roscovitine is delayed to 8 h p.i., cdk9 RNA levels in the roscovitine-treated virus-infected samples are comparable to those in the untreated viral samples at 16 h p.i., although the levels were more variable in individual experiments. In contrast, when roscovitine addition is delayed until 8 h p.i. in the mock-infected samples, the reduction in cdk9 RNA levels is comparable to those when roscovitine is added at the time of infection.
FIG. 7.

Effect of roscovitine on cdk9 and cdk7 RNA levels. G0-synchronized cells were released into G1 and infected with HCMV Towne at an MOI of 5 or with mock supernatant. Cells were treated with 16 μM roscovitine or DMSO at the time of infection or at 8 h p.i. and were harvested at 4, 8, and 16 h p.i. Total RNA was isolated and analyzed by quantitative real-time RT-PCR using primers and probes listed in Table 1 for cdk9 (A) and cdk7 (B) RNAs. The values for cdk9 and cdk7 were normalized to that of G6PD RNA for each sample. For each time point, two separate experiments were performed and duplicate reactions were analyzed. A standard curve for each gene was used to calculate the relative amount of specific RNA present in a sample, which was then converted to fold activation relative to DMSO-treated, mock-infected 8 h p.i. samples. The graphs shows the averages from the two experiments, and the range bars indicate the highest and lowest values. G0, cells released from G0; M, mock treated; V, HCMV infected; −, DMSO treated; +, roscovitine treated at the time of infection; 8, roscovitine treated at 8 h p.i.
Surprisingly, the decrease in cdk7 expression during roscovitine treatment was much greater at the RNA level than that observed at the protein level (Fig. 7B). In untreated samples, cdk7 RNA does not change in mock-infected cells and increases approximately 1.4-fold in infected cells by 8 h p.i. When cells are infected in the presence of roscovitine, cdk7 RNA levels decrease approximately three- to fourfold in both mock- and virus-infected samples relative to untreated samples by 4 h p.i., and the levels are approximately 12- to 13-fold lower in the roscovitine-treated samples by 8 h p.i. When roscovitine addition is delayed until 8 h p.i., the mock-infected and infected cells have higher levels of cdk7 RNA than the cells that are treated with the inhibitor at the beginning of the infection, but the levels are still lower than those in the untreated samples. Taken together, these results indicate that the decrease in cdk9 and cdk7 expression in the presence of roscovitine is primarily at the RNA level. However, we cannot exclude additional effects on protein synthesis or stability, since the effect of the drug on the cdk9 protein level was significantly greater than that for cdk7 protein.
When cells are transitioning from G0 to G1, there is a general increase in the basal level of cellular gene expression. Thus, it is possible that some of the effects of roscovitine described above could be due to an inefficient entry of the cells from G0 into G1. We examined this possibility by repeating the infections described above, but the cells were infected 8 h after release into G1. Cells were collected at the indicated time points and processed for Western blot analysis. The overall pattern of expression of cdk9, cdk7, and viral genes in the presence and absence of roscovitine treatment (whether roscovitine was added at the time of infection or 8 h later) was similar to that for the G0 infection (data not shown).
Overexpression of cdk9 does not compensate for the effects of roscovitine on its localization in HCMV-infected cells.
Our previous studies have implicated cdk9 as being particularly important in establishing a productive infection. In a paper by Tamrakar et al. (39), we showed that RNAP II phospho-Ser2 (cdk9 substrate) levels decreased to a greater extent than RNAP II phospho-Ser5 (cdk7 substrate) levels when cells were infected in the presence of roscovitine. We also found that infection in the presence of roscovitine (or flavopiridol, which inhibits cdk9/cyclin T1 more potently than other cdk complexes) led to changes in the differential splicing and polyadenylation of HCMV IE transcripts (31). This was particularly relevant since phosphorylation of RNAP II Ser2 by cdk9 is believed to play a key role in the recruitment of factors for RNA processing. The above-described experiments showing a marked decrease in the amount of cdk9 protein in infected cells treated with roscovitine raised the possibility that these lower levels might be responsible for the deficit in cdk9 recruitment to the transcriptosome and the observed effects on IE transcription and entry into viral early gene expression.
To address the above possibility, we used the following strategy to construct a cell line that overexpressed HA-tagged cdk9. Based on our finding that the RNA levels of the endogenous cdk9 were affected by roscovitine, we reasoned that the drug treatment might be selectively affecting the transcription directed by the endogenous promoter or the processing of the genomic transcript. We therefore placed a cDNA copy of cdk9HA into a lentiviral vector where its transcription was driven by an EF1α promoter. We have used this promoter in other experiments and have found it to be constitutively active in infected cells. This vector was then used to transduce HFFs. We first established that the HA-tagged cdk9 colocalized appropriately with IE2 at the viral transcriptosome by IFA with anti-HA and anti-IE2 MAbs (Fig. 8A). Next, we confirmed that the exogenously expressed cdk9 is active kinase by immunoprecipitating cell lysates with control IgG or anti-HA and anti-cdk9 antibodies and assessing the kinase activity in the precipitate with GST-CTD as the substrate and the amount of protein in the precipitate (Fig. 8B and C, respectively).
FIG. 8.

Roscovitine treatment affects steady-state levels of endogenous cdk9 and not those of overexpressed cdk9HA. (A) The G0-synchronized cdk9HA-overexpressing HFF were released into G1, HCMV infected at an MOI of 5, and seeded onto coverslips. At 12 h p.i., the cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with IE2 (IgG1) and HA (IgG2a). As a control, as second anti-HA antibody (rabbit polyclonal) was used to confirm specific staining (not shown). Specific antibody staining was detected with fluorescein isothiocyanate- or tetramethyl rhodamine isocyanate-conjugated isotype-specific secondary antibodies. Nuclei were stained with Hoechst dye. The white arrow in each panel indicates a region of colocalization, although more are present. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 under oil immersion. (B and C) The immunocomplexes from mock cells containing the exogenously expressed cdk9 were assayed for kinase activity towards purified GST-CTD (B), and the corresponding immunoprecipitates were run on an 8% SDS-polyacrylamide gel and subjected to Western blot with an antibody specific for cdk9 (C). Pre, preimmunoprecipitate; IP, immunoprecipitate; Post, postimmunoprecipitate. (D) The G0-synchronized cdk9HA-overexpressing HFF and untransduced HFF were HCMV infected at an MOI of 5 or mock infected in the presence of 16 μM roscovitine or DMSO. Samples were collected at 8 and 24 h p.i. Lysates consisting of an equal number of cells were loaded on a 10% SDS-polyacrylamide gel. Western blot analysis was performed as described above for cdk9. M, mock treated; V, HCMV infected; −, DMSO treated; +, roscovitine treated at the time of infection; F, untransduced HFF cells; 9, cdk9HA-overexpressing cells; *, endogenous cdk9; **, cdk9HA.
Since the results of the above-described experiments showed that the exogenous cdk9HA was an active kinase and colocalized with IE2 at the transcriptosome, we then infected these cdk9-overexpressing cells in the presence of roscovitine (or DMSO) and analyzed the accumulation of endogenous and exogenous cdk9 protein, which can be differentiated by the slower migration of the HA-tagged cdk9 on a 10% SDS-polyacrylamide gel (Fig. 8D). The endogenous cdk9 protein levels reflected the changes in the absence and presence of roscovitine that were described above. More specifically, the endogenous cdk9 protein in mock- and virus-infected samples decreased when roscovitine was present at the time of infection. However, the exogenous cdk9HA that was expressed from the integrated lentiviral construct showed no observable decrease at 8 and 24 h p.i. in the mock- and virus-infected cells treated with roscovitine at the time of infection.
We reported previously that infection in the presence of roscovitine differentially alters viral protein expression. In particular, the ratio of IE1 and IE2 protein accumulation is switched, UL44 protein is moderately decreased, and UL57 protein and pp28 protein expression are completely inhibited (31). To determine if the overexpression of cdk9 would overcome some of these effects of roscovitine, we assessed viral protein synthesis by Western blot analysis. As shown in Fig. 9A, the effects of roscovitine on viral IE1/2 protein expression were comparable in control cells and cdk9HA-overexpressing cells. Several viral early (UL44 and UL57) and late (pp28) protein levels were also found to be comparable in the control and cdk9HA-overexpressing cells (data not shown).
FIG. 9.

Overexpression of cdk9 does not compensate for alterations in the expression of viral IE proteins and RNAP II during infection in the presence of roscovitine. The G0-synchronized cdk9HA-overexpressing HFF and untransduced HFF were HCMV infected at an MOI of 5 or mock treated in the presence of 16 μM roscovitine or DMSO. Samples were collected at 8 and 24 h p.i. and analyzed by Western blotting using antibodies against the viral proteins IE1/2 (CH16.0) and β-actin (loading control) (A) or antibodies against total RNAP II (ARNA3) and serine 2-phosphorylated RNAP II CTD (H5) (B). β-Catenin was used as a loading control. M, mock treated; V, HCMV infected; −, DMSO treated; +, roscovitine treated at the time of infection; F, untransduced HFF cells; 9, cdk9HA-overexpressing cells.
In addition, RNAP II CTD phosphorylation was examined with an antibody against the Rpb1 subunit (ARNA3 antibody) and an antibody against the cdk9 phosphorylation site, serine 2 residues of the CTD (H5 antibody) (Fig. 9B). The virus-specific loss of RNAP II CTD phosphorylation was also not recovered by cdk9 overexpression.
Although the above results show that overexpression of cdk9 does not counteract the effects of roscovitine on viral protein expression, we were also interested in whether localization of cdk9 was also affected. This question was addressed by infecting HFF cells and cdk9-overexpressing cells in the presence of roscovitine and checking the localization by IFA (Fig. 10). As expected, in the absence of roscovitine, the cdk9HA-overexpressing cells showed an intense cdk9 stain and formed nuclear aggregates that colocalized with IE2 protein (panels 1 to 3). In the presence of roscovitine, most of the cdk9HA-overexpressing cells exhibited a similar impairment in cdk9 aggregation (panels 5 to 7) as control HFF (panels 12 to 14), although in a small subset of cells, a few cdk9 aggregates comparable in size to untreated cells were observed (panels 9 to 11). These data indicate that during an infection in the presence of roscovitine, where expression of endogenous cdk9 is decreased, overexpression of cdk9 from a retrovirus vector does not overcome the impaired accumulation of cdk9 at the sites of viral IE transcription. Therefore, the decrease in the amount of cdk9 protein is not the primary cause of an unproductive HCMV infection in the presence of roscovitine. Rather, it appears that the recruitment of cdk9 to the sites of IE transcription, despite the presence of IE2, is impaired.
FIG. 10.

Impaired cdk9 localization in the presence of cdk inhibitor occurs in infected cells overexpressing cdk9HA. The G0-synchronized cdk9HA-overexpressing HFF and untransduced HFF were HCMV-infected at an MOI of 5 or mock treated in the presence of 16 μM roscovitine or DMSO. At 8 h p.i., cells were washed with PBS, fixed in formaldehyde, permeabilized, and immunostained with cdk9 antibody followed by fluorescein isothiocyanate- or Cy3-conjugated isotype-specific secondary antibodies. For controls, one of the specific antibodies of the pair was replaced with isotype-specific normal IgG (not shown). Nuclei were stained with Hoechst dye. The white arrow in each panel indicates a region of colocalization, but more are present. All of the images are confocal optical sections of 0.2 μm at a magnification of ×1,000 under oil immersion. M, mock treated; V, HCMV infected; −R, DMSO treated; +R, roscovitine treated at the time of infection.
DISCUSSION
Transcription is a multistep process that requires several regulatory proteins involved in chromatin remodeling, promoter recognition and clearance, initiation, elongation, and RNA processing. Viral IE transcription must be robust for initiation of a productive viral infection, and there is increasing evidence that HCMV establishes at the beginning of the infection transcriptosomes consisting of cellular and viral proteins. This expanded list includes RNAP II, TBP, TFIIB, cdk7, cdk9, cyclin T1, Brd4, DSIF, HDAC1, HDAC2, the input viral genome, and the viral proteins IE2, UL112-113, and UL69 (1, 2, 17, 39, 48). By selectively recruiting specific cellular proteins to where the viral input genome is deposited and viral proteins are localizing, subnuclear transcriptosomes where viral transcripts are preferentially synthesized and efficiently processed are created. Of the known cellular proteins that are involved at the viral IE transcription sites, the RNAP II CTD kinases, cdk9, and cdk7 are recruited very early in the infection (39). Since cellular RNAP II directs HCMV transcription, and phosphorylation of RNAP II regulates its transcriptional activity, cdk9 and cdk7 are likely to be important components in establishing viral IE transcription.
Roscovitine has an approximate IC50 of 0.6 μM for both cdk9 and cdk7 in vitro, and it works by competing with ATP for the active sites of those kinases (24). While it is reasonable to expect that the activities of cdk9 and cdk7 are inhibited in the presence of roscovitine, it was surprising to see that the steady-state levels of cdk9 and cdk7 protein and transcripts decreased. It is interesting that cdk9 showed a greater decrease in protein levels than cdk7 did. This implies that although the activities of cdk9 and cdk7 are inhibited by a similar concentration of roscovitine in vitro, the effect of the drug on cdk9 function in the context of the cell may be greater due to protein turnover.
Tracking cdk9 relocalization during the infection is one way to monitor the progression of viral transcriptosome assembly, an event that seems to be associated with a productive HCMV infection. cdk9 accumulates at the sites of viral IE transcription as early as 2 h p.i., and as the infection proceeds, the cdk9 aggregates increase in both size and abundance (39). HCMV infection in the presence of roscovitine dramatically impairs cdk9 relocalization to viral transcriptosomes. In a high multiplicities of infection, the majority of cells display prominent cdk9 aggregates, whereas treatment with roscovitine at the time of the infection prevents the formation of these aggregates in almost all of the cells. This was not simply due to a delay in the creation of the aggregates, as they were not observed even at later times. Surprisingly, IE2 protein localization at the viral nuclear bodies is not affected by roscovitine treatment, which indicates that newly synthesized IE2 can localize at the viral transcriptosomes in the absence of cdk9 and cdk7 accumulation. This also supports the existing data that IE2 comprises only part of the viral transcriptosome, and IE2 localization at these sites alone is not sufficient for a productive infection.
Infection in the presence of roscovitine also inhibited cdk7 localization to the IE transcription sites. The observation that recruitment of several of the other proteins that accumulate at the viral nuclear bodies was not affected by roscovitine treatment highlights the specificity of inhibition by roscovitine. Roscovitine inhibits cdk9 and cdk7 activity through its binding at the ATP-binding sites of those kinases. Perhaps binding of roscovitine to cdk9 and cdk7 confers a conformational change of the kinases, preventing their recruitment to the viral transcriptosomes. It is also possible that functional cdk1 or cdk2 is required for this recruitment at the beginning of the infection. However, preliminary data with a specific inhibitor of cdk1 suggest that this kinase is not involved.
Many of the effects of roscovitine in HCMV-infected cells are limited to the first 6 to 8 h of infection. A delay in the treatment of roscovitine until 8 h p.i. does not cause the switch in the ratio of the IE1/2 transcripts, permits viral early and late gene expression, and maintains the hyperphosphorylation of RNAP II (31, 39). In this study, delaying the addition of roscovitine in infected cells by 8 h also prevented the inhibition of cdk9 expression and localization to the transcriptosome. This decrease in sensitivity to roscovitine with respect to expression of cdk9 occurs only in the infected cells, suggesting that the cellular environment in HCMV-infected cells has been altered from that in uninfected cells within 8 h. We propose that the establishment of the viral transcriptosome during this time is the virus-specific event that contributes to the lack of sensitivity towards roscovitine after 8 h p.i. This possibility is consistent with the kinetics of HCMV infection, since this is the time during which IE transcription is coordinated and localization of proteins at IE transcription sites is detected by IFA. By 8 h p.i., cdk9 and cdk7 localization at the viral transcriptosomes may allow resistance to the binding of roscovitine.
Upon discovering that treatment with roscovitine at the time of infection decreases cdk9 protein levels, we were concerned that this decrease in cdk9 availability prevented cdk9 accumulation at the IE transcription sites. However, further experiments show that this is not the case; overexpression of cdk9 in the infected cells did not overcome the inhibitory effects of roscovitine. One possibility is that inhibition of cdk9 alone is not responsible for the alterations in viral gene expression and that several cdks work synergistically in the early hours of the infection to initiate viral IE transcription. Another possibility is that the level of cdk9 protein present is not the determining factor, but rather the ability of cdk9 to localize to abundant levels at the viral IE transcription sites by a certain time of the infection. In its active state, cdk9 is part of a complex with cyclin T1 and Brd4 (PTEF-b complex). We showed that these components of PTEF-b all localize at the sites of IE transcription.
The identification of Brd4 with the viral transcriptosome is interesting because Brd4 has been shown to play a key role in infections of other DNA viruses. Papillomavirus E2 protein and Kaposi's sarcoma-associated herpesvirus LANA-1 protein both interact with Brd4 to tether the viral genome with the cellular chromatin (6, 23, 50, 51). The role that Brd4 may be serving at the site of HCMV IE transcription still needs to be elucidated. However, it may play a role in recruitment of the cdk9/cyclin T1 complex to this site. A study by Yang et al. describes that although Brd4 and cyclin T1 can directly interact, the serine 175 residue on cdk9 is important to maintain the interaction of Brd4 with the cdk9/cyclin T1 complex. This residue is also necessary for kinase activity due to its location near the active site (49). Perhaps binding of roscovitine to cdk9 creates some steric hindrance at the active site, preventing the association of cdk9/cyclin T1 to Brd4 and therefore recruitment of cdk9 to the IE transcription sites. Cyclin T1 may not be affected in this process, because there is non-cdk9-associated cyclin T1 available in cells, including infected cells (39). Interestingly, serine 175 is a conserved residue in the T loops of cdk9 and cdk7, so similar structural changes during inhibitor binding may be taking place with cdk7 as well.
Taken together, the studies presented here highlight the importance of cdk9 and cdk7 recruitment and accumulation at the sites of viral IE transcription. During the first hours of the infection, the virus is highly dependent on the existing cellular environment and host factors to initiate efficient IE transcription, including where the input viral genome is deposited and active recruitment of the host transcription machinery. However, by 8 h p.i., the cellular environment (especially in the nucleus) is sufficiently altered to optimize for viral replication, after which the dependence on the host factors, such as cdk9 and cdk7, that were initially required has greatly diminished. The kinetics of establishing IE transcription is also an important consideration with regard to the potency of roscovitine as an antiviral drug.
Acknowledgments
We are grateful to Keiko Ozato for the Brd4 antibody and to William Britt for the UL69 antibody. We are also thankful to Inder Verma for the EF1α promoter-driven lentivector, to William Dynan for the plasmid containing GST-CTD, and to Xavier Graña for the pRc-CMV-cdk9HA plasmid. We also appreciate the use of the Deltavision microscope and SoftWorx software at the UCSD Cancer Center Digital Imaging Shared Resource. We thank Rebecca Sanders for helpful discussions and feedback during the preparation of the manuscript.
This work was supported by NIH grants CA73490 and CA34729.
Footnotes
Published ahead of print on 17 October 2007.
REFERENCES
- 1.Ahn, J.-H., W.-J. Jang, and G. S. Hayward. 1999. The human cytomegalovirus IE2 and UL112-113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein-associated nuclear bodies (PODs or ND10). J. Virol. 7310458-10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn, J. H., E. R. Brignole, and G. S. Hayward. 1998. Disruption of PML subnuclear domains by the acidic IE1 protein of human cytomegalovirus is mediated through interaction with PML and may modulate a RING finger-dependent cryptic transactivator function of PML. Mol. Cell. Biol. 184899-4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn, J. H., and G. S. Hayward. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 714599-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baek, M.-C., P. M. Krosky, A. Pearson, and D. M. Coen. 2004. Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virol. 324184-193. [DOI] [PubMed] [Google Scholar]
- 5.Bain, J., H. McLauchlan, M. Elliott, and P. Cohen. 2003. The specificities of protein kinase inhibitors: an update. Biochem. J. 371199-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baxter, M. K., M. G. McPhillips, K. Ozato, and A. A. McBride. 2005. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 794806-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bresnahan, W. A., E. A. Thompson, and T. Albrecht. 1997. Human cytomegalovirus infection results in altered Cdk2 subcellular localization. J. Gen. Virol. 781993-1997. [DOI] [PubMed] [Google Scholar]
- 8.Cheng, B., and D. H. Price. 2007. Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J. Biol. Chem. 28221901-21912. [DOI] [PubMed] [Google Scholar]
- 9.Clayton, A. L., C. A. Hazzalin, and L. C. Mahadevan. 2006. Enhanced histone acetylation and transcription: a dynamic perspective. Mol. Cell 23289-296. [DOI] [PubMed] [Google Scholar]
- 10.Davido, D. J., D. A. Leib, and P. A. Schaffer. 2002. The cyclin-dependent kinase inhibitor roscovitine inhibits the transactivating activity and alters the posttranslational modification of herpes simplex virus type 1 ICP0. J. Virol. 761077-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davido, D. J., W. F. von Zagorski, G. G. Maul, and P. A. Schaffer. 2003. The differential requirement for cyclin-dependent kinase activities distinguishes two functions of herpes simplex virus type 1 ICPO. J. Virol. 7712603-12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Azevedo, W. F., S. Leclerc, L. Meijer, L. Havlicek, M. Strnad, and S. H. Kim. 1997. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 243518-526. [DOI] [PubMed] [Google Scholar]
- 13.de Ruijter, A. J., A. H. van Gennip, H. N. Caron, S. Kemp, and A. B. van Kuilenburg. 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370737-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diwan, P., J. J. Lacasse, and L. M. Schang. 2004. Roscovitine inhibits activation of promoters in herpes simplex virus type 1 genomes independently of promoter-specific factors. J. Virol. 789352-9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edamatsu, H., C. L. Gau, T. Nemoto, L. Guo, and F. Tamanoi. 2000. Cdk inhibitors, roscovitine and olomoucine, synergize with farnesyltransferase inhibitor (FTI) to induce efficient apoptosis of human cancer cell lines. Oncogene 193059-3068. [DOI] [PubMed] [Google Scholar]
- 16.Fortunato, E. A., A. K. McElroy, V. Sanchez, and D. H. Spector. 2000. Exploitation of cellular signaling and regulatory pathways by human cytomegalovirus. Trends Microbiol. 8111-119. [DOI] [PubMed] [Google Scholar]
- 17.Ishov, A. M., R. M. Stenberg, and G. G. Maul. 1997. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 1385-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang, M. K., K. Mochizuki, M. Zhou, H. S. Jeong, J. N. Brady, and K. Ozato. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19523-534. [DOI] [PubMed] [Google Scholar]
- 19.Kudoh, A., T. Daikoku, Y. Sugaya, H. Isomura, M. Fujita, T. Kiyono, Y. Nishiyama, and T. Tsurumi. 2004. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J. Virol. 78104-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavoie, S. B., A. L. Albert, H. Handa, M. Vincent, and O. Bensaude. 2001. The peptidyl-prolyl isomerase Pin1 interacts with hSpt5 phosphorylated by Cdk9. J. Mol. Biol. 312675-685. [DOI] [PubMed] [Google Scholar]
- 21.Lischka, P., and T. Stamminger. 2006. Regulation of viral mRNA export from the nucleus, p. 185-204. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Horizon Scientific Press, Hethersett, Norwich, United Kingdom.
- 22.Majello, B., and G. Napolitano. 2001. Control of RNA polymerase II activity by dedicated CTD kinases and phosphatases. Front. Biosci. 6D1358-D1368. [DOI] [PubMed] [Google Scholar]
- 23.McPhillips, M. G., J. G. Oliveira, J. E. Spindler, R. Mitra, and A. A. McBride. 2006. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 809530-9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meijer, L., A. Borgne, O. Mulner, J. P. J. Chong, J. J. Blow, N. Inagaki, M. Inagaki, J. G. Delcros, and J. P. Molinoux. 1997. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243527-536. [DOI] [PubMed] [Google Scholar]
- 25.Meinhart, A., T. Kamenski, S. Hoeppner, S. Baumli, and P. Cramer. 2005. A structural perspective of CTD function. Genes Dev. 151401-1415. [DOI] [PubMed] [Google Scholar]
- 26.Muller, S., and A. Dejean. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 735137-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson, P. J., I. H. Gelman, and P. E. Klotman. 2001. Suppression of HIV-1 expression by inhibitors of cyclin-dependent kinases promotes differentiation of infected podocytes. J. Am. Soc. Nephrol. 122827-2831. [DOI] [PubMed] [Google Scholar]
- 28.Ping, Y. H., and T. M. Rana. 2001. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 27612951-12958. [DOI] [PubMed] [Google Scholar]
- 29.Prelich, G. 2002. RNA polymerase II carboxy-terminal domain kinases: emerging clues to their function. Euk. Cell 1153-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salvant, B. S., E. A. Fortunato, and D. H. Spector. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 723729-3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez, V., A. K. McElroy, J. Yen, S. Tamrakar, C. L. Clark, R. A. Schwartz, and D. H. Spector. 2004. Cyclin-dependent kinase activity is required at early times for accurate processing and accumulation of the human cytomegalovirus UL122-123 and UL37 immediate-early transcripts and at later times for virus production. J. Virol. 7811219-11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez, V., and D. H. Spector. 2006. Cyclin-dependent kinase activity is required for efficient expression and posttranslational modification of human cytomegalovirus proteins and for production of extracellular particles. J. Virol. 805886-5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schang, L. M., A. Bantly, M. Knockaert, F. Shaheen, L. Meijer, M. H. Malim, N. S. Gray, and P. A. Schaffer. 2002. Pharmacological cyclin-dependent kinase inhibitors inhibit replication of wild-type and drug-resistant strains of herpes simplex virus and human immunodeficiency virus type 1 by targeting cellular, not viral, proteins. J. Virol. 767874-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schang, L. M., J. Phillips, and P. A. Schaffer. 1998. Requirement for cellular cyclin-dependent kinases in herpes simplex virus replication and transcription. J. Virol. 725626-5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schang, L. M., A. Rosenberg, and P. A. Schaffer. 2000. Roscovitine, a specific inhibitor of cellular cyclin-dependent kinases, inhibits herpes simplex virus DNA synthesis in the presence of viral early proteins. J. Virol. 742107-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schang, L. M., A. Rosenberg, and P. A. Schaffer. 1999. Transcription of herpes simplex virus immediate-early and early genes is inhibited by roscovitine, an inhibitor specific for cellular cyclin-dependent kinases. J. Virol. 732161-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schang, L. M., M. R. S. Vincent, and J. J. Lacasse. 2006. Five years of progress on cyclin-dependent kinases and other cellular proteins as potential targets for antiviral drugs. Antivir. Chem. Chemother. 17293-320. [DOI] [PubMed] [Google Scholar]
- 38.Tamashiro, J. C., L. J. Hock, and D. H. Spector. 1982. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169). J. Virol. 42547-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamrakar, S., A. J. Kapasi, and D. H. Spector. 2005. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of cdk9 and cdk7. J. Virol. 7915477-15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor, S. L., P. R. Kinchington, A. Brooks, and J. F. Moffat. 2004. Roscovitine, a cyclin-dependent kinase inhibitor, prevents replication of varicella-zoster virus. J. Virol. 782853-2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wada, T., T. Takagi, Y. Yamaguchi, A. Ferdous, T. Imai, S. Hirose, S. Sugimoto, K. Yano, G. A. Hartzog, F. Winston, S. Buratowski, and H. Handa. 1998. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 12343-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wada, T., T. Takagi, Y. Yamaguchi, D. Watanabe, and H. Handa. 1998. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 177395-7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, D., C. de la Fuente, L. Deng, L. Wang, I. Zilberman, C. Eadie, M. Healey, D. Stein, T. Denny, L. E. Harrison, L. Meijer, and F. Kashanchi. 2001. Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors. J. Virol. 757266-7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkinson, G. W., C. Kelly, J. H. Sinclair, and C. Rickards. 1998. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J. Gen. Virol. 791233-1245. [DOI] [PubMed] [Google Scholar]
- 45.Wu, S. Y., and C. M. Chiang. 2007. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 28213141-13145. [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi, Y., N. Inukai, T. Narita, T. Wada, and H. Handa. 2002. Evidence that negative elongation factor represses transcription elongation through binding to a DRB sensitivity-inducing factor/RNA polymerase II complex and RNA. Mol. Cell. Biol. 222918-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi, Y., T. Wada, D. Watanabe, T. Takagi, J. Hasegawa, and H. Handa. 1999. Structure and function of the human transcription elongation factor DSIF. J. Biol. Chem. 2748085-8092. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto, T., S. Suzuki, K. Radsak, and K. Hirai. 1998. The UL112/113 gene products of human cytomegalovirus which colocalize with viral DNA in infected cell nuclei are related to efficient viral DNA replication. Virus Res. 56107-114. [DOI] [PubMed] [Google Scholar]
- 49.Yang, Z., J. H. Yik, T. Chen, N. He, M. K. Jang, K. Ozato, and Q. Zhou. 2005. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19535-545. [DOI] [PubMed] [Google Scholar]
- 50.You, J., J. L. Croyle, A. Nishimura, K. Ozato, and P. M. Howley. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117349-360. [DOI] [PubMed] [Google Scholar]
- 51.You, J., V. Srinivasan, G. V. Denis, W. J. Harrington, Jr., M. E. Ballestas, K. M. Kaye, and P. M. Howley. 2006. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 808909-8919. [DOI] [PMC free article] [PubMed] [Google Scholar]