

Abstract
K-cyclin, encoded by Kaposi's sarcoma-associated herpesvirus, has previously been demonstrated to activate cyclin-dependent kinase 6 (Cdk6) to induce the phosphorylation of various cell cycle regulators. In this study, we identified Cdk9 as a new K-cyclin-associated Cdk and showed that K-cyclin interacted with Cdk9 through its basic domain. We hypothesized that K-cyclin served as a regulatory subunit for the activity of Cdk9. Recent reports show that Cdk9 phosphorylates tumor suppressor p53, and we found that the K-cyclin/Cdk9 interaction greatly enhanced the kinase activity of Cdk9 toward p53. The phosphorylation site(s) of K-cyclin/Cdk9 kinase complexes was mapped in the transactivation domain of p53. We showed that the ectopic expression of K-cyclin led to a sustained increase of p53 phosphorylation on Ser33 in vivo, and the phosphorylation could be inhibited by a dominant negative Cdk9 mutant, dn-Cdk9. Using p53-positive U2OS and p53-null SaOS2 cells, we demonstrated that K-cyclin-induced growth arrest was associated with the presence of p53. In addition, K-cyclin-induced p53-dependent growth arrest was rescued by the dn-Cdk9- or Cdk9-specific short hairpin RNA in SaOS2 cells. Together, our findings for the first time demonstrated the interaction of K-cyclin and Cdk9 and revealed a new molecular link between K-cyclin and p53.
Kaposi's sarcoma-associated herpesvirus (KSHV) or human herpesvirus 8 is a DNA tumor virus and etiologic agent of Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD) (13, 53, 72). KS is one of the most common malignancies in AIDS patients and the fourth most common cancer caused by infection worldwide (4). PEL is a high-grade non-Hodgkin B-cell lymphoma with poor prognosis and short survival time (6). MCD is a lymphoproliferative disorder that has a very high risk of developing into PEL (58). The majority of tumor cells in these neoplasms are latently infected with KSHV, suggesting that viral latent genes are important for KSHV-associated malignancies (5, 29, 79). K-cyclin, encoded by orf72, is one of the few KSHV latent proteins that are expressed in the tumors, suggesting its pathological role in oncogenesis (12, 19, 21).
KSHV encodes several homologues of cellular proteins, including K-cyclin, vFLIP, vIRF, v-Bcl-2, and vIL-6. These viral proteins enable the virus to manipulate and “hijack” cellular machineries that regulate cell signaling pathways, gene expression, cell growth, and apoptosis (38, 51). K-cyclin shares about 30% sequence identity with cellular D-type cyclins and preferentially interacts with cyclin-dependent kinase 6 (Cdk6) (11). The K-cyclin/Cdk6 complexes show several unique differences with their cellular counterparts, Cdk6/cyclin D complexes. First, the K-cyclin/Cdk6 complexes exhibit higher kinase activity toward Rb and histone H1 (32, 46). Second, this kinase activity is not regulated by Cdk-activating kinase (40) or Cdk inhibitors p16Ink4, p21Cip1, and p27Kip1 (25, 48, 73). Therefore, the K-cyclin/Cdk6 complexes in part mimic constitutively activated Cdk/cyclin kinases of G1 and S phases. Third, K-cyclin interacts with a broad range of Cdks, including Cdk2, Cdk4, and Cdk5 (32, 43, 46, 48, 63, 75). Fourth, K-cyclin/Cdk6 complexes have an extended spectrum of substrates. These include histone H1 (32, 46), p27Kip1 (25, 48, 68, 75), p21Cip1 (37, 75), Bcl-2 (57), Id-2 (48), Cdc25A (48), Cdc6 (43), Orc1(43), and caldesmon (17). Finally, K-cyclin is involved in the deregulation of cellular gene expression. K-cyclin inhibits STAT3-mediated gene activation (47) and upregulates proproliferative genes, such as the cyclin A gene (23). These properties demonstrate a great potential for K-cyclin to disrupt the regulatory mechanisms of cellular growth control and support the notion that it is an oncogenic protein of KSHV. Indeed, ectopic expression of K-cyclin prevents G1 arrest imposed by many cell cycle inhibitors (48, 73) and stimulates cell cycle progression in quiescent fibroblasts (14, 73).
Similar to many oncogenic proteins, K-cyclin can also trigger apoptosis and induce cell growth arrest under certain circumstances. For example, K-cyclin induces apoptosis in the presence of high levels of Cdk6 (56, 57) or stress signals (78). The proapoptotic effect is at least in part associated with its ability to induce the phosphorylation and inhibition of the antiapoptotic protein Bcl-2 (57). K-cyclin-associated apoptosis and aberrant cell growth are also closely related to tumor suppressor p53 during tumorigenesis (76, 78). Tumor suppressor p53 is a crucial component of the cellular machinery that mediates cell growth arrest and apoptosis in response to a wide range of internal and external stress signals. The ectopic expression of K-cyclin in mouse embryonic fibroblasts (MEFs) causes p53 accumulation, which is likely to contribute to K-cyclin-induced growth arrest and apoptosis in MEFs (78). In transgenic-mouse models, K-cyclin induces tumor formation in p53−/− mice but not in mice expressing functional wild-type p53 (76). These results indicate the existence of interplay between p53 and K-cyclin.
p53 is tightly controlled by multiple posttranslational modifications. Among them, phosphorylation plays a special role in fine-tuning the activity of p53. A variety of cellular kinases can phosphorylate serine or threonine residues at the N and C termini of p53 (3, 7). Recently, Cdk9 was found to phosphorylate p53 on serine 33, 315, and 392 (15, 64). Cdk9 belongs to the category of Cdk that is involved in transcriptional regulation (16, 20). The major function of Cdk9 is to regulate mRNA synthesis during initiation or elongation steps (24). In complexes with cyclin T1, T2a, T2b, or K, Cdk9 can phosphorylate the C-terminal domain of the large subunit of RNA polymerase II (RNA pol II) (28, 49, 60) and inhibitory elongation factors, such as 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole sensitivity-inducing factor and the negative elongation factor (30). In addition, the Cdk9 gene is located on chromosome 9q34.1, a region involved in nonrandom chromosome alterations, suggesting a possible involvement of Cdk9 in some human neoplasms (10).
In the search for new K-cyclin partners, we found that K-cyclin interacts with Cdk9. This led us to hypothesize that K-cyclin regulates the kinase activity of Cdk9 to phosphorylate p53. We found that the K-cyclin/Cdk9 interaction activated Cdk9 to induce the phosphorylation of p53. We demonstrated that the phosphorylation site was located within the transactivation domain (TAD) of p53, specifically on Ser33. Moreover, cell growth arrest caused by K-cyclin was linked to the presence of p53 and apparently required the kinase activity of K-cyclin/Cdk9 complexes. Our study demonstrated that Cdk9 is a K-cyclin target and revealed a novel link between K-cyclin and the p53 pathway.
MATERIALS AND METHODS
Plasmids.
Plasmids encoding human cyclin T1 (pCI-86) and a dominant-negative mutant of human Cdk9 (dn-Cdk9; pFlag-CMV2-hCdk9-D167N) were generously provided by Andrew P. Rice (Baylor College of Medicine, Houston, TX). pEF1/myc-his A and pcDNA3 plasmids were purchased from Invitrogen. pEF1A-K-cyc-myc-his, pEF1A-K-cyc-D1/2-myc-his, pEF1A-K-cyc-D2-myc-his, pEF1A-K-cyc-D2/3-myc-his, and pEF1A-K-cyc-D1/3-myc-his were constructed using a standard PCR cloning technique. pGEX-4T-p53, pGEX-4T-p53-N2, pGEX-4T-p53-C2, pGEX-4T-p53-TAD, pGEX-4T-p53-SH3, pGEX-4T-p53-DBD, pGEX-4T-p53-TD, and pGEX-4T-p53-BD plasmids, expressing full-length human p53 and its series of deletion mutants, were constructed as previously described (54).
Cell culture and DNA transfection.
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), Opti-MEM, and other routinely used cell culture reagents were purchased from Invitrogen. 293FT, AD-293, U2OS, and SaOS2 cells were maintained in DMEM supplemented with 10% heat-inactivated FBS at 37°C with 5% CO2. For the coimmunoprecipitation assay, 6 × 105 293FT cells were seeded in a six-well plate and cotransfected with either 0.4 μg of pEF1A-K-cyc-myc-his, 0.4 μg of pcDNA3-flag-Cdk9, or both. A control plasmid, pEF1A-myc-his or pcDNA3, was added to maintain equivalent (total, 0.8 μg) DNA inputs. Transfections were performed with TransFectin reagent according to the manufacturer's instructions (Bio-Rad).
Western blot analysis.
Transfected or adenovirus-infected cells were collected after being washed once with ice-cold phosphate-buffered saline (PBS). To prepare total cell lysate (TCL), cells were lysed in 500 μl of total cell lysis buffer (1× PBS, 1 μg/ml pepstatin A, 0.5 μg/ml leupeptin, 0.5 μg/ml aprotinin, 0.5 mM phenylmethylsulfonyl fluoride, 0.3 μg/ml E-64, 8 mM NaF, 25 mM NaVO3, and 20 mM sodium pyrophosphate) containing 0.5% NP-40 and sonicated for 10 s. Supernatants were then collected after centrifugation at 14,000 × g for 20 min at 4°C. Standard Western blotting was performed by using the TCL, and the targeted proteins were visualized with an enhanced chemiluminescence detection system. The primary antibodies used in the Western blots were purchased from the following companies: anti-Flag-horseradish peroxidase (HRP) (M2) antibody and antitubulin monoclonal antibody (MAb), Sigma; rabbit antibodies against p53, phospho-p53 (Ser33), phospho-p53 (Ser315), and phospho-p53 (Ser392), Cell Signaling Technology; and anti-Cdk9 (D-7) MAb, Santa Cruz Biotechnology. Anti-myc MAb was produced from the MYC 1-9E10.2 hybridoma cell line (American Tissue Culture Collection). HRP-conjugated secondary antibodies were purchased from Rockland Immunochemicals (Gilbertsville, PA).
Coimmunoprecipitation.
For the transfected 293FT cells, TCL was prepared as described above and then was gently mixed with an appropriate antibody on a rotator for 2 h at 4°C. Five microliters of Protein G Plus-agarose beads (Santa Cruz Biotechnology) was then added, and samples were continuously mixed for an additional 2 h at 4°C. The agarose beads were washed three times with cold total cell lysis buffer, and the immunoprecipitated protein complexes were eluted in sodium dodecyl sulfate (SDS) sample buffer for Western blot analysis. For the KSHV-infected BCBL-1 and control BJAB cells, 107 cells were treated with 1 mM of butyric acid for 48 h and then lysed in total cell lysis buffer containing 1% NP-40. Five hundred microliters of the lysates was diluted with 1,000 μl of PBS containing protease inhibitors. Coimmunoprecipitation was performed in two steps. First, 5 μl of rat anti-K-cyclin antibody (Santa Cruz Biotechnology) was added and the samples were mixed on a rotator at 4°C for 3 h. Second, 1 μl of rabbit anti-rat antibody (Santa Cruz Biotechnology) was added, followed by mixing for another 3 h. Ten microliters of protein A-agarose beads (Sigma) was then added, and the coimmunoprecipitation was performed overnight at 4°C. The immune complexes were analyzed by Western blotting as described above.
In vitro kinase assay.
Transfected 293FT cells were lysed on ice for 10 min in 500 μl of kinase lysis buffer (50 mM Tris-HCl, pH 8.0, 120 mM NaCl, 5 mM dithiothreitol, 0.5% NP-40, 1 μg/ml pepstatin A, 0.5 μg/ml leupeptin, 0.5 μg/ml aprotinin, 0.5 mM phenylmethylsulfonyl fluoride, 0.3 μg/ml E-64, 20 mM sodium pyrophosphate, and 1 mM NaF). The cell lysates were clarified by centrifugation, and subjected to immunoprecipitation with anti-Flag or anti-Cdk9 antibody as described above. The agarose beads were rinsed three times with the kinase lysis buffer and once with a kinase reaction buffer (100 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 10 mM dithiothreitol, and 5 μM ATP). The beads were resuspended in 20 μl of the kinase reaction buffer containing 0.5 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) and purified glutathione S-transferase (GST)-p53. The kinase reaction was performed at 30°C for 1 h. The samples were resolved by SDS-polyacrylamide gel electrophoresis, and autoradiography was performed at −70°C with an intensifying screen.
Recombinant adenovirus.
Recombinant adenoviruses Ad-GFP and Ad-p53 were described previously (54). Recombinant adenovirus expressing Cdk9, dn-Cdk9, or K-cyclin was constructed using the AdEasy system (Quantum Biotechnologies). Briefly, the PCR products of Cdk9 and dn-Cdk9 were cloned into the pAdTrack-CMV shuttle vector at BglII and XhoI sites, and Flag-tagged K-cyclin was cloned at HindIII and XbaI sites. After being linearized with PmeI, the pAd-Cdk9, pAd-dn-Cdk9, or pAd-F-K-cyclin plasmid was individually cotransformed with pAdeasy-1 DNA into BJ5183 cells by electroporation. Homologous recombinants were selected with kanamycin and confirmed by PacI or BstXI digestion. The plasmid DNA was then amplified in MJ109 cells and purified by the standard CsCl2 density gradient method. To produce adenovirus, the PacI-linearized recombinant plasmid was transfected into AD-293 cells (Stratagene) with TransFectin. Seven to 10 days after the transfection, the AD-293 cells and media were collected, and the recombinant viruses were isolated by three repeated freeze-thaw cycles. Virus-containing supernatants were subjected to further amplification. The titer of stock virus was determined by a plaque formation assay after a series of dilutions.
Lentivirus production and transduction.
Empty control lentiviral vector pLKO.1 and pLKO.1-shRNA-Cdk9 (oligonucleotide identification: TRCN0000000496) were purchased from Addgene and Open Biosystems, respectively. Lentivirus production was performed with the four-plasmid transfection system (66). Briefly, 106 293FT cells were seeded in six-well plates. pLKO.1 or pLKO.1-shRNA-Cdk9 (0.3 μg), pVSV-G (0.1 μg), pRSV-Rev (0.1 μg), and pMDL-gag/pol (0.2 μg) Rev-responsive element packaging plasmids were cotransfected into 293FT cells with TransFectin reagent. Forty-eight and 72 h after transfection, the virus-containing media were harvested, clarified at 20,000 × g for 5 min, and used for transduction. To generate Cdk9 knockdown cells, 2 × 105 SaOS2 cells were seeded in six-well plates and transduced with 4 ml of lentivirus and 10 μg/ml of Polybrene (Sigma) by spin inoculation (1,000 × g for 2 h at room temperature). After spinning, cells were incubated at 37°C with 5% CO2 for 2 h. The virus-containing media were then removed and replaced with fresh medium. Forty-eight hours posttransduction, cells were subjected to puromycin (0.5 μg/ml) selection over a period of 5 days. The SaOS2 cells were then plated in 96-well plates at a density of 2.5 × 103 cells per well and infected with recombinant adenoviruses for cell proliferation assays.
Cell proliferation assay.
Sixteen to 18 hours before infection, U2OS or SaOS2 cells were plated in 96-well culture plates at a density of 2.5 × 103 cell per well. Cells were infected with recombinant adenoviruses at the desired multiplicity of infection (MOI). Cell proliferation was assessed using the CellTiter-Glo luminescent cell viability assay kit (Promega), according to the manufacturer's protocol. The luminescence signal was measured with the Clarity luminescence microplate reader (BioTek Instruments Inc., Winooski, VT). Five wells were measured for each time point. The average value and the standard deviation were calculated and graphed.
RESULTS
KSHV K-cyclin interacts with Cdk9.
It has been well studied that KSHV K-cyclin can interact with and activate cellular Cdk6, a Cdc2-related kinase. To identify a new K-cyclin-regulated kinase partner, we examined the ability of K-cyclin to interact with Cdk9, another member of Cdc2-related kinases, by coimmunoprecipitation. myc-tagged K-cyclin and Flag-tagged Cdk9 were transiently expressed in 293FT cells, and immunoprecipitation was performed with anti-myc antibody to pull down K-cyclin-associated complexes. The immunocomplexes were then analyzed by Western blotting with anti-Flag antibody to determine the existence of Cdk9. As shown in Fig. 1A, top, Cdk9 was readily detected in K-cyclin-associated immunocomplexes. Reciprocally, K-cyclin was detected in Cdk9 kinase complexes (Fig. 1A, second panel from the top). The successful immunoprecipitation was confirmed by Western blotting with anti-K-cyclin or anti-Cdk9 antibody (data not shown). The expression levels of Cdk9 and K-cyclin in TCLs are shown in Fig. 1A, third and fourth panels, respectively. These results demonstrated the molecular interaction between K-cyclin and Cdk9. The interaction of K-cyclin with endogenous Cdk9 was also demonstrated in KSHV-infected BCBL-1 cells. The expression of K-cyclin in BCBL-1 cells was detectable, but at a low level. To increase the expression of K-cyclin, we pretreated BCBL-1 cells with 1 mM of butyric acid for 48 h. The butyric acid-treated BCBL-1 and control BJAB cells were subjected to coimmunoprecipitation with anti-K-cyclin antibody. As shown in Fig. 1B, Cdk9 was detected in K-cyclin-associated complexes, confirming K-cyclin/Cdk9 complex formation in the virally infected cells.
FIG. 1.

Interaction of KSHV K-cyclin and Cdk9. (A) 293FT cells were transfected with myc-tagged K-cyclin and/or Flag-tagged Cdk9 expression plasmids. Forty-eight hours after transfection, TCLs were used for immunoprecipitation (IP), followed by Western blotting (WB) with the antibodies indicated (top two panels). TCLs of transfected 293FT cells were analyzed by Western blotting with anti-Flag or anti-myc antibody to show the expression of Cdk9 or K-cyclin (bottom two panels). α, anti. (B) BJAB and BCBL-1 cells were treated with 1 mM of butyric acid. Forty-eight hours after treatment, Co-IP and WB were performed with the antibodies indicated above (top panel). TCLs of BJAB and BCBL-1 cells were analyzed by WB to show the expression levels of endogenous Cdk9 and K-cyclin (bottom two panels).
KSHV K-cyclin contains three major domains, domain 1 (neutral domain), domain 2 (basic domain [BD]), and domain 3 (acidic domain). To determine the domain(s) of K-cyclin involved in the interaction with Cdk9, myc-tagged K-cyclin variants containing full-length, domains 1 and 2, domain 2, domains 2 and 3, and domains 1 and 3 were constructed (Fig. 2A). They were used for coimmunoprecipitation with anti-Flag antibody. As shown in Fig. 2B, only the deletion of domain 2 disrupted the interaction of K-cyclin with Cdk9 (Fig. 2B, lanes 3 to 6). This result suggested that domain 2 (BD) of K-cyclin was involved in K-cyclin/Cdk9 complex formation.
FIG. 2.

Identification of K-cyclin domain(s) that binds to Cdk9. (A) Schematic representation of the K-cyclin domains and deletion mutants. Positive and negative binding of Cdk9 with the K-cyclin mutants is denoted by + and −, respectively, on the right. (B) Interactions of Cdk9 and the deletion mutants of K-cyclin. myc-tagged full-length K-cyclin (lane 2) or its mutants (lanes 3 to 6) were transiently coexpressed with Flag-tagged Cdk9 in 293FT cells. TCLs from transfected cells were used for immunoprecipitation (IP) with anti-myc antibody, followed by Western blotting (WB) with anti-Flag antibody (top). Expression of K-cyclin, its mutants, and Cdk9 was confirmed by Western blotting with anti-myc or anti-Flag antibody as indicated (middle and bottom). α, anti.
KSHV K-cyclin enhances the phosphorylation of p53 by Cdk9.
The identification of the K-cyclin/Cdk9 interaction suggests that K-cyclin may serve as a regulatory subunit in Cdk9 kinase complexes. Two recent studies showed that Cdk9 is able to phosphorylate tumor suppressor p53 (15, 64), implying that the K-cyclin/Cdk9 interaction may alter the kinase activity of Cdk9 toward p53. Therefore, we performed in vitro kinase assays to determine the effect of K-cyclin on Cdk9-mediated p53 phosphorylation. The kinase assay was carried out with Cdk9 kinase complexes immunoprecipitated from lysates of Cdk9- and/or K-cyclin-transfected 293FT cells. Recombinant GST or GST-p53 was used as a substrate in the reaction. Consistent with previous reports from others (15, 64), our results confirmed that Cdk9 phosphorylated GST-p53 but not the GST control (Fig. 3A). In the presence of K-cyclin, the phosphorylation of p53 was noticeably increased, indicating the enhancement of Cdk9 kinase activity by K-cyclin (Fig. 3A). To further confirm the capability of K-cyclin to activate Cdk9, endogenous Cdk9 complexes immunoprecipitated with anti-Cdk9 antibody were used for in vitro kinase assays. The phosphorylation of p53 by endogenous Cdk9 was observed (Fig. 3B, lane 2), and a significant increase of p53 phosphorylation was found in the presence of K-cyclin (Fig. 3B, lane 4). These results showed that K-cyclin specifically enhanced Cdk9 kinase activity in the phosphorylation of p53.
FIG. 3.

Enhancement of p53 phosphorylation by K-cyclin/Cdk9 complexes. (A) Phosphorylation of p53 by exogenously expressed Cdk9 and K-cyclin. A Flag-tagged Cdk9 expression plasmid was transfected alone (lanes 1 and 2) or with a myc-tagged K-cyclin expression plasmid (lanes 3 and 4) into 293FT cells. Forty-eight hours after transfection, Cdk9-associated kinase complexes were immunoprecipitated from TCLs with anti-Flag antibody and assayed for the kinase activity. Recombinant GST (lanes 1 and 3) and GST-p53 (lanes 2 and 4) were used as substrates. Samples were subjected to SDS-polyacrylamide gel electrophoresis and autoradiography. (B) Phosphorylation of p53 by endogenous Cdk9 in the presence of K-cyclin. Endogenous Cdk9 kinase complexes were immunoprecipitated with anti-Cdk9 antibody from TCLs of 293FT cells (lanes 1 and 2) or 293FT cells transfected with myc-tagged K-cyclin expression plasmid (lanes 3 and 4). Kinase assays were performed as described for panel A.
To further demonstrate that Cdk9 was the kinase responsible for p53 phosphorylation by K-cyclin/Cdk9 complexes, we included a kinase-inactive dominant-negative mutant of Cdk9, dn-Cdk9, in our study. dn-Cdk9 contains a mutation that replaces Asp167 with Asn (31, 33). As has been demonstrated in many publications, this mutant specifically blocks the kinase activity of endogenous Cdk9. Though we found that dn-Cdk9 still maintained its ability to interact with K-cyclin (Fig. 4A), its ability to phosphorylate p53 was significantly diminished in comparison with that of wild-type Cdk9 (Fig. 4B, top). Similarly, when serving as a negative control, dn-Cdk9 inhibited p53 phosphorylation in a complex with its major regulator, cyclin T1 (Fig. 4B, bottom). Together, these results demonstrated that dn-Cdk9 still maintained its ability to interact with K-cyclin but lost the ability to phosphorylate p53 efficiently. This feature implied that the kinase activity of Cdk9 is important for p53 phosphorylation.
FIG. 4.

Requirement of Cdk9 kinase activity for p53 phosphorylation by K-cyclin/Cdk9 complexes. (A) Association of K-cyclin and dn-Cdk9 in vivo. 293FT cells were transfected with myc-tagged K-cyclin and/or Flag-tagged dn-Cdk9 expression plasmid. Forty-eight hours after transfection, TCLs of transfected 293FT cells were used for immunoprecipitation (IP), followed by Western blotting (WB) with the antibodies indicated (top two panels). TCLs were also analyzed by Western blotting with anti-Flag or anti-myc antibody to show the expression of dn-Cdk9 or K-cyclin (bottom two panels). α, anti. (B) Inhibition of p53 phosphorylation by dn-Cdk9. A myc-tagged K-cyclin expression plasmid was cotransfected with Flag-tagged Cdk9 (top panel, lane 1) or dn-Cdk9 (top panel, lane 2) expression plasmid into 293FT cells. The kinase activities of K-cyclin/Cdk9 and K-cyclin/dn-Cdk9 were analyzed as described for Fig. 3. Similarly, Cdk9/cyclin T1 (bottom panel, lane 1) and dn-Cdk9/cyclin T1 (bottom panel, lane 2) were used as a positive and negative controls for p53 phosphorylation.
K-cyclin enhances Cdk9-mediated phosphorylation of p53 on Ser33.
To determine the K-cyclin/Cdk9 phosphorylation site(s) on p53, we first investigated the ability of the K-cyclin/Cdk9 holoenzyme to phosphorylate different domains of p53 in in vitro kinase assays. In this experiment, a series of bacterially produced GST fusion proteins, including different domains of p53 and full-length p53, were used as substrates (Fig. 5A). The K-cyclin/Cdk9 holoenzyme clearly phosphorylated GST-p53, GST-p53-N2, GST-p53-C2, and GST-p53-TAD (Fig. 5B). In addition, GST-p53-TD and GST-p53-BD showed a weak but detectable phosphorylation by the K-cyclin/Cdk9 holoenzyme (Fig. 5B). A similar phosphorylation pattern of these GST-p53 fusion proteins was also obtained when Cdk9 alone was used (data not shown). These results indicated that K-cyclin/Cdk9 or Cdk9 phosphorylation sites are located within the TAD, tetramerization domain (TD), and BD of p53. Next, the kinase activities of Cdk9 and K-cyclin/Cdk9 complexes on different domains of p53 were compared. Coexpression of Cdk9 and K-cyclin led to an increase in phosphorylation of full-length GST-p53 and GST-p53-TAD (Fig. 5C). In contrast, no significant changes of phosphorylation were observed in GST-p53-SH3, GST-p53-DBD, GST-p53-TD, and GST-p53-BD in the presence of K-cyclin. These results indicated that the phosphorylation site(s) within the TAD of p53 was mainly influenced by the K-cyclin/Cdk9 holoenzyme.
FIG. 5.

Mapping of the p53 domains responsible for K-cyclin-enhanced Cdk9 phosphorylation. (A) Schematic presentation of p53 domains, including the transaction domain (TAD; amino acids [aa] 1 to 60), Src homology 3-like domain (SH3; aa 60 to 100), DNA-binding domain (DBD; aa 100 to 300), tetramerization domain (TD; aa 300 to 356), and basic domain (BD; aa 357 to 393), and a series of GST-p53 fusion constructs used in the experiment. (B) Phosphorylation of recombinant GST-p53 fusion proteins by K-cyclin/Cdk9 kinase complexes. An in vitro kinase assay was performed as described for Fig. 3A (top). A series of GST-p53 deletion mutants, as indicated, were used as substrates. The amount of input GST-tagged p53 proteins was confirmed on a Coomassie blue-stained gel (bottom). (C) Comparison of the kinase activities of Cdk9 and K-cyclin/Cdk9 complexes with domains of p53. Cdk9 or K-cyclin/Cdk9 complexes were prepared as described for Fig. 3A, and in vitro kinase assays were performed using the indicated GST-p53 fusion proteins as substrates.
Previous reports have showed that p53 was phosphorylated by Cdk9 on Ser33, Ser315, and Ser392 (15, 64). The phosphorylation of these sites was correlated with the phosphorylation of the TAD, TD, and BD of p53 by K-cyclin/Cdk9 complexes in our in vitro kinase assays. We therefore examined if K-cyclin enhances the Cdk9-mediated phosphorylation on any of these sites in vivo. To do this, different levels of K-cyclin were expressed in the p53-positive U2OS cells. As shown in Fig. 6A, increased levels of Ser33 phosphorylation on p53 were found to correlate with increased expression of K-cyclin. In contrast, we detected no significant changes of the phosphorylation on Ser315 and Ser392 in the presence of increasing K-cyclin (Fig. 6A). These results were consistent with the findings that only the phosphorylation of TAD (but not of TD and BD) was increased by K-cyclin/Cdk9 complexes in our in vitro kinase assays. Increased phosphorylation of Ser33 induced by K-cyclin was also demonstrated in 293FT cells (Fig. 6B). Interestingly, slight increases of total p53 were also observed, and this appeared to relate to the expression of K-cyclin (Fig. 6A and B). To confirm the specific role of Cdk9 in K-cyclin-induced p53 phosphorylation, we expressed dn-Cdk9 in U2OS cells to block the kinase activity of endogenous Cdk9. We detected a decreased phosphorylation of Ser33 in cells expressing dn-Cdk9, while the other two Cdk9 phosphorylation sites, Ser315 and Ser392, were unaffected (Fig. 6C). These results showed that K-cyclin specifically enhanced Cdk9-mediated phosphorylation of p53 on Ser33.
FIG. 6.

Identification of K-cyclin-enhanced Cdk9 phosphorylation sites within p53. (A) Increased Ser33 phosphorylation of p53 by K-cyclin in U2OS cells. U2OS cells were transfected with an empty plasmid (lane 1) or with different amounts of K-cyclin expression plasmid (lanes 2 to 4). Twenty-four hours after transfection, TCLs were analyzed by Western blotting (WB) with the indicated antibodies. α, anti. (B) Increased Ser33 phosphorylation of p53 by K-cyclin in 293FT cells. Similar to the experiments described for panel A, levels of Ser33 phosphorylation were analyzed in the presence (lane 2) and absence (lane 1) of K-cyclin in 293FT cells. (C) Inhibition of Ser33 phosphorylation by dn-Cdk9. The K-cyclin expression plasmid was cotransfected with either an empty plasmid (lane 1) or the dn-Cdk9 expression plasmid (lane 2) into U2OS cells, and phosphorylation of p53 was analyzed with the antibodies indicated.
K-cyclin induces a p53-dependent growth suppression.
Verschuren and colleagues previously demonstrated that constitutive expression of K-cyclin induced growth arrest in a p53-dependent manner in MEF cells (78). To elucidate mechanisms involved in K-cyclin-induced growth suppression, we used an adenoviral expression system to facilitate gene expression in two osteosarcoma-derived cell lines, U2OS (p53-expressing cells) and SaOS2 (p53-null cells). Titers of stock recombinant adenoviruses that express K-cyclin, p53, Cdk9, and dn-Cdk9 were first determined by standard plaque assay to ensure accurate viral input in all experiments. Figure 7 shows protein expression levels of p53, K-cyclin, Cdk9, and dn-Cdk9 in U2OS and SaOS2 cells after cells were infected with recombinant adenoviruses at different MOIs. Interestingly, we found that expression levels of recombinant proteins are generally higher in U2OS cells than in SaOS2 cells. Thus, we used higher MOIs in the later studies of SaOS2 cell proliferation. An adenovirus, Ad-GFP, which expresses only green fluorescent protein was constructed to serve as a negative control. Infections of U2OS and SaOS2 cells with Ad-GFP virus do not affect cell growth in comparison with uninfected cells (data not shown).
FIG. 7.
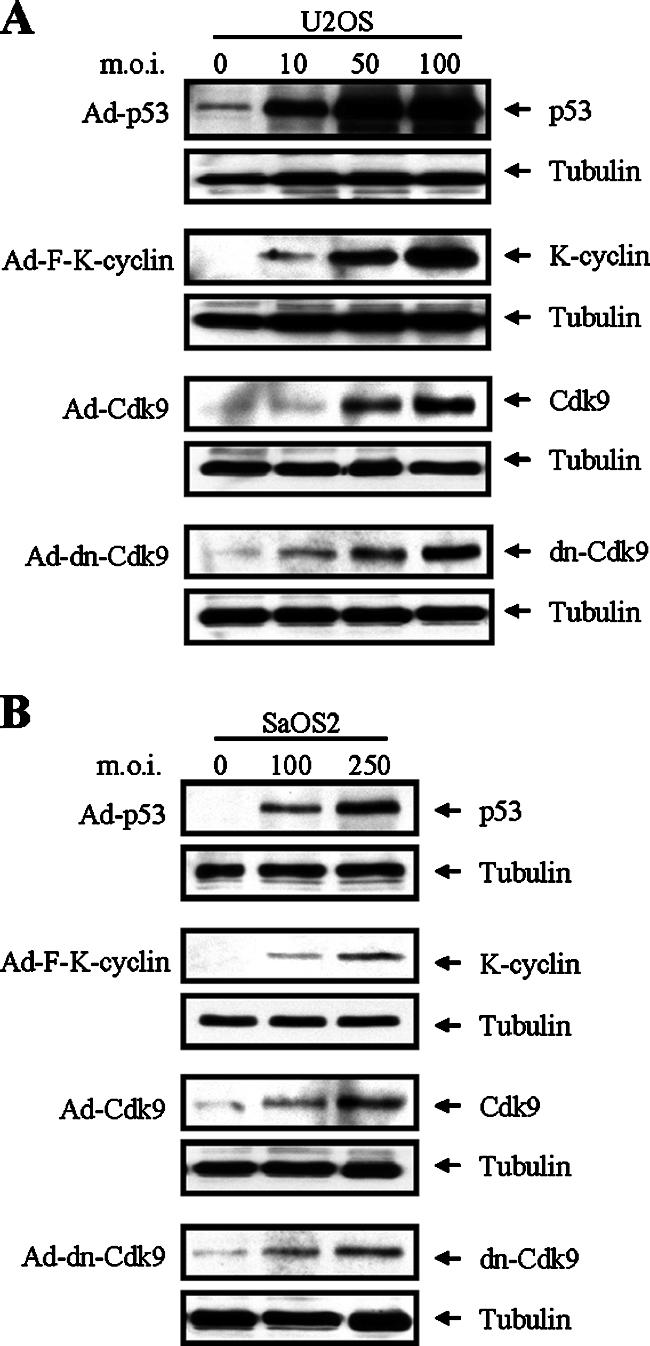
Dose-dependent expression of p53, K-cyclin, Cdk9, and dn-Cdk9 with recombinant adenoviruses. U2OS (A) and SaOS2 (B) cells were infected with Ad-GFP, Ad-p53, Ad-F-K-cyclin, Ad-Cdk9, or Ad-dn-Cdk9 recombinant adenovirus. Twenty-four to 48 h after infection, TLCs were analyzed by Western blotting with the indicated antibodies. Levels of tubulin were used as a loading control.
We first evaluated the dose-related effects of K-cyclin on the growth of U2OS and SaOS2 cells. Recombinant adenoviruses at different MOIs were used to infect the cells in 96-well culture plates. Four days postinfection, the total number of viable cells was analyzed using the CellTiter-Glo luminescent cell viability assay kit. As shown in Fig. 8A, K-cyclin inhibited U2OS cell growth in a dose-dependent manner. However, K-cyclin had no significant effect on the growth of SaOS2 cells, even when higher MOIs were used for Ad-F-K-cyclin infection (Fig. 8B). As described by many groups, the p53-null SaOS2 cells are sensitive to the exogenous expression of wild-type p53. Overexpression of p53 in SaOS2 cells often induces cell growth arrest and apoptosis. In this experiment, Ad-p53 was included as a positive control in SaOS2 cell experiments (Fig. 8B). We next examined the growth of U2OS and SaOS2 cells on five consecutive days in the presence of K-cyclin. Since there was no significant growth difference between SaOS2 cells infected with Ad-F-K-cyclin at MOIs of 100 and 250, we chose an MOI of 100 as the viral input in the experiments. In U2OS cells, K-cyclin suppressed cell growth, as evidenced by a slower cell proliferation rate from the second day postinfection (Fig. 8C). In contrast, there was no significant growth suppression observed in the Ad-F-K-cyclin-infected SaOS2 cells (Fig. 8D).
FIG. 8.

Effects of K-cyclin on the growth of U2OS and SaOS2 cells. (A) Dose-dependent growth suppression by K-cyclin expression in U2OS cells. U2OS cells were infected at different MOIs with Ad-GFP or Ad-F-K-cyclin as indicated. Four days after infection, cell numbers were analyzed with the CellTiter-Glo luminescent cell viability assay kit. Results shown at each MOI point represent the means ± standard deviations (SD) from five experiments. (B) Effects of K-cyclin on the growth of SaOS2 cells. SaOS2 cells were infected with Ad-GFP, Ad-F-K-cyclin, or Ad-p53 at MOIs of 100 and 250. Four days after infection, cell numbers were analyzed as described for panel A. (C) Growth suppression by K-cyclin in U2OS cells. U2OS cells were infected with Ad-GFP or Ad-F-K-cyclins at an MOI of 100. Cell numbers were analyzed over five consecutive days. Results shown represent the means ± SD from five experiments. (D) Effects of K-cyclin on the growth rate of SaOS2 cells. Proliferations of SaOS2 cells after infection with Ad-GFP, Ad-F-K-cyclins, or Ad-p53 at an MOI of 100 were analyzed as for panel C. RLU, relative light units.
To further determine whether p53 was required for the growth suppression induced by K-cyclin, p53 and K-cyclin were coexpressed in p53-null SaOS2 cells. In the experiments, the control Ad-GFP was added to maintain equivalent viral inputs in the samples that were infected with Ad-p53 or Ad-F-K-cyclin alone. With the fixed input of Ad-p53 (MOI, 50), we infected SaOS2 cells with different amounts of Ad-F-K-cyclin (MOIs, 100, 200, and 300). Under this condition, the inputs of Ad-F-K-cyclin at MOIs of 100 and 200 showed no and moderate inhibition of the growth of SaOS2 cells, respectively (data not shown). Notably, when Ad-K-cyclin at an MOI of 300 was used, it markedly enhanced the p53-induced growth repression in SaOS2 cells (Fig. 9). In contrast, K-cyclin alone did not affect cell growth. Our results demonstrated that K-cyclin-induced growth repression was linked to the presence of p53.
FIG. 9.

Inhibition of SaOS2 cell growth by K-cyclin in the presence of p53. SaOS2 cells were infected with either Ad-p53 (MOI, 50) and Ad-F-K-cyclin (MOI, 300) alone or with both. Cell growth of infected SaOS2 cells was analyzed over five consecutive days. Ad-GFP was added to equalize the total viral input when single adenovirus infection was performed. Results shown represent the means ± standard deviations from five experiments. RLU, relative light units.
Cdk9 is required for K-cyclin-induced p53-dependent growth suppression.
Given the interaction between K-cyclin and Cdk9, the enhancement of p53 phosphorylation by K-cyclin/Cdk9 holoenzyme, and the necessity of p53 in K-cyclin-induced growth suppression, we determined the role of Cdk9 in the interplay between K-cyclin and p53 in growth regulation by overexpression of Cdk9 in p53-null SaOS2 cells. To examine if additional Cdk9 further enhanced the K-cyclin-induced growth suppression in the presence of p53, pAd-p53 (MOI, 50) and Ad-F-K-cyclin (MOI, 200) were used to induce a medium level of growth suppression in SaOS2 cells. Under this condition, Ad-Cdk9 (MOI, 100) was added to determine the effect of additional Cdk9 on cell growth. As shown in Fig. 10A, exogenous Cdk9 did not significantly further enhance the growth suppression induced by K-cyclin in the presence of p53 in SaOS2 cells. This result was consistent with a previous report from Granan's group, who showed that overexpression of Cdk9 at up to four times the endogenous level was not sufficient to increase the total kinase activity of Cdk9 in the cell (31). They suggested that some limiting cellular factors must be required for modulating Cdk9 kinase activity. Taking these results together, we speculated that endogenous Cdk9 may be enough for the growth suppression induced by K-cyclin and p53.
FIG. 10.

Role of Cdk9 on the growth suppression induced by the collaboration of K-cyclin and p53 in SaOS2 cells. (A and B) SaOS2 cells were infected with recombinant adenoviruses as indicated. The viral inputs of Ad-p53 (MOI, 50), Ad-F-K-cyclin (MOI, 200), and Ad-Cdk9 (MOI, 100) or Ad-dn-Cdk9 (MOI, 1) were used in the experiments. Ad-GFP was added to equalize the total viral input. The effect of Cdk9 (A) and dn-Cdk9 (B) on the growth rate of SaOS2 cells was analyzed as described for Fig. 8. (C) Inhibition of endogenous Cdk9 expression by shRNA-Cdk9 in SaOS2 cells. Five days after puromycin selection, TCLs were analyzed by Western blotting with anti-Cdk9 (top) and antitubulin antibodies (bottom). (D) Control and Cdk9-knockdown SaOS2 cells were infected with recombinant adenoviruses, and the cell growth rates were analyzed as described for panels A and B.
To further elucidate the role of Cdk9 in K-cyclin-induced growth arrest, we applied two approaches to inhibit the activity of endogenous Cdk9. First, we used recombinant adenovirus that expressed dn-Cdk9 to block Cdk9 activity. Our previous experiments above showed that K-cyclin augmented p53-induced growth suppression in SaOS2 cells (Fig. 9). Here, we found that overexpression of dn-Cdk9 reversed the growth inhibition induced by K-cyclin in the presence of p53 (Fig. 10B). This was apparently due to the inhibition of endogenous Cdk9 by this dominant negative mutant. Second, we used a lentiviral vector to introduce short hairpin RNA (shRNA) that specifically knocked down the expression of endogenous Cdk9 in SaOS2 cells. Forty-eight hours after lentiviral transduction, SaOS2 cells were subjected to puromycin selection for 5 days. We then performed cell proliferation experiments. As shown in Fig. 10C, the shRNA-Cdk9 successfully knocked down endogenous Cdk9 expression in SaOS2 cells. Consistent with the result from the dn-Cdk9 study (Fig. 10B), shRNA-Cdk9 reversed the K-cyclin-induced cell growth arrest (Fig. 10D). Together, these results demonstrated the pivotal role of Cdk9 that links K-cyclin to the p53 pathway.
DISCUSSION
Similar to many oncogenic proteins, KSHV K-cyclin has dual opposite effects on cell growth: cell cycle progression and growth suppression (73, 78). The proproliferation role of K-cyclin has been illustrated by its ability to activate cell cycle-related Cdk6 and promote cell cycle G1/S transition (8, 32, 39, 46, 77). The growth suppression induced by K-cyclin is apparently linked to a p53-dependent but p21-independent pathway (78). The coexistence of these two opposite effects on cell growth leads to different outcomes, largely depending on the influences from other cellular or environmental factors. For example, the activation of the p53 pathway or the high-level expression of Cdk6 can induce growth arrest and apoptosis in the K-cyclin-expressing cells (56, 78). In other cases, K-cyclin sensitizes cells to apoptosis induced by various proapoptotic stimuli (78). Based on current evidence, apparently K-cyclin expression alone mainly induces growth arrest and additional factors may be required for triggering apoptosis under normal conditions. The molecular mechanisms involved in K-cyclin-associated cell growth arrest are not well understood. In this study, we identified Cdk9, a non-cell cycle-related Cdk, as a new member of K-cyclin-interacting Cdks. The interaction between K-cyclin and Cdk9 suggests the involvement of K-cyclin in regulating Cdk9 kinase activity. Recent studies show that Cdk9 can phosphorylate tumor suppressor p53 (15, 64). Here we provide the first evidence that KSHV K-cyclin regulates the activity of Cdk9 toward p53 and reveal the underlying molecular interplay between K-cyclin and the p53 pathway.
Cdk9 plays an important role in basic transcription control and is critically involved in human immunodeficiency virus gene activation (20, 65). In addition to the major function of Cdk9 in transcriptional initiation and elongation, we found that the Cdk9 kinase activity toward p53 was upregulated by K-cyclin in vitro and in vivo. Infections with viruses, including cytomegalovirus (74), herpes simplex virus type 1 (22), and Epstein-Barr virus (1) upregulate Cdk9 kinase activity to phosphorylate RNA pol II. Whether the K-cyclin/Cdk9 interaction affects the function of Cdk9 on RNA pol II-mediated gene activation is worth further investigation.
Previous reports showed that Cdk9 phosphorylates p53 on Ser33, Ser315, and Ser392 (15, 64). Consistent with the location of these three Cdk9 phosphorylation sites on p53, we found that substrates containing TAD, TD, or BD of p53 can be phosphorylated by Cdk9 or K-cyclin/Cdk9 complexes. However, only the phosphorylation of the TAD was enhanced by K-cyclin/Cdk9 complexes. Ser33 is located in the TAD of p53, and its phosphorylation induced by K-cyclin was confirmed in vivo by Western blotting. The phosphorylation status of Ser315 and Ser392 was not significantly changed by K-cyclin. Although Ser33 appears to be the primary site for K-cyclin/Cdk9 phosphorylation, we cannot exclude the possibility that another site(s) may also be phosphorylated by these complexes under different cell growth conditions or in different cells. Phosphorylation plays a special role in regulating the activity of p53 (3, 7). Many oncogenic DNA virus-encoded proteins alter the function of p53 by modulating its phosphorylation status. For example, simian virus 40 (SV40) large T antigen (52) and Epstein-Barr virus BZLF1 (50) upregulate the phosphorylation of p53. Here we demonstrate that K-cyclin modulates the p53 phosphorylation on Ser33. Phosphorylation of this site is associated with cell responses to DNA-damaging agents and many other stress signals (9, 42, 67). Therefore, Ser33 phosphorylation might be linked to abnormal cell cycle progression and genome instability resulting from K-cyclin expression (76, 78). Interestingly, we also found that the level of p53 was slightly elevated in parallel with K-cyclin expression (Fig. 6). An increased p53 level in MEFs that express K-cyclin was also observed by other groups (76, 78). So far, it is not clear whether the elevation of p53 is caused by the activation of p53 gene expression or by the increased stability of p53. Since the phosphorylation of Ser33 was associated with an increased stability of p53 (9, 44), we speculate that K-cyclin may increase the stability of p53 by inducing the phosphorylation of its Ser33 residue. However, more studies are needed to address this issue.
Verschuren and colleagues showed that expression of K-cyclin in MEFs resulted in a dramatic suppression in cell growth (78). They demonstrated that such growth suppression was mainly caused by p53-dependent growth arrest, rather than increased apoptosis, under the normal cell growth condition (78). To dissect the connection between K-cyclin and p53, we compared the biological effects of K-cyclin in the p53-positive U2OS and p53-null SaOS2 cells. Our data show that K-cyclin inhibits cell growth in U2OS cells but not in SaOS2 cells. Furthermore, K-cyclin showed the growth suppression effect in SaOS2 cells only when p53 was transiently expressed. Together, these observations suggest that p53 participates in the K-cyclin signaling cascade, leading to cell growth suppression. To determine if the p53 target gene, encoding p21Cip1, was involved in K-cyclin-induced growth arrest, we examined the level of p21Cip1 protein in the presence and absence of K-cyclin. We found that K-cyclin did not affect p53-induced p21Cip1 expression in SaOS2 cells (data not shown). Our findings suggested that p21Cip1 appeared not to contribute to the p53-dependent growth suppression induced by K-cyclin. Identification of p53 targets is worth future investigation.
To demonstrate the role of Cdk9 in K-cyclin induced p53 phosphorylation and growth suppression, we utilized dn-Cdk9 in the study. The specificity of this mutant to inhibit the kinase activity of endogenous Cdk9 has been well demonstrated by A. P. Rice's and X. Grana's groups in their publications (see, e.g., references 31 and 33). Although dn-Cdk9 still interacts with K-cyclin, its kinase activity was diminished in accordance with the results of in vitro kinase assays. The in vivo inhibition of p53 phosphorylation on Ser33 by dn-Cdk9 was observed in U2OS cells, providing evidence that Cdk9 was responsible for K-cyclin-induced p53 phosphorylation. The Cdk9/K-cyclin/p53 interplay was further illustrated in the cell proliferation studies, in which dn-Cdk9 or shRNA-Cdk9 was able to rescue the p53-dependent growth suppression induced by K-cyclin in SaOS2 cells. Together, these results highlight the role of Cdk9 in connecting K-cyclin to the p53 pathway.
In the context of KSHV infection, the relationship of K-cyclin and the p53 pathway is more complicated. Unlike the frequent mutation and inactivation of p53 in many human cancers (35), more than one-half of KS and PEL samples express wild-type p53 and at least maintain certain p53 functions (41, 61, 69, 70). However, the expression of p53 varies in different stages of KS progression. The expression of p53 was hardly detectable in the early stage of KS, but the percentage of p53-positive cells increased in the more advanced stage (2, 18, 34, 45, 55, 62, 71). Apparently, the absence of p53 results in a disadvantage for cells in controlling the aberrant proliferation that is induced by KSHV viral proteins, such as K-cyclin. Evidence supporting this presumption was provided by Verschuren and colleagues. They demonstrated that K-cyclin induces tumor formation in all p53−/− transgenic mice with a median latency of 2.5 to 3 months. However, in the p53+/+ mice, only 17% of mice develop tumors by 9 months of age and all these tumors exhibit loss of p53 (76, 78). On the other hand, K-cyclin promotes genomic instability, which induces expression and activation of p53 (78). This may partially explain why increased p53-positive cells were seen in the advanced stage of KS. Despite the upregulation of p53, its ability to induce full-scale cell growth arrest is apparently impaired by multiple viral proteins that interfere with p53 activity (27, 54, 59). The partially functioning p53 is probably still able to slow down the K-cyclin-induced aberrant cell cycle progression. Studies have suggested that a certain degree of cell cycle control benefits gene expression and viral replication of KSHV and other herpesviruses (26, 36).
In summary, we identified Cdk9 as a novel KSHV K-cyclin-interacting Cdk and demonstrated that K-cyclin upregulated the Cdk9 kinase activity to phosphorylate p53. The phosphorylation site was mapped to Ser33 of p53. We also showed that K-cyclin induced p53-dependent growth arrest and defined the role of Cdk9 in cell growth regulation. Our study revealed Cdk9 as a novel molecular link between K-cyclin and the p53 pathway.
Acknowledgments
This study was supported by the grant DE016644-02 from the National Institutes of Health.
We thank Andrew P. Rice (Baylor College of Medicine, Houston, TX) for the plasmids expressing cyclin T1 and Cdk9 mutant dn-Cdk9. We also thank Craig S. Miller, Jeff Ebersole, and Robert J. Jacob for critically reading the manuscript.
Footnotes
Published ahead of print on 17 October 2007.
REFERENCES
- 1.Bark-Jones, S. J., H. M. Webb, and M. J. West. 2006. EBV EBNA 2 stimulates CDK9-dependent transcription and RNA polymerase II phosphorylation on serine 5. Oncogene 251775-1785. [DOI] [PubMed] [Google Scholar]
- 2.Bergman, R., M. Ramon, S. Kilim, C. Lichtig, and R. Friedman-Birnbaum. 1996. An immunohistochemical study of p53 protein expression in classical Kaposi's sarcoma. Am. J. Dermatopathol. 18367-370. [DOI] [PubMed] [Google Scholar]
- 3.Bode, A. M., and Z. Dong. 2004. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4793-805. [DOI] [PubMed] [Google Scholar]
- 4.Boshoff, C. 2003. Kaposi virus scores cancer coup. Nat. Med. 9261-262. [DOI] [PubMed] [Google Scholar]
- 5.Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 11274-1278. [DOI] [PubMed] [Google Scholar]
- 6.Boulanger, E., L. Gerard, J. Gabarre, J. M. Molina, C. Rapp, J. F. Abino, J. Cadranel, S. Chevret, and E. Oksenhendler. 2005. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J. Clin. Oncol. 234372-4380. [DOI] [PubMed] [Google Scholar]
- 7.Brooks, C. L., and W. Gu. 2003. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr. Opin. Cell Biol. 15164-171. [DOI] [PubMed] [Google Scholar]
- 8.Brooks, L. A., A. J. Wilson, and T. Crook. 1997. Kaposi's sarcoma-associated herpesvirus (KSHV)/human herpesvirus 8 (HHV8)—a new human tumour virus. J. Pathol. 182262-265. [DOI] [PubMed] [Google Scholar]
- 9.Bulavin, D. V., S. Saito, M. C. Hollander, K. Sakaguchi, C. W. Anderson, E. Appella, and A. J. Fornace, Jr. 1999. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 186845-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullrich, F., T. K. MacLachlan, N. Sang, T. Druck, M. L. Veronese, S. L. Allen, N. Chiorazzi, A. Koff, K. Heubner, C. M. Croce, et al. 1995. Chromosomal mapping of members of the cdc2 family of protein kinases, cdk3, cdk6, PISSLRE, and PITALRE, and a cdk inhibitor, p27Kip1, to regions involved in human cancer. Cancer Res. 551199-1205. [PubMed] [Google Scholar]
- 11.Cannell, E., and S. Mittnacht. 1999. Viral encoded cyclins. Semin. Cancer Biol. 9221-229. [DOI] [PubMed] [Google Scholar]
- 12.Cesarman, E., R. G. Nador, F. Bai, R. A. Bohenzky, J. J. Russo, P. S. Moore, Y. Chang, and D. M. Knowles. 1996. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J. Virol. 708218-8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 2661865-1869. [DOI] [PubMed] [Google Scholar]
- 14.Child, E. S., and D. J. Mann. 2001. Novel properties of the cyclin encoded by human herpesvirus 8 that facilitate exit from quiescence. Oncogene 203311-3322. [DOI] [PubMed] [Google Scholar]
- 15.Claudio, P. P., J. Cui, M. Ghafouri, C. Mariano, M. K. White, M. Safak, J. B. Sheffield, A. Giordano, K. Khalili, S. Amini, and B. E. Sawaya. 2006. Cdk9 phosphorylates p53 on serine 392 independently of CKII. J. Cell Physiol. 208602-612. [DOI] [PubMed] [Google Scholar]
- 16.Coqueret, O. 2002. Linking cyclins to transcriptional control. Gene 29935-55. [DOI] [PubMed] [Google Scholar]
- 17.Cuomo, M. E., A. Knebel, G. Platt, N. Morrice, P. Cohen, and S. Mittnacht. 2005. Regulation of microfilament organization by Kaposi sarcoma-associated herpes virus-cyclin.CDK6 phosphorylation of caldesmon. J. Biol. Chem. 28035844-35858. [DOI] [PubMed] [Google Scholar]
- 18.Dada, M. A., R. Chetty, S. C. Biddolph, J. W. Schneider, and K. C. Gatter. 1996. The immunoexpression of bcl-2 and p53 in Kaposi's sarcoma. Histopathology 29159-163. [DOI] [PubMed] [Google Scholar]
- 19.Davis, M. A., M. A. Sturzl, C. Blasig, A. Schreier, H. G. Guo, M. Reitz, S. R. Opalenik, and P. J. Browning. 1997. Expression of human herpesvirus 8-encoded cyclin D in Kaposi's sarcoma spindle cells. J. Natl. Cancer Inst. 891868-1874. [DOI] [PubMed] [Google Scholar]
- 20.De Falco, G., and A. Giordano. 2002. CDK9: from basal transcription to cancer and AIDS. Cancer Biol. Ther. 1342-347. [PubMed] [Google Scholar]
- 21.Dittmer, D. P. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 632010-2015. [PubMed] [Google Scholar]
- 22.Durand, L. O., S. J. Advani, A. P. Poon, and B. Roizman. 2005. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing cdk9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 796757-6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duro, D., A. Schulze, B. Vogt, J. Bartek, S. Mittnacht, and P. Jansen-Durr. 1999. Activation of cyclin A gene expression by the cyclin encoded by human herpesvirus-8. J. Gen. Virol. 80549-555. [DOI] [PubMed] [Google Scholar]
- 24.Dynlacht, B. D. 1997. Regulation of transcription by proteins that control the cell cycle. Nature 389149-152. [DOI] [PubMed] [Google Scholar]
- 25.Ellis, M., Y. P. Chew, L. Fallis, S. Freddersdorf, C. Boshoff, R. A. Weiss, X. Lu, and S. Mittnacht. 1999. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin-cdk6 complex. EMBO J. 18644-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flemington, E. K. 2001. Herpesvirus lytic replication and the cell cycle: arresting new developments. J. Virol. 754475-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friborg, J., Jr., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402889-894. [DOI] [PubMed] [Google Scholar]
- 28.Fu, T. J., J. Peng, G. Lee, D. H. Price, and O. Flores. 1999. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J. Biol. Chem. 27434527-34530. [DOI] [PubMed] [Google Scholar]
- 29.Ganem, D. 1997. KSHV and Kaposi's sarcoma: the end of the beginning? Cell 91157-160. [DOI] [PubMed] [Google Scholar]
- 30.Garriga, J., and X. Grana. 2004. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene 33715-23. [DOI] [PubMed] [Google Scholar]
- 31.Garriga, J., X. Mayol, and X. Grana. 1996. The CDC2-related kinase PITALRE is the catalytic subunit of active multimeric protein complexes. Biochem. J. 319293-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Godden-Kent, D., S. J. Talbot, C. Boshoff, Y. Chang, P. Moore, R. A. Weiss, and S. Mittnacht. 1997. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 714193-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gold, M. O., X. Yang, C. H. Herrmann, and A. P. Rice. 1998. PITALRE, the catalytic subunit of TAK, is required for human immunodeficiency virus Tat transactivation in vivo. J. Virol. 724448-4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodak, E., I. Hammel, M. Feinmesser, A. Zelinger, L. Maron, J. Sulkes, and M. David. 1999. Differential expression of p53 and Ki-67 proteins in classic and iatrogenic Kaposi's sarcoma. Am. J. Dermatopathol. 21138-145. [DOI] [PubMed] [Google Scholar]
- 35.Hollstein, M., D. Sidransky, B. Vogelstein, and C. C. Harris. 1991. p53 mutations in human cancers. Science 25349-53. [DOI] [PubMed] [Google Scholar]
- 36.Izumiya, Y., S. F. Lin, T. J. Ellison, A. M. Levy, G. L. Mayeur, C. Izumiya, and H. J. Kung. 2003. Cell cycle regulation by Kaposi's sarcoma-associated herpesvirus K-bZIP: direct interaction with cyclin-CDK2 and induction of G1 growth arrest. J. Virol. 779652-9661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jarviluoma, A., E. S. Child, G. Sarek, P. Sirimongkolkasem, G. Peters, P. M. Ojala, and D. J. Mann. 2006. Phosphorylation of the cyclin-dependent kinase inhibitor p21Cip1 on serine 130 is essential for viral cyclin-mediated bypass of a p21Cip1-imposed G1 arrest. Mol. Cell. Biol. 262430-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarviluoma, A., and P. M. Ojala. 2006. Cell signaling pathways engaged by KSHV. Biochim. Biophys. Acta 1766140-158. [DOI] [PubMed] [Google Scholar]
- 39.Jung, J. U., M. Stager, and R. C. Desrosiers. 1994. Virus-encoded cyclin. Mol. Cell. Biol. 147235-7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaldis, P., P. M. Ojala, L. Tong, T. P. Makela, and M. J. Solomon. 2001. CAK-independent activation of CDK6 by a viral cyclin. Mol. Biol. Cell 123987-3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katano, H., Y. Sato, and T. Sata. 2001. Expression of p53 and human herpesvirus-8 (HHV-8)-encoded latency-associated nuclear antigen with inhibition of apoptosis in HHV-8-associated malignancies. Cancer 923076-3084. [DOI] [PubMed] [Google Scholar]
- 42.Kishi, H., K. Nakagawa, M. Matsumoto, M. Suga, M. Ando, Y. Taya, and M. Yamaizumi. 2001. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem. 27639115-39122. [DOI] [PubMed] [Google Scholar]
- 43.Laman, H., D. Coverley, T. Krude, R. Laskey, and N. Jones. 2001. Viral cyclin-cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol. Cell. Biol. 21624-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee, J. H., and K. T. Kim. 2007. Regulation of cyclin-dependent kinase 5 and p53 by ERK1/2 pathway in the DNA damage-induced neuronal death. J. Cell Physiol. 210784-797. [DOI] [PubMed] [Google Scholar]
- 45.Li, J. J., Y. Q. Huang, C. J. Cockerell, W. G. Zhang, A. Nicolaides, and A. E. Friedman-Kien. 1997. Expression and mutation of the tumor suppressor gene p53 in AIDS-associated Kaposi's sarcoma. Am. J. Dermatopathol. 19373-378. [DOI] [PubMed] [Google Scholar]
- 46.Li, M., H. Lee, D. W. Yoon, J. C. Albrecht, B. Fleckenstein, F. Neipel, and J. U. Jung. 1997. Kaposi's sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol. 711984-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lundquist, A., B. Barre, F. Bienvenu, J. Hermann, S. Avril, and O. Coqueret. 2003. Kaposi sarcoma-associated viral cyclin K overrides cell growth inhibition mediated by oncostatin M through STAT3 inhibition. Blood 1014070-4077. [DOI] [PubMed] [Google Scholar]
- 48.Mann, D. J., E. S. Child, C. Swanton, H. Laman, and N. Jones. 1999. Modulation of p27Kip1 levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus. EMBO J. 18654-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marshall, N. F., J. Peng, Z. Xie, and D. H. Price. 1996. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 27127176-27183. [DOI] [PubMed] [Google Scholar]
- 50.Mauser, A., S. Saito, E. Appella, C. W. Anderson, W. T. Seaman, and S. Kenney. 2002. The Epstein-Barr virus immediate-early protein BZLF1 regulates p53 function through multiple mechanisms. J. Virol. 7612503-12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore, P. S., and Y. Chang. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57609-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muller, E., and K. H. Scheidtmann. 1995. Purification and characterization of a protein kinase which is activated by SV40 large T-antigen and phosphorylates the tumor suppressor protein p53. Oncogene 101175-1185. [PubMed] [Google Scholar]
- 53.Nador, R. G., E. Cesarman, A. Chadburn, D. B. Dawson, M. Q. Ansari, J. Sald, and D. M. Knowles. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88645-656. [PubMed] [Google Scholar]
- 54.Nakamura, H., M. Li, J. Zarycki, and J. U. Jung. 2001. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 757572-7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noel, J. C., F. De Thier, T. Simonart, J. Andre, P. Hermans, J. P. Van Vooren, and M. Heenen. 1997. p53 protein overexpression is a common but late event in the pathogenesis of iatrogenic and AIDS-related Kaposi's sarcoma. Arch. Dermatol. Res. 289660-661. [DOI] [PubMed] [Google Scholar]
- 56.Ojala, P. M., M. Tiainen, P. Salven, T. Veikkola, E. Castanos-Velez, R. Sarid, P. Biberfeld, and T. P. Makela. 1999. Kaposi's sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. 594984-4989. [PubMed] [Google Scholar]
- 57.Ojala, P. M., K. Yamamoto, E. Castanos-Velez, P. Biberfeld, S. J. Korsmeyer, and T. P. Makela. 2000. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat. Cell Biol. 2819-825. [DOI] [PubMed] [Google Scholar]
- 58.Oksenhendler, E., E. Boulanger, L. Galicier, M. Q. Du, N. Dupin, T. C. Diss, R. Hamoudi, M. T. Daniel, F. Agbalika, C. Boshoff, J. P. Clauvel, P. G. Isaacson, and V. Meignin. 2002. High incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood 992331-2336. [DOI] [PubMed] [Google Scholar]
- 59.Park, J., T. Seo, S. Hwang, D. Lee, Y. Gwack, and J. Choe. 2000. The K-bZIP protein from Kaposi's sarcoma-associated herpesvirus interacts with p53 and represses its transcriptional activity. J. Virol. 7411977-11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peng, J., Y. Zhu, J. T. Milton, and D. H. Price. 1998. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 12755-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petre, C. E., S. H. Sin, and D. P. Dittmer. 2007. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J. Virol. 811912-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pillay, P., R. Chetty, and R. Reddy. 1999. Bcl-2 and p53 immunoprofile in Kaposi's sarcoma. Pathol. Oncol. Res. 517-20. [DOI] [PubMed] [Google Scholar]
- 63.Platt, G. M., E. Cannell, M. E. Cuomo, S. Singh, and S. Mittnacht. 2000. Detection of the human herpesvirus 8-encoded cyclin protein in primary effusion lymphoma-derived cell lines. Virology 272257-266. [DOI] [PubMed] [Google Scholar]
- 64.Radhakrishnan, S. K., and A. L. Gartel. 2006. CDK9 phosphorylates p53 on serine residues 33, 315 and 392. Cell Cycle 5519-521. [DOI] [PubMed] [Google Scholar]
- 65.Romano, G., M. Kasten, G. De Falco, P. Micheli, K. Khalili, and A. Giordano. 1999. Regulatory functions of Cdk9 and of cyclin T1 in HIV tat transactivation pathway gene expression. J. Cell. Biochem. 75357-368. [PubMed] [Google Scholar]
- 66.Rubinson, D. A., C. P. Dillon, A. V. Kwiatkowski, C. Sievers, L. Yang, J. Kopinja, D. L. Rooney, M. Zhang, M. M. Ihrig, M. T. McManus, F. B. Gertler, M. L. Scott, and L. Van Parijs. 2003. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 33401-406. [DOI] [PubMed] [Google Scholar]
- 67.Sanchez-Prieto, R., J. M. Rojas, Y. Taya, and J. S. Gutkind. 2000. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 602464-2472. [PubMed] [Google Scholar]
- 68.Sarek, G., A. Jarviluoma, and P. M. Ojala. 2006. KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 107725-732. [DOI] [PubMed] [Google Scholar]
- 69.Sarek, G., S. Kurki, J. Enback, G. Iotzova, J. Haas, P. Laakkonen, M. Laiho, and P. M. Ojala. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Investig. 1171019-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scinicariello, F., M. J. Dolan, I. Nedelcu, S. K. Tyring, and J. K. Hilliard. 1994. Occurrence of human papillomavirus and p53 gene mutations in Kaposi's sarcoma. Virology 203153-157. [DOI] [PubMed] [Google Scholar]
- 71.Simonart, T., and J. C. Noel. 2000. Increased p53 staining in non-sun-exposed epidermis overlying Kaposi sarcoma. Am. J. Dermatopathol. 22373-374. [DOI] [PubMed] [Google Scholar]
- 72.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 861276-1280. [PubMed] [Google Scholar]
- 73.Swanton, C., D. J. Mann, B. Fleckenstein, F. Neipel, G. Peters, and N. Jones. 1997. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390184-187. [DOI] [PubMed] [Google Scholar]
- 74.Tamrakar, S., A. J. Kapasi, and D. H. Spector. 2005. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of cdk9 and cdk7. J. Virol. 7915477-15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Dross, R., S. Yao, S. Asad, G. Westlake, D. J. Mays, L. Barquero, S. Duell, J. A. Pietenpol, and P. J. Browning. 2005. Constitutively active K-cyclin/cdk6 kinase in Kaposi sarcoma-associated herpesvirus-infected cells. J. Natl. Cancer Inst. 97656-666. [DOI] [PubMed] [Google Scholar]
- 76.Verschuren, E. W., J. G. Hodgson, J. W. Gray, S. Kogan, N. Jones, and G. I. Evan. 2004. The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res. 64581-589. [DOI] [PubMed] [Google Scholar]
- 77.Verschuren, E. W., N. Jones, and G. I. Evan. 2004. The cell cycle and how it is steered by Kaposi's sarcoma-associated herpesvirus cyclin. J. Gen. Virol. 851347-1361. [DOI] [PubMed] [Google Scholar]
- 78.Verschuren, E. W., J. Klefstrom, G. I. Evan, and N. Jones. 2002. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2229-241. [DOI] [PubMed] [Google Scholar]
- 79.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 936641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]