

Abstract
Previous studies have indicated that the replication of the RNA genome of hepatitis delta virus (HDV) involves redirection of RNA polymerase II (Pol II), a host enzyme that normally uses DNA as a template. However, there has been some controversy about whether in one part of this HDV RNA transcription, a polymerase other than Pol II is involved. The present study applied a recently described cell system (293-HDV) of tetracycline-inducible HDV RNA replication to provide new data regarding the involvement of host polymerases in HDV transcription. The data generated with a nuclear run-on assay demonstrated that synthesis not only of genomic RNA but also of its complement, the antigenome, could be inhibited by low concentrations of amanitin specific for Pol II transcription. Subsequent studies used immunoprecipitation and rate-zonal sedimentation of nuclear extracts together with double immunostaining of 293-HDV cells, in order to examine the associations between Pol II and HDV RNAs, as well as the small delta antigen, an HDV-encoded protein known to be essential for replication. Findings include evidence that HDV replication is somehow able to direct the available delta antigen to sites in the nucleoplasm, almost exclusively colocalized with Pol II in what others have described as transcription factories.
During the replication of human hepatitis delta virus (HDV), three RNAs are generated by RNA-directed RNA transcription and posttranscriptional processing. The genome and its exact complement, the antigenome, are 1,679-nucleotide circular RNAs that fold into a rod-like structure with 74% of their nucleotides base paired (20, 38). It is considered that these RNAs are derived from longer than unit length primary transcripts that are processed to unit length by the HDV ribozymes and then ligated into circles (35). The third RNA is of the same polarity as the antigenome but only about 800 nucleotides in length. It has a defined 5′ end that is capped and a defined 3′ end that is polyadenylated (14). This mRNA contains the open reading frame for the only protein of HDV, the small delta antigen (δAg). This protein is essential for HDV replication but at 195 amino acids in length is too small to have polymerase activity (7).
Several lines of evidence implicate the host RNA polymerase II (Pol II) as being required for the transcription of HDV RNAs (reviewed in references 21, 35, and 36). However, data from Lai and coworkers has been interpreted as evidence that the synthesis of antigenomic RNA is resistant to high doses of amanitin and therefore may be more consistent with being directed by RNA Pol I, the enzyme involved in the transcription of rRNAs (23, 25, 27). In order to resolve this controversy and also to obtain more information about HDV RNA-directed transcription, we have made use of the following experimental system.
As previously described, we first established 293-δAg cells, a line of 293 cells in which the essential δAg is provided by an integrated cDNA, with expression inducible by tetracycline (TET). These cells were in turn transfected with an HDV RNA to produce the line referred to as 293-HDV (4). These cells are such that in the absence of TET a low level of HDV replication is maintained, at this time, for more than 3 years (unpublished observations). However, upon the addition of TET there occurs within 16 h a significant synthesis of δAg, along with an accumulation of large amounts of newly transcribed HDV RNAs. Another advantage of this system is the avoidance of a transfection procedure and the introduction to the cell of large amounts of nucleic acid, whether cDNA constructs or in vitro-transcribed RNA. In contrast, all of the HDV transcription in the inducible system is from replication-competent HDV RNA. However, another advantage is that the only form of δAg present is the small form essential for the accumulation of HDV RNAs. The replicating genome is modified and does not produce a translatable mRNA, and so there arises no large δAg nor any mutated forms of the δAg.
Recently, we used this new experimental system to examine the effect of various antivirals added during the period of TET induction on the ability to accumulate processed HDV RNAs (5). We found that low concentrations of amanitin that block the accumulation of host Pol II transcripts but not those of Pol I or Pol III were also able to block the accumulation of HDV genomic RNA, antigenomic RNA, and the mRNA. This, however, is only indirect evidence that Pol II might be required for all aspects of HDV RNA-directed transcription.
The aim of the present study was to use the 293-HDV cells to provide more direct evidence relating to HDV RNA transcription. The strategy involved using them at 16 h after TET induction. At this time HDV replication is still increasing, with there being around 1 million molecules of δAg per cell along with 20,000 molecules of HDV genomic RNA and severalfold less of antigenomic RNA (4). Studies by others have determined that a mammalian cell should contain about 320,000 molecules of Pol II, ca. 25% of which at a given time are estimated to be active in DNA-directed RNA transcription (11). As described here, we performed transcriptional run-on assays using nuclei isolated from the cells after induction of HDV genome replication and demonstrated that the synthesis of both genomic and antigenomic RNAs was sensitive to low-dose amanitin inhibition. Furthermore, in order to examine the intracellular associations between Pol II, HDV RNAs, and δAg, we used immunoprecipitation (IP) and rate-zonal sedimentation analyses of nuclear extracts and immunostaining of cells. Among other things, these studies provide evidence that HDV replication is somehow able to direct the available delta antigen to sites in the nucleoplasm, extensively colocalized with Pol II in what others have described as transcription factories (11).
MATERIALS AND METHODS
Cells.
The cell culture system that supports inducible HDV genome replication has been previously described (4). Briefly, we first established a cell clone, 293-δAg that expresses a single copy of δAg cDNA under TET control. Next, HDV genome replication was initiated in these cells by transfection of HDV RNA that has a frameshift mutation and does not express δAg by itself. A clone derived from a single cell was identified by its high level of HDV replication and is designated as 293-HDV cells. Both 293-HDV and 293-δAg cells were maintained in Dulbecco modified Eagle medium with 10% fetal bovine serum and the selective antibiotics blasticidin and hygromycin (Invitrogen).
RNA extraction Northern analysis and quantitative real-time PCR assays.
Total cell RNA was routinely extracted with Tri Reagent (Molecular Research Center). Detection and quantitation of HDV genomic and antigenomic RNAs was either by Northern analysis with quantitation using a Bioimager (Fuji) (28) or by real-time reverse transcription-PCR (15).
Nuclear run-on assay.
Extraction of nuclei and the in vitro transcription procedure were carried out as described previously (10) with several modifications. At 16 h after TET induction, 2 × 107 cells were pelleted, resuspended in cell lysis buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40), and incubated on ice for 5 min. After a wash with the same buffer, the nuclei were resuspended in 100 μl of nuclear storage buffer (50 mM Tris-HCl [pH 8.3], 40% glycerol, 5 mM MgCl2, 0.1 mM EDTA). For in vitro nuclear run-on, the nuclei were mixed with an equal volume of 2× run-on transcription buffer (300 mM KCl, 10 mM Tris-HCl [pH 8.0], 5 mM MgCl2, 0.5 mM concentrations each of ATP, GTP, and CTP) with or without 1 μg of amanitin/ml as indicated and incubated on ice for 10 min. Then, 100 μCi of [α-32P]UTP (800 Ci/mmol; Perkin-Elmer) was added to the nuclei, followed by incubation at 28°C for 10 min. Total RNA was immediately extracted by using Tri-Reagent. In some cases, the resulting run-on RNA was treated with alkali to reduce the size before hybridization.
Slot blot hybridization was performed as previously described (28), except that different nucleic acids were applied and fixed to the membrane. To assay Pol I transcription, we used 1 μg of 50-mer antisense oligonucleotide of 18S rRNA (TTTCAAAGTAAACGCTTCGGGCCCCGCGGGACACTCAGCTAAGAGCATCG). For Pol II transcription, 2 μg of DNA fragments from constructs expressing woodchuck actin and mouse GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used (both are reactive with human genes). We used, as a control for Pol III transcription, 5 μg of a 50-mer antisense oligonucleotide of snU6 RNA (TGGAACGCTTCACGAATTTGCGTGTCATCCTTGCGCAGGGGCCATGCTAA). For detection of HDV genomic and antigenomic RNAs, we used in vitro-transcribed HDV RNAs corresponding to nucleotides 4 to 660 in the sequence of Kuo et al. (20). The slot assays were performed in parallel using the same hybridization conditions.
Immunoselection of HDV RNAs.
At 16 h after TET induction of 3.4 × 107 293-HDV cells, the medium was removed, the cells were washed with ice-cold phosphate-buffered saline (PBS), and then cross-linking was achieved with 1% formaldehyde for 10 min at room temperature with gentle rocking (3). The cells were then lysed and spun down to collect nuclei. A nuclear extract was obtained by sonication followed by low-speed centrifugation to remove debris. For immunoselection, protein A-Sepharose beads (Sigma) were prepared as described using NTE-NDS buffer (34) and incubated with antibodies at 4°C overnight with gentle rocking. The antibodies to human Pol II were 8WG16 (Covance) and 4H8 (Abcam). δAb was a rabbit polyclonal antibody against HDV δAg. After a washing step, beads with bound antibodies were incubated with nuclear extract at 4°C for 1 h with gentle rocking and then washed with NTE-NDS buffer. Elution from the beads was done with 10 mM Tris (pH 8.1)-10 mM EDTA-1% sodium dodecyl sulfate at 65°C for 5 min. Where applicable, formaldehyde cross-linking was reversed by heating at 65°C for 1 h. Eluted RNA was extracted with Tri-Reagent. Extracted unbound and bound RNAs were subjected to Northern analysis.
Nuclear protein complex IP.
IP was performed by using a kit (Active Motif). 293-HDV cells were first induced for 16 h. Nuclear extraction was performed using low-salt buffers and gentle DNA digestion conditions (4°C for 90 min) to protect protein complexes with weak association. For the experiment whose results are given in Table 2, RNase A was also added during this digestion. The nuclei from approximately 5 × 106 cells were used per reaction. For HDV δAg we used a 1:200 dilution of a rabbit polyclonal antibody, with preimmune serum as a negative control. For Pol II we used 5 μg of the mouse monoclonal antibody 4H8 and omitted antibody for the negative control. For the binding reaction we used 50 μl of protein A-agarose (Invitrogen). Binding and washing were both performed in low-stringency buffers. For the experiment in Table 2, just prior to elution, an aliquot was extracted for RNA and then assayed by quantitative PCR (qPCR). Elution was performed with gel sample buffer. Immunoblots were performed and probed with a series of different antibodies. Note that to detect δAg the sample was not reduced prior to electrophoresis; this allowed the separation of δAg away from what otherwise would have been an overwhelming signal due to the light chain of the rabbit antibody used for IP.
TABLE 2.
Sensitivity to RNase pretreatment of the association between Pol II and δAg in 293-HDV cells
| Assay | % IP with antibody to Pol II or δAG or no antibodya:
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| None | δAg
|
None
|
Pol II (4H8)
|
|||||||
| 0 | 0 | 0.1 | 1 | 10 | 0 | 0 | 0.1 | 1 | 10 | |
| Assay for HDV genomic RNA | 3.0 | 20.9 | 27.4 | 0.02 | <0.02 | 0.47 | 11.6 | 15.0 | 0.02 | <0.02 |
| Assay for δAg | 4.2 | 29.1 | 27.6 | 28.2 | 21.3 | <0.16 | 4.1 | 2.8 | 0.21 | 0.12 |
| Assay for Pol II (4H8) | 0.2 | 1.7 | 1.9 | 3.5 | 2.3 | 2.3 | 37.5 | 34.5 | 27.6 | 23.2 |
Aliquots of the nuclear fraction of TET-induced 293T-HDV cells were subjected to DNase treatment, with or without the presence of RNase at the concentrations indicated (in μg/ml) in the subheadings. Samples were subjected to IP using either antibody to δAg or Pol II (4H8). The negative controls (None) were preimmune rabbit serum instead of rabbit anti-δAg and empty beads for anti-Pol II. Samples were assayed by immunoblotting for δAg and Pol II and by qPCR for genomic HDV RNA. The values for the percentage of the total collected by IP were deduced and are shown in the table.
Immunoblot analysis.
Samples were heated at 95°C for 5 min in Laemmli buffer without or with dithiothreitol, prior to analysis on precast gels of either 3 to 8 or 12% polyacrylamide (NuPage; Invitrogen). After electrophoresis and electrotransfer to nitrocellulose membranes, proteins were detected with a variety of rabbit and mouse primary antibodies. These antibodies were as indicated for Fig. 4. The specific antibodies used to detect Pol II are summarized in Table 1. This was followed by goat anti-rabbit and anti-mouse secondary antibodies that were infrared dye labeled (LI-COR). Detection and quantitation was done with an Odyssey apparatus (LI-COR).
FIG. 4.
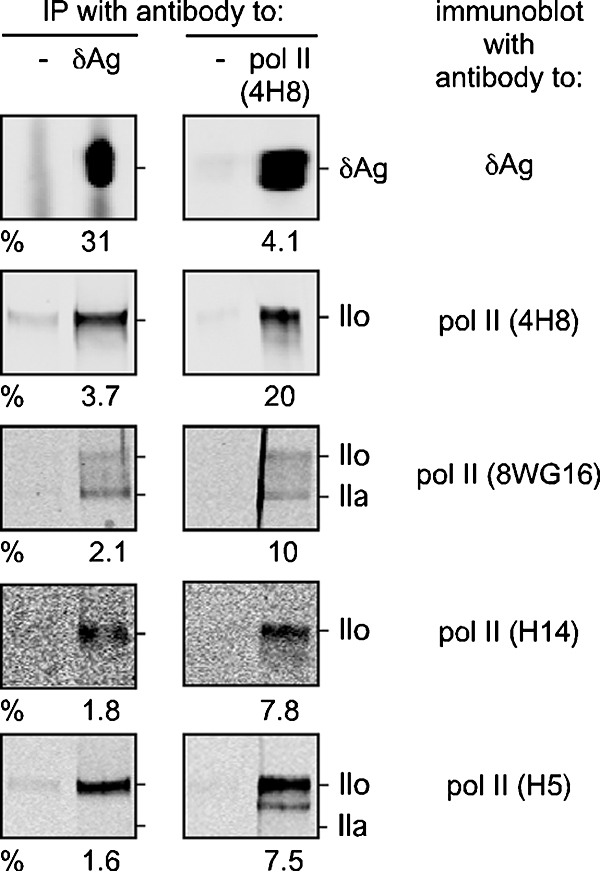
Association between δAg and Pol II transcription complex. Nuclear IPs were performed with rabbit antibody to δAg or, as a negative control, normal rabbit serum. Similarly, we used 4H8, a mouse monoclonal specific for Pol II or, as a negative control, no added antibody (4H8). Immune complexes were selected by using protein A-agarose and then released for gel electrophoresis and immunoblotting. The latter, as indicated at the left side, were developed using antibody to δAg and four antibodies that recognize different forms of Pol II. For each sample, four dilutions of the total unbound material were examined in parallel in order to help determine the indicated percentages of the detected protein that was selected by IP.
TABLE 1.
Reported specificities of Pol II antibodies used in this studya
| Antibody | Commercial source | Reported specificity within CTD | Source or reference(s) |
|---|---|---|---|
| 4H8 | Abcam | All forms of Pol II | 3 |
| Phosphorylated serine-5 | 33 | ||
| 8WG16 | Covance | All unphosphorylated forms | 3 |
| Unphosphorylated serine-2 | 37 | ||
| H14 | Covance | Phosphorylated serine-5 | 9, 26 |
| Weak response to unphosphorylated forms | Covance | ||
| H5 | Abcam | Phosphorylated serine-2 | 9, 26 |
| Phosphorylated serine 2 and/or serine-5 | 29 | ||
| Weak response to unphosphorylated forms | Covance |
This summary of specificities reported for each antibody represent an updated form of data tabulated by Palancade and Bensaude (31). Some of the discrepancies may have arisen because the antibodies were used in different experimental situations.
Rate-zonal sedimentation.
Nuclear extract samples were diluted with STE buffer (100 mM NaCl, 10 mM Tris-HCl [pH 8.0], 1 mM EDTA) to 0.2 ml and then applied to gradients of 10 to 30% sucrose in STE. Centrifugation was in a Beckman SW61 rotor at 60 krpm for 40 min at 4°C. Fractions of 0.3 ml were collected from above and assayed by immunoblotting for δAg and Pol II or by qPCR to detect HDV genomic and antigenomic RNA. Procedures, along with the preparation of subviral particles (SVPs), HDV, HBV, and HDV ribonucleoprotein, were otherwise done as previously described (15, 34).
Indirect immunostaining.
293-δAg and 293-HDV cells grown on coverslips were induced with TET for 16 h. Cells were fixed with paraformaldehyde (4% in PBS) for 15 min at room temperature, followed by permeabilization with Triton X-100 (0.1% in PBS) on ice for 15 min. After a blocking step (5% bovine serum albumin in PBS) at room temperature for 1 h, primary staining was carried out at 37°C for 1 h with combinations of rabbit anti-δAg primary antibody (17), mouse anti-Pol II (4H8), or mouse anti-SC35 antibody (Abcam). This was followed by incubation at 37°C for 30 min with Alexa 488 chicken anti-mouse and Alexa 594 goat anti-rabbit antibodies. Cells then were stained with DAPI (Sigma) to visualize the nucleus and mounted with antifade mounting solution (Molecular Probes). Images were examined by using a Zeiss 510 LSM confocal microscope with ×63 PlanApo objective lens. Colocalization values were computed by using Zeiss LSM510 META v3.2.
RESULTS
Nuclear run-on assay.
As previously reported, the 293-HDV cells, even in the absence of TET induction, expressed a level of δAg that supported a basic level of HDV genome replication (4). However, after the addition of TET (1 μg/ml) δAg was induced, and there was a major increase in HDV genome replication. Within 16 h of induction, the accumulation of HDV genome reached half-maximal amount, and all HDV RNAs were directed from natural circular RNA templates. In the present study we used 293-HDV cells 16 h after TET induction, as a model system to study HDV RNA-directed RNA transcription.
Shown in Fig. 1 is an outline of the nuclear run-on assay. The 293-HDV cells were induced with TET for 16 h before the extraction of nuclei. The in vitro run-on assay was initially performed in the presence of 32P-labeled precursors, with or without 1 μg of amanitin/ml. We added amanitin to the in vitro run-on reaction rather than to the cells prior to nuclear isolation. This was done to exclude possible indirect effects of Pol II inhibition on HDV transcription.
FIG. 1.

Outline of nuclear run-on assay. HDV replication was initiated in 293-HDV cells by induction with TET for 16 h. Nuclei were then extracted followed by in vitro nuclear run-on transcription in the presence of [32P]UTP. To inhibit Pol II transcription, 1 μg of amanitin/ml was added to the in vitro reaction. Radioactively labeled run-on RNAs were extracted and hybridized to a membrane on which were immobilized nuclei acids complementary to 18SrRNA, actin mRNA, GAPDH mRNA, U6 RNA, and HDV genomic and antigenomic RNA. Bound 32P was quantitated by using a Bioimager.
After the in vitro transcription, total RNAs were extracted. This total RNA containing 32P-labeled nascent HDV and host RNAs was then hybridized to a membrane on which were immobilized nucleic acids complementary to 18S rRNA, actin mRNA, GAPDH mRNA, and U6 RNA, as standards for transcription directed by polymerases I to III. Also used were RNAs to detect genomic and antigenomic transcripts. In the latter case, the immobilized RNA was such that it should not detect antigenomic mRNA produced during HDV replication or mRNA transcribed from the DNA copy integrated into the genome of the 293-HDV cells. This expectation was confirmed in parallel run-on transcription assays using nuclei from 293-δAg cells (data not shown).
After hybridization the 32P signal was quantitated by using a Bioimager to determine the effect of amanitin on the HDV genomic and antigenomic RNA transcription compared to that of the host RNA transcriptions. Typical results are shown at the left of Fig. 2. At the right side of Fig. 2 are listed the average values and standard deviations for such inhibition, as obtained from multiple experiments.
FIG. 2.

Summary of nuclear run-on assays. A representative slot blot hybridization is shown at the left, without or with 1 μg of amanitin/ml added during in vitro transcription, as indicated. The transcripts on the membrane are listed in the first column of the table. The second panel shows the percent resistance to amanitin. This refers to the residual signals in the presence of amanitin relative to untreated controls. These are mean values, with standard deviations, and the number of independent experiments is indicated in parentheses. The third column indicates the host RNA polymerase used in transcription.
Note first that we detected a significant signal for both genomic and antigenomic RNAs. In previous studies, prior to the development of this 293-HDV cell system, the genomic signal could be barely detected, and no antigenomic signal was detected (28). Note also that in both cases there was inhibition to 14% by the low dose of amanitin. Under the same conditions, actin and GAPDH mRNAs, which are accepted as Pol II transcripts, were inhibited to about the same extent (16 and 7%, respectively). In contrast, 18S rRNA and U6 RNA, which are accepted as Pol I and Pol III transcripts, respectively, were not inhibited. If anything, 18S rRNA and U6 transcription were somewhat enhanced, and we suggest this might have been because amanitin blocked host Pol II transcription, leaving more precursors available for Pol I and Pol III transcription.
As a control for these studies, similar nuclear run-on assays were also performed with 293-δAg cells that stably express δAg but without HDV replication. In these cells, no specific HDV genomic and antigenomic signals were detected. This confirmed the specificity of our ability to detect genomic and antigenomic HDV RNAs. At the same time, the results obtained for the host Pol I, II, and III transcripts were essentially as shown in Fig. 2 (data not shown).
Taken together, we have demonstrated that with a low concentration of amanitin that only inhibited Pol II transcription and did not affect host Pol I or Pol III transcription there was a significant inhibition for HDV genomic and antigenomic RNAs. Thus, we believe that Pol II is required for the transcription of both genomic and antigenomic HDV RNAs.
Association between Pol II and HDV RNAs detected by IP.
Given the above results for the involvement of Pol II, we undertook to determine whether IP procedures could detect associations between Pol II and HDV RNAs. We used a modification of chromatin IP, an approach widely used to study interactions between Pol II transcription complexes and DNA templates (3). Our aim was to determine whether HDV RNA species could be detected that were bound to Pol II. Moreover, using a Northern assay for unit-length genomic and antigenomic RNA, a positive result would be an indication that such a posttranscriptionally processed RNA was possibly acting as an RNA template.
For this IP the mouse monoclonal antibodies to Pol II were 4H8 and 8WG16. As indicated in Table 1, these bind to the Pol II carboxyl-terminal domain (CTD) and are considered to preferentially recognize the elongation and initiation forms, respectively, of Pol II. As a positive control, we also used antibody against δAg. As a negative control we used protein A but no antibody.
As shown in Fig. 3, antibody to δAg was able to select significant amounts of both antigenomic and genomic HDV RNAs. This provides in vivo support for previous in vitro studies that have shown that δAg is able to bind both strands of HDV RNAs (8). It also confirms and extends a prior IP study by Niranjanakumari et al. (30).
FIG. 3.

Association between Pol II and HDV RNAs detected by using IP assay. Cultures of 293-HDV cells after 16 h of TET induction, either with or without formaldehyde cross-linking, as indicated, were used to prepare nuclear extracts for immunoselection. Monoclonal antibodies 4H8 and 8WG16 were used to select for Pol II. A rabbit polyclonal was used to select for δAg. RNAs from fractions that were bound (B) or unbound (u) by protein A beads were (treated to reverse cross-linking, if applicable before being) extracted, and subjected to Northern assay to detect antigenomic and genomic RNAs. The amount of unbound RNA analyzed was 1.25% relative to the bound RNA. After quantitation, we obtained the percentages bound, as indicated.
For the two Pol II antibodies, with prior cross-linking using 1% formaldehyde, both were able to select relatively small but real amounts of antigenomic and genomic unit-length HDV RNAs. Totals of 2.5% of the antigenomic and 1% of the genomic RNAs were selected by the 4H8 antibody. We also found that 1.6% of the antigenomic and 0.4% of the genomic RNAs were selected by the 8WG16.
These data were consistent with the interpretation that HDV genomic and antigenomic RNAs were in complexes not only with δAg but also with Pol II. However, since the detected HDV RNAs were already processed to unit-length, they could not be nascent transcripts. That is, it remained possible but unproven that some were acting as RNA templates for Pol II.
Association between δAg and Pol II transcription detected by IP.
The above studies indicated associations between δAg and HDV RNAs and, to a lower extent, between HDV RNAs and Pol II. Therefore, we next sought to determine whether we could detect associations between δAg and Pol II. For this we used TET-induced 293-HDV cells and a standard IP procedure, under low-stringency conditions, without any prior cross-linking. We tested IP with both antibodies to δAg and Pol II relative to appropriate negative controls. Samples of both total and eluted materials were assayed by immunoblotting. This allowed us to deduce the percentage of each protein that was brought down by the IP. The results for such an experiment are shown in Fig. 4. From the top four panels it can be seen that IP with anti-δAg brought down 3.7% of the total Pol II and, conversely, that anti-Pol II brought down 4.1% of the δAg. These would be underestimates of the total associated material, since the homologous IP procedure was only 20 to 31% efficient.
As shown in the next six panels, we also performed immunoblot assays with three other monoclonal antibodies: 8WG16, H14, and H5, the specificities of which are described in Table 1. Our aim was to determine which forms of Pol II were associated with HDV RNA-directed transcription. However, within the errors, we found no evidence that any one form of Pol II was more associated with the IP using anti-δAg than with the IP using anti-Pol II.
The results depicted in Fig. 4 support the interpretation that a small fraction of the Pol II might be associated with δAg and a similar small fraction of the Pol II is associated with δAg. Such results do not address to what extent such complexes are active in transcription. Furthermore, it is not clear whether the observed associations are direct or indirect. Since δAg is known to be an RNA-binding protein (8), an obvious possibility for indirect association would be via the HDV RNA template. To test for this possibility, we next examined whether a treatment of the affinity-selected complexes with increasing amounts of RNase would separate the δAg from the Pol II. The results of such an experiment are summarized in Table 2.
As a measure of the RNase digestion, we used qPCR to detect the HDV RNA after the RNase treatment and immediately before the elution of the IP material. As shown in the table, with increasing concentrations of RNase during the pretreatment, we reduced the RNA level to <0.02%. We presume that HDV and host RNAs were equally sensitive to the RNase treatment.
Consider next the effect of RNase on the ability to IP via anti-δAg. It can be seen that this had no detectable effect on either the amount of selected Pol II or that of δAg. This led to the interpretation that the ability of Pol II to bind to δAg was not dependent upon HDV RNA. We also tested the converse. That is, we examined the effect of RNase on the ability to immunoprecipitate via Pol II. As shown, the RNase had no effect on the ability to immunoprecipitate Pol II, but there was a progressive decrease (from 4.1 to 0.12%) in the ability to immunoprecipitate δAg. This supported the interpretation that the majority of δAg associated with Pol II was actually associated via RNA, presumably HDV RNA.
In summary these IP studies did provide data consistent with interactions between Pol II and δAg. The association of δAg with Pol II was at a low level (<4.1%) and RNA mediated. The association of Pol II and δAg was also of a low level (<4%) but not RNA mediated. As discussed later, there are numerous caveats that apply to the interpretation of these data. However, we next chose to determine whether HDV RNA species in any way facilitated these observed associations.
Role of HDV genome replication in detected association between δAg and Pol II.
As a strategy to detect whether there were interactions between δAg and Pol II independent of HDV RNA, we made a comparison between 293-HDV and 293-δAg cells. The latter cells are identical in terms of TET induction of δAg, but only the former contains an HDV genome that can replicate. Therefore, we took both cell types, induced with TET, and then made nuclear extracts for IP, using antibodies specific for Pol II. Table 3 summarizes the results of such an experiment. As expected, the Pol II antibody was able to select Pol II from both cell types, and the signal was at least 10 times more than that obtained in the absence of the antibody. However, also shown in the table is that the Pol II antibody was able to select a significant fraction of the δAg from both cell types. This observed association between Pol II and δAg in 293-δAg cells was thus independent of HDV RNA. It could have been a direct interaction, as has been suggested by Handa and coworkers from in vitro studies (41) and yeast two-hybrid analyses (40). Alternatively, the association could have been indirect, such as via IP of multicomponent protein complexes. At another level, as discussed below in terms of immunostaining studies, it remains possible that there might have occurred during the extraction and analysis some level of reorganization of δAg relative to Pol II that influences both these data and those from Fig. 4.
TABLE 3.
Comparison of association between Pol II and δAg in 293-HDV and 293-δAg cellsa
| Source of cell extract | Assay | % IP with antibody Pol II (4H8)
|
|
|---|---|---|---|
| − | + | ||
| 293-HDV | Pol II (4H8) | 1.2 | 13.7 |
| 293-HDV | δAg | 0.02 | 6.1 |
| 293-δAg | Pol II (4H8) | 0.4 | 4.7 |
| 293-δAg | δAg | 0.06 | 1.8 |
Nuclear extracts were made of TET-induced 293-HDV and 293-δAg cells. Samples were subjected to IP and immunoblot assays as in Table 2. The percentages shown are the average of two experiments.
Rate-zonal sedimentation analyses of δAg and Pol II complexes from 293-HDV and 293-δAg cells.
In order to further characterize multicomponent complexes containing Pol II and δAg, we next made use of rate-zonal sedimentation of the same nuclear extracts that had been used in the IP. After sedimentation, gradient fractions were assayed by immunoblotting to detect Pol II and δAg and, when relevant, qPCR was used to assay HDV genomic and antigenomic RNAs. Typical results are shown in Fig. 5C to G. Figure 5A shows a sedimentation control SVPs of HBV with a peak at fractions 5 and 6. Based on a recent report, SVPs are predominantly roughly spherical particles 25 nm in diameter containing 48 molecules of species of at least 30 kDa (12). Thus, the combined molecular mass is about 1.2 MDa. Also shown in Fig. 5A are the sedimentation of HDV and HBV particles, as detected by qPCR. Again, based on structural studies, we estimate these to have molecular masses of about 4.2 and 7.9 MDa, respectively. Figure 5B shows the sedimentation of the ribonucleoprotein of HDV, as released from HDV particles by nonionic detergent treatment, and estimated to have a molecular mass of about 2.1 MDa (34).
FIG. 5.

Rate-zonal sedimentation of nuclear extracts of HDV-293 and 293-δAg cells. Sedimentation was performed on gradients of 10 to 30% sucrose in STE. Fractions were collected and assayed by immunoblot for proteins and qPCR for nucleic acids. The results are expressed in arbitrary units. (A and B) Profiles for SVP, HDV, HBV, and HDV ribonucleoprotein; (C to G) profiles for the indicated nuclear extracts, prepared largely as described for Fig. 4, with Pol II represented by circles and δAg represented by shaded squares. While all extracts underwent a prior DNase treatment, those in panels D and G were also treated with RNase.
We detected for the nuclear extracts from TET-induced 293-HDV cells both genomic and antigenomic RNA (Fig. 5C). The observation that the genomic RNA in these nuclei was more abundant than the antigenomic RNA argues against the report that most antigenomic RNA never leave the nucleus, with the majority of genomic RNA rapidly transported out of the nucleus into the cytoplasm (24). Also, note that the majority of the HDV genomic and antigenomic RNAs sedimented consistent with a size of the viral RNP (Fig. 5B). This was also true for ca. 50% of the δAg, with the remainder sedimenting even further down the gradient. All of the Pol II sedimented consistent with masses of at least 2 MDa. This size was consistent with reports that the Pol II holoenzyme contains at least 100 proteins, with a combined molecular mass of 2 MDa. However, the majority of the Pol II sedimented even further, as far as the 8-MDa control (Fig. 5A). This larger size is consistent with the report that, of the 320,000 molecules of Pol II per eukaryotic cell, most are distributed in the nucleoplasm and exist as clusters of about eight active polymerases (11).
In Fig. 5C, as part of the standard protocol, these extracts were treated with DNase prior to sedimentation. However, in Fig. 5D, an additional treatment with RNase was included. The δAg now sedimented much more slowly, a finding consistent with the interpretation that it was previously in RNA-containing complexes. However, the majority of the Pol II remained in high-molecular-weight complexes, a finding consistent with the interpretation that these were not held together by either RNA or DNA.
We next tested the distribution for extracts made from 293-HDV cells not induced with TET. Such cells contain only about 40,000 molecules of δAg, much less than the 1 million molecules detected in induced cells. As seen in Fig. 5E, most of the δAg was now in complexes of around 2 MDa, a finding consistent with it being in RNP complexes (Fig. 5B). Again, some of the Pol II was still in high-molecular-weight complexes; it would seem such complexes exist independent of extensive HDV genome replication or even the presence of large amounts of δAg.
In Fig. 5F we examined nuclear extracts made from induced 293-δAg cells. These cells contained no HDV genomic or antigenomic RNA. Surprisingly, the δAg sedimented as ≥2 MDa complexes, just as in panel C. Figure 5G shows that when the extract was pretreated with RNase, most of the higher MW complexes were no longer present. Thus, since HDV RNA was not present in these cells, the higher-molecular-mass complexes seen in Fig. 5F must have involved, to some extent, RNAs of the host cell. As in Fig. 5C and D, the Pol II was in nuclease-resistant complexes of ≥2 MDa.
In summary, these sedimentation studies provided sobering information about the existence of high-molecular-weight complexes for Pol II, δAg, and HDV RNAs, as present in the nuclear extracts on which the IP studies (Fig. 3 and 4; Tables 2 and 3) were based. The detection of genomic and antigenomic RNAs sedimenting just as for the RNP released from detergent-disrupted virions (Fig. 5B and C) was as might be expected. Nevertheless, for these sedimentation data and with the IP data of Fig. 3 and Table 2, there is the concern that associations may to some extent be disrupted and/or created during the extraction and analysis procedures. Therefore, to exclude the possibility of such reorganization, we turned to the use of immunostaining using cells that were fixed prior to analysis.
Immunostaining of 293-HDV and 293-δAg cells.
Previous studies by us and by others have examined the intracellular distribution of δAg in cells, with or without the association of HDV genome replication (2, 4, 23). In order to better understand the IP and sedimentation results described thus far, we considered it important to carry out a parallel study by immunostaining, with assays for δAg, Pol II, and the splicing factor SC35. In previous studies we have observed that in the absence of HDV replication, δAg accumulates in nucleoli. In contrast, when HDV genome replication is present, δAg accumulation is predominantly localized to SC35-containing structures in the nucleoplasm, referred to as SC35 speckles (2, 4). Such structures, when examined by electron microscopy, are referred to as interchromatin granule clusters (22). Although SC35 speckles (and interchromatin granule clusters) are known to contain Pol II, it remains a somewhat controversial issue as to whether some or none of this Pol II is currently active in transcription (11, 39). It is clear, however, that in speckles or in adjacent structures, referred to as paraspeckles, there can occur processing of Pol II RNA transcripts (22) and sometimes the accumulation of unprocessed transcripts (32). Moreover, these paraspeckles, which in the electron microscope are referred to as perichromatin fibrils, are probably also sites of Pol II DNA-directed RNA transcription (22).
Some of our double immunostaining results are shown in Fig. 6A. In the 293-HDV cells at 16 h after TET induction, both the δAg and the Pol II (as detected with 4H8) were nucleoplasmic. The results of computer-based analyses of the merged images are summarized in Table 4. There was 80% colocalization of Pol II with δAg and almost 100% colocalization of δAg with Pol II. A quite different result was obtained for the 293-δAg cells. For these, the δAg was strictly nucleolar and the colocalization of Pol II with δAg was only 3%, while that of δAg with Pol II was 10%.
FIG. 6.

Immunostaining of HDV-293 and 293-δAg cells. After fixation cells were subjected to double immunostaining as indicated above panels A and B. Confocal images are shown along with the indicated merges, and the separate staining was done with DAPI. In all cases, the detected staining was nuclear, with partitions between the nucleoplasm and nucleoli.
TABLE 4.
Colocalizations computed from double immunostaininga
| Cell type | % Colocalization
|
|||
|---|---|---|---|---|
| Pol II with δAg | δAg with Pol II | SC35 with δAg | δAg with SC35 | |
| 293-HDV | 80 | 99.6 | 95 | 47 |
| 293-δAg | 3 | 10 | 2 | 3 |
The digital data are from Fig. 6, and the computer analysis for colocalization was obtained by using the META program.
We also examined the double immunostaining for δAg and SC35 with results as shown in Fig. 6B. In the 293-HDV cells both δAg and SC35 were predominantly in the nucleoplasm, and for the merged images the computed colocalizations were 95% of the SC35 with the δAg and 47% of the δAg with SC35. Thus, almost half of the δAg showed association with SC35 consistent with previous studies from this lab, including studies with Huh7 cells transiently transfected with expression constructs (2). As expected, in the 293-δAg cells, there was no obvious colocalization, and the computed values were both ≤3%.
In summary, these immunostaining studies indicated that in the 293-HDV cells, but not in the 293-δAg cells, there was major colocalization of δAg with Pol II and significant but less colocalization with SC35. Nevertheless, such colocalization could be explained by direct or indirect associations. Furthermore, we should expect that only perhaps 20% of the detected Pol II was actually active (11), and we do not know what subfraction of that was involved in RNA-directed transcription.
In other studies (data not shown), we also examined the distributions of nucleolin and fibrillarin. The nucleolar localization of these two proteins was not disturbed in any detectable way by either the expression of δAg in 293-δAg cells or this combined with HDV genome replication in 293-HDV cells.
DISCUSSION
Of the three HDV RNAs accumulated during HDV replication, it has been generally accepted that the transcription of mRNA is via Pol II, since this mRNA has all of the typical properties of Pol II-transcribed host mRNA, including the 5′ cap (16) and 3′ poly(A) tail (19). In addition, the accumulation of HDV mRNA has been shown to be consistently sensitive to a low concentration of amanitin (27). Previously, we have reported the development of 293-HDV cells that support exclusively HDV RNA-directed RNA transcription with high efficiency (4). Using this system, we have demonstrated that the accumulations of all three HDV RNAs were differentially inhibited by amanitin, with HDV mRNA being the most sensitive (6). However, we realized that in this kind of in vivo inhibitor treatment experiment, we were assaying the accumulation of steady-state levels of HDV RNAs, that is, the combined consequences of transcription, processing, and stability.
In order to directly measure HDV transcription, in particular, the HDV genomic and antigenomic RNA transcription under the treatment of amanitin, we and Macnaughton et al. have reported nuclear run-on experiments (25, 28). Both studies agree that transcription of genomic RNA is sensitive to amanitin. However, we were unable to obtain data for the relatively lower amounts of antigenomic RNA. In contrast, Macnaughton et al. obtained data that antigenomic RNA synthesis seemed resistant to amanitin, but we consider that the signals they detected were weak and specificity controls were missing. Therefore, given the advantages of the 293-HDV inducible system, we returned in the present study to the issue of run-on transcription. We thus obtained direct evidence that the transcription of not only new genomic RNA but also that of new antigenomic RNA were inhibited by amanitin at a concentration that was consistent with host Pol II (Fig. 2). After the run-on studies we used several additional approaches to investigate the intracellular associations between Pol II, δAg, and HDV RNAs. First, we used an IP selection, analogous to how chromatin IP is used to study DNA-directed RNA transcription. The data were obtained both with or without the use of formaldehyde cross-linking prior to preparing the nuclear extracts. In this way it was possible to detect significant associations between δAg and HDV genomic and antigenomic RNAs. However, the levels of association between Pol II and HDV genomic and antigenomic RNAs were much less (Fig. 3). In all of these studies the HDV RNAs were detected by Northern assay and were of unit-length. Thus, these were already the products of posttranscriptional processing and could therefore not be nascent RNA transcripts. Nascent transcripts, if present, could be expected to be heterogeneous in length and thus more difficult to detect according to size. Thus, we are left with the possibility, but not the proof, that what we detected might have been Pol II associated with HDV RNA templates.
In a somewhat different approach, we used IP to detect protein-protein interactions. Specifically, we used antibodies to δAg to test for associations with Pol II and vice versa. Small but significant levels of association were detected (Fig. 4). We used four monoclonal antibodies to the CTD of the large subunit of Pol II. As summarized in Table 1, these antibodies have a range of specificities for detecting variations in the serine-5 and serine-2 phosphorylation within the 7-amino-acid repeat of the CTD. Such reversible phosphorylation changes are linked to different stages of Pol II transcription on DNA templates. However, our studies did not in this way detect any significant difference between the forms of Pol II associated with DNA-directed relative to putative HDV RNA-directed transcription (Fig. 4).
As an extension of these IP studies, we investigated whether RNA species were needed for the detected interaction between Pol II and δAg. Using pretreatment of IP selected material, we showed that for the δAg that was selected with antibody to Pol II the HDV RNA was needed for the majority, but not all. In contrast, for the Pol II that was selected by antibody to δAg the HDV was apparently not needed (Table 2). That is, for most of the δAg associated with Pol II the interaction was mediated via RNA, while for the Pol II associated with δAg RNA was not involved.
These findings raised the question of whether or not HDV RNA at some earlier time might have been involved in the formation of the complexes. To test this, we compared 293-HDV cells with 293-δAg cells, since the latter contain δAg but no replicating HDV RNA. We found, as summarized in Table 3, that such associations between δAg and Pol II could be detected with similar efficiency. This finding supported the possibility that Pol II could interact with δAg in a manner independent of HDV RNA. However, we considered it still possible that host RNAs and/or multiprotein complexes were formed, either naturally and/or during the IP.
To shed some light on these possibilities, we next made use rate-zonal sedimentation of the nuclear extracts (Fig. 5) and immunostaining of intact cells (Fig. 6). The sedimentation studies showed that at least half of the genomic and antigenomic RNA was of a size comparable to the genomic ribonucleoprotein released from HDV by nonionic detergent treatment (Fig. 5B and C). This is consistent with the interactions detected by IP between δAg and unit-length genomic and antigenomic RNAs (Fig. 3).
We also found that the majority of the Pol II, independent of HDV genome replication or even of the presence of large amounts of δAg, sedimented in complexes of 2 MDa and larger that were resistant to both DNase and RNase (Fig. 5C, D, F, and G). One interpretation is that any natural associations between Pol II and δAg and HDV RNAs did not survive the sedimentation analysis procedure. An equally possible alternative is that the majority of the Pol II was not associated with HDV replication.
In the IP studies of Fig. 4 and Table 3, as well as in the sedimentation studies of Fig. 5, there is the specter that to some unknown extent there could have occurred during the extraction and analysis procedures some level of reorganization of δAg and Pol II, either to produce or disrupt associations. (The caveat does not apply to the data in Fig. 3, where we were able confirm the results both with or without prior cross-linking.) Therefore, we turned to immunostaining of cells fixed prior to further analysis. This showed that when δAg was expressed in the absence of HDV replication, that is, in 293-δAg cells, the δAg was strictly nucleolar, whereas the Pol II was strictly nucleoplasmic (Fig. 6A and Table 4). This result argues against the biological relevance of what by IP seemed to be even a low frequency of associations in 293-δAg cells between δAg and Pol II (Table 3). It also casts doubt on the comparable low frequency of IP interactions detected in 293-HDV cells (Fig. 4 and Table 3). A parallel but not mutually exclusive interpretation comes from the immunostaining of the 293-HDV cells (Fig. 6A and Table 4). Here there was clear and extensive colocalization of δAg and Pol II.
In search of an explanation for the colocalization, it is tempting to link our results to those of Faro-Trindade and Cook (11), who favor the interpretation that in the nuclei of eukaryotic cells specific RNA transcripts, whether by Pol I, II, or II, are assembled in distinct “factories.” These authors consider that the DNA templates diffuse and bind to these factories prior to RNA transcription and processing. According to this hypothesis, our data for HDV might be interpreted as evidence that unit-length HDV RNAs, whether genomic or antigenomic, in association with δAg to form a ribonucleoprotein, diffuse and bind to Pol II factories in the nucleoplasm, thereby increasing the chance of somehow achieving RNA-directed RNA transcription. In contrast, in the absence of HDV RNAs, the δAg diffuses to the nucleoli and binds to rRNA Pol I factories.
We are aware that others have used in vitro studies to detect a direct interaction between Pol II and δAg (41). However, such an interaction did not occur in 293-δAg cells, where there was little if any colocalization between δAg and Pol II (Fig. 6A and Table 4).
After all of these studies, especially the nuclear run-on assays (Fig. 2), we are now confident that Pol II is needed for the transcription of both genomic and antigenomic RNAs. Other questions now need to be resolved. (i) We have known for some time that δAg is essential for the accumulation of the HDV RNAs (7), but we are still unsure whether it is a direct participant of in vivo transcription. (ii) Another question regards the existence of promoters. That is, how does Pol II recognize HDV genomic and antigenomic RNAs as templates for RNA-directed transcription? Some attempts have been reported (1, 18), and it should be noted that recent in vitro studies indicate that the ends of the rod-like folding of HDV genomic and antigenomic RNA can be sites for Pol II binding (13). (iii) An equally important question concerns locating the initiation sites for such transcription. We think the 5′ end of the δAg mRNA is one such site (16), but we do not know the sites leading to the synthesis of genomes and antigenomes. Furthermore, it has not yet been possible to demonstrate the initiation of transcription in vitro. In summary, while we may be certain that Pol II can be redirected to copy RNA, we have yet to understand how this is achieved.
Acknowledgments
J.T. was supported by grants AI-26522 and CA-06927 from the National Institutes of Health and by an appropriation from the Commonwealth of Pennsylvania. J.C. was supported in part by a pilot grant from American Cancer Society.
We thank Emmanuelle Nicolas and the Fox Chase Biotechnology Facility for the real-time PCR assays. Confocal imaging was performed at the Biomedical Imaging Core, Department of Pathology and Laboratory Medicine, University of Pennsylvania, with the assistance of Xinyu Zhao. HBV and HDV were provided by Severin Gudima. Cheng-Ming Chiang gave advice and encouragement on the RNA transcription studies. Constructive comments on the manuscript were given by Severin Gudima, Richard Katz, and William Mason.
Footnotes
Published ahead of print on 21 November 2007.
REFERENCES
- 1.Beard, M. R., T. B. Macnaughton, and E. J. Gowans. 1996. Identification and characterization of a hepatitis delta virus RNA transcriptional promoter. J. Virol. 704986-4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bichko, V. V., and J. M. Taylor. 1996. Redistribution of the delta antigens in cells replicating the genome of hepatitis delta virus. J. Virol. 708064-8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodsky, A. S., C. A. Meyer, I. A. Swinburne, G. Hall, B. J. Keenan, X. S. Liu, E. A. Fox, and P. A. Silver. 2005. Genomic mapping of RNA polymerase II reveals sites of co-transcriptional regulation in human cells. Genome Biol. 6R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, J., S. O. Gudima, C. Tarn, X. Nie, and J. M. Taylor. 2005. Development of a novel system to study hepatitis delta virus genome replication. J. Virol. 798182-8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang, J., X. Nie, S. Gudima, and J. Taylor. 2006. Replication of the hepatitis delta virus genome, p. 195-212. In K. L. Hefferon (ed.), Recent advances in RNA virus replication. Transworld Research Network, Kerala, India.
- 6.Chang, J., X. Nie, S. O. Gudima, and J. M. Taylor. 2006. Action of inhibitors on the accumulation of processed hepatitis delta virus RNAs. J. Virol. 803204-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao, M., S.-Y. Hsieh, and J. Taylor. 1990. Role of two forms of the hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome replication. J. Virol. 645066-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chao, M., S.-Y. Hsieh, and J. Taylor. 1991. The antigen of hepatitis delta virus: examination of in vitro RNA-binding specificity. J. Virol. 654057-4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corden, J. L. 1993. RNA polymerase II transcription cycles. Curr. Opin. Genet. Dev. 3213-218. [DOI] [PubMed] [Google Scholar]
- 10.Eick, D., F. Kohlhuber, D. A. Wolf, and L. J. Strobl. 1994. Activation of pausing RNA polymerases by nuclear run-on experiments. Anal. Biochem. 218347-351. [DOI] [PubMed] [Google Scholar]
- 11.Faro-Trindade, I., and P. R. Cook. 2006. Transcription factories: structures conserved during differentiation and evolution. Biochem. Soc. Trans. 341133-1137. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert, R. J., L. Beales, D. Blond, M. N. Simon, B. Y. Lin, F. V. Chisari, D. I. Stuart, and D. J. Rowlands. 2005. Hepatitis B small surface antigen particles are octahedral. Proc. Natl. Acad. Sci. USA 10214783-14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greco-Stewart, V. S., P. Miron, A. Abrahem, and M. Pelchat. 2007. The human RNA polymerase II interacts with the terminal stem-loop regions of the hepatitis delta virus RNA genome. Virology 35768-78. [DOI] [PubMed] [Google Scholar]
- 14.Gudima, S., K. Dingle, T.-T. Wu, G. Moraleda, and J. Taylor. 1999. Characterization of the 5′-ends for polyadenylated RNAs synthesized during the replication of hepatitis delta virus. J. Virol. 736533-6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gudima, S., Y. He, A. Meier, J. Chang, R. Chen, M. Jarnik, E. Nicolas, V. Bruss, and J. Taylor. 2007. Assembly of hepatitis delta virus: particle characterization including ability to infect primary human hepatocytes. J. Virol. 813608-3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gudima, S., S.-Y. Wu, C.-M. Chiang, G. Moraleda, and J. Taylor. 2000. Origin of the hepatitis delta virus mRNA. J. Virol. 747204-7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gudima, S. O., J. Chang, G. Moraleda, A. Azvolinsky, and J. Taylor. 2002. Parameters of human hepatitis delta virus replication: the quantity, quality, and intracellular distribution of viral proteins and RNA. J. Virol. 763709-3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gudima, S. O., J. Chang, and J. M. Taylor. 2004. Features affecting the ability of hepatitis delta virus RNAs to initiate RNA-directed RNA synthesis. J. Virol. 785737-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsieh, S.-Y., M. Chao, L. Coates, and J. Taylor. 1990. Hepatitis delta virus genome replication: a polyadenylated mRNA for delta antigen. J. Virol. 643192-3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuo, M. Y.-P., J. Goldberg, L. Coates, W. Mason, J. Gerin, and J. Taylor. 1988. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: sequence, structure, and applications. J. Virol. 621855-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai, M. M. 2005. RNA replication without RNA-dependent RNA polymerase: surprises from hepatitis delta virus. J. Virol. 797951-7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamond, A. I., and D. L. Spector. 2003. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell. Biol. 4605-612. [DOI] [PubMed] [Google Scholar]
- 23.Li, Y. J., T. Macnaughton, L. Gao, and M. M. Lai. 2006. RNA-templated replication of hepatitis delta virus: genomic and antigenomic RNAs associate with different nuclear bodies. J. Virol. 806478-6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macnaughton, T. B., and M. M. Lai. 2002. Genomic but not antigenomic hepatitis delta virus RNA is preferentially exported from the nucleus immediately after synthesis and processing. J. Virol. 763928-3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macnaughton, T. B., S. T. Shi, L. E. Modahl, and M. M. Lai. 2002. Rolling circle replication of hepatitis delta virus RNA is carried out by two different cellular RNA polymerases. J. Virol. 763920-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meininghaus, M., R. D. Chapman, M. Horndasch, and D. Eick. 2000. Conditional expression of RNA polymerase II in mammalian cells. Deletion of the carboxyl-terminal domain of the large subunit affects early steps in transcription. J. Biol. Chem. 27524375-24382. [DOI] [PubMed] [Google Scholar]
- 27.Modahl, L. E., T. B. Macnaughton, N. Zhu, D. L. Johnson, and M. M. C. Lai. 2000. RNA-dependent replication and transcription of hepatitis delta virus RNA involve distinct cellular RNA polymerases. Mol. Cell. Biol. 206030-6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moraleda, G., and J. Taylor. 2001. Host RNA polymerase requirements for transcription of the human hepatitis delta virus genome. J. Virol. 7510161-10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris, D. P., G. A. Michelotti, and D. A. Schwinn. 2005. Evidence that phosphorylation of the RNA polymerase II carboxyl-terminal repeats is similar in yeast and humans. J. Biol. Chem. 28031368-31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niranjanakumari, S., E. Lasda, R. Brazas, and M. A. Garcia-Blanco. 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26182-190. [DOI] [PubMed] [Google Scholar]
- 31.Palancade, B., and O. Bensaude. 2003. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. 2703859-3870. [DOI] [PubMed] [Google Scholar]
- 32.Prasanth, K. V., S. G. Prasanth, Z. Xuan, S. Hearn, S. M. Freier, C. F. Bennett, M. Q. Zhang, and D. L. Spector. 2005. Regulating gene expression through RNA nuclear retention. Cell 123249-263. [DOI] [PubMed] [Google Scholar]
- 33.Reid, J., and J. Q. Svejstrup. 2006. An assay for studying ubiquitylation of RNA polymerase II and other proteins in crude yeast extracts. Methods Enzymol. 408264-273. [DOI] [PubMed] [Google Scholar]
- 34.Ryu, W. S., H. J. Netter, M. Bayer, and J. Taylor. 1993. Ribonucleoprotein complexes of hepatitis delta virus. J. Virol. 673281-3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor, J. M. 2006. Hepatitis delta virus. Virology 34471-76. [DOI] [PubMed] [Google Scholar]
- 36.Taylor, J. M. 2006. Structure and replication of HDV RNA, p. 1-23. In J. L. Casey (ed.), Hepatitis delta virus, vol. 307. Springer, Heidelberg, Germany. [Google Scholar]
- 37.Thompson, N. E., T. H. Steinberg, D. B. Aronson, and R. R. Burgess. 1989. Inhibition of in vivo and in vitro transcription by monoclonal antibodies prepared against wheat germ RNA polymerase II that react with the heptapeptide repeat of eukaryotic RNA polymerase II. J. Biol. Chem. 26411511-11520. [PubMed] [Google Scholar]
- 38.Wang, K.-S., Q.-L. Choo, A. J. Weiner, J.-H. Ou, C. Najarian, R. M. Thayer, G. T. Mullenbach, K. J. Denniston, J. L. Gerin, and M. Houghton. 1986. Structure, sequence and expression of the hepatitis delta viral genome. Nature 323508-513. [DOI] [PubMed] [Google Scholar]
- 39.Xie, S. Q., S. Martin, P. V. Guillot, D. L. Bentley, and A. Pombo. 2006. Splicing speckles are not reservoirs of RNA polymerase II, but contain an inactive form, phosphorylated on serine-2 residues of the C-terminal domain. Mol. Biol. Cell 171723-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamaguchi, K., and H. Handa (ed.). 2006. Hepatitis delta antigen and RNA polymerase II. Landes Bioscience, Georgetown, TX.
- 41.Yamaguchi, Y., T. Mura, S. Chanarat, S. Okamoto, and H. Handa. 2007. Hepatitis delta antigen binds to the clamp of RNA polymerase II and affects transcriptional fidelity. Genes Cells 12863-875. [DOI] [PubMed] [Google Scholar]