

Abstract
We compared in vitro transcription-initiated folding of the ribozyme from Bacillus subtilis RNase P to refolding from the full-length, denatured state by monitoring the appearance of its catalytic activity. At 37°C, Mg2+-initiated refolding of the wild type and a circularly permutate ribozyme takes minutes and is limited by a kinetic trap. Transcription by T7 RNA polymerase alters the folding pathway of both RNAs and introduces new kinetic traps. Transcription by the core Escherichia coli RNA polymerase yields the same result, in spite of its 4-fold-slower elongation rate. However, the presence of its elongation factor NusA accelerates more than 10-fold the transcription-initiated folding of the circularly, permutated ribozyme by E. coli RNA polymerase. The effect of NusA likely is caused by its enhancement of transcriptional pausing because NusA did not accelerate transcription-initiated folding using a mutant RNA polymerase that failed to pause or respond to NusA during ribozyme synthesis. We conclude that both transcription and specific pausing therein can alter RNA-folding pathways.
Keywords: cotranscriptional RNA folding, pausing
Most studies of RNA and protein folding are carried out in vitro by using full-length, denatured molecules. Two relevant issues are whether the folding pathway is altered during biosynthesis and to what extent this change depends on the mechanistic details of the RNA polymerase or the ribosome. In the case of highly cooperative, single-domain proteins whose stability requires the presence of >90% of the chain, significant folding probably does not occur during synthesis. On the other hand, a multidomain protein can fold sequentially as a nascent polypeptide emerges from the ribosome (1).
The RNA component of the B. subtilis RNase P (denoted P RNA), which generates the 5′ end of all mature tRNAs in the cell through a site-specific endonucleolytic reaction (2, 3), has been studied extensively in vitro as a model system for large ribozyme folding (4, 5). We showed previously that upon addition of micromolar amounts of Mg2+, the full-length P RNA rapidly forms highly stable helices and hairpins. However, within seconds or less after the addition of millimolar amounts of Mg2+, a kinetic trap is created that requires a minutes-long structural reorganization to reach a catalytically active structure. The folding pathway of P RNA subsequently was demonstrated to be malleable and changed easily by circular permutation and altered initial conditions. Based on the results from both tRNA folding and the first structural transition of P RNA, we suggested previously that the time scale for the error-free RNA folding should be no more than a few seconds at 37°C if the kinetic traps observed in the Mg2+-initiated refolding can be avoided. Consistent with this hypothesis, refolding of the Tetrahymena group I ribozyme takes minutes in vitro, but is complete in less than 6 sec in vivo (6–8), implying the existence of a cellular mechanism that changes the folding pathway of this group I ribozyme.
One obvious difference between cotranscriptional folding and Mg2+-induced refolding is that during extension of the RNA chain, upstream regions of RNA can fold before synthesis of downstream regions. RNA secondary structures form on a submillisecond time scale in vitro, and their formation during transcription may be limited by elongation rate of RNA polymerase (denoted RNAP) along the template. RNA has a propensity to become kinetically trapped because even a short hairpin can have a folding free energy of −10 kcal/mol and is able to form as soon as RNA emerges from the elongating RNAP. Depending on the folding pathway, transcription can affect folding either positively, if sequestering upstream RNA makes it not available for kinetic trap formation, or negatively, if the stable secondary structure formed during transcription represents a nonnative intermediate that has to be disrupted before the active conformation forms. This model for cotranscriptional folding was invoked to explain folding defects of Escherichia coli ribosomal RNA during transcription by T7 RNAP (9).
An alternative mechanism of influencing RNA folding during transcription can be envisioned to involve transcriptional pausing. The transcript elongation is known to occur at an uneven rate because RNAP pauses at specific sites along the template with elongation rate decreasing by as much as 10,000-fold (10). Pausing at some sites in bacterial genome depends in part on interaction of a nascent RNA hairpin with the RNAP; this interaction appears to stabilize RNA hairpin and, thus, can facilitate the formation of structures critical for (or inhibiting) proper RNA folding. The dwell time of RNAP at these hairpin-dependent pause sites is enhanced strongly by the transcription elongation factor NusA and can be modulated by mutations in RNAP (11, 12).
To compare cotranscriptional folding of a biologically important and highly structured ribozyme with its refolding in vitro, we have studied both the 409-nt P RNA and a circularly permutated construct that alters the order of RNA synthesis (CP RNA). We found that synthesis of either P or CP RNA by T7 or E. coli RNAP changed their folding pathways from those observed during Mg2+-initiated refolding. The presence of the E. coli NusA accelerated folding of CP RNA more than 10-fold when it was transcribed by the wild type but not a mutant E. coli RNAP that was deficient in pausing and NusA response. These results demonstrate that both the process of transcription and the transcriptional machinery can control RNA-folding pathways.
MATERIALS AND METHODS
Sources of Proteins.
The E. coli RNAP and NusA were isolated as described (13, 14). Mutant RNAP [rpoB5101(P560S, T563I) in ref. 14] was purified as described by Hager et al. (15). T7 RNAP was purified from an overexpression clone as described (16).
Transcription in Vitro.
Transcription was performed under standard conditions with minor modifications. The P RNA and CP RNA constructs under the control of T7 RNAP promoter have been described (17, 18). The CP RNA template for E. coli RNAP was obtained by PCR. The PCR product contained the immediate 73 nt upstream of the native B. subtilis P RNA gene (19) followed by the 5′ end of CP RNA and the precise 3′ end for run-off transcription. Transcription using T7 RNAP was performed in 40 mM Tris⋅HCl, pH 8.1/1 mM spermidine/50 μg/ml BSA/2 mM each of ATP, GTP, CTP, and UTP/5–10 μCi [α-32P]CTP/14 mM MgCl2/60 μg/ml plasmid DNA/40 μg/ml T7 RNAP. Transcription using E. coli RNAP was performed in 40 mM Tris⋅HCl, pH 7.9/10 mM 2-mercaptoethanol/4 mM spermidine/1 mM each of ATP, GTP, CTP, and UTP/100 μg/ml BSA/10 mM MgCl2/120 mM KCl/0.05–0.1 μM DNA template/0.2 μM RNAP holoenzyme/0 or 0.4 μM E. coli NusA protein. Transcription was carried out at 37°C for 8–10 min with removal of aliquots beginning at 20 sec.
To assay for transcriptional pausing, ternary complexes were formed as described (20) at 50 nM in transcription buffer (20 mM Tris⋅HCl/20 mM NaCl/14 mM MgCl2/14 mM 2-mercaptoethanol/0.1 mM Na2EDTA) with 32P derived from [α-32P]CTP (NEN; 3,000 Ci/mmol). Halted complexes were formed with 150 nM GpC/10 μM CTP/25 μM ATP and GTP; omission of UTP allows stalling at A14 in the initial transcribed region of the CP RNA (GCGAGAAACCCAAA UUU). Elongation was resumed with addition of 20 μM GTP/150 μM each of ATP, CTP, and UTP (Amersham Pharmacia)/100 μg heparin/ml. Samples were taken at the desired time points and mixed with the equal volume of 2× stop solution, denatured for 2 min at 90°C, and electrophoresed through 9% denaturing gels [19:1 (wt/wt) acrylamide to bis-acrylamide/7 M urea] in 1× TBE (44 mM Tris-borate, pH 8.3/7.5 mM EDTA).
Folding Monitored by Catalytic Activity.
Two parameters were needed to calculate the folding rate of RNA transcripts: the amount of synthesized transcript and the amount of catalytically active ribozyme. The fraction of [α-32P]CTP incorporated into the full-length RNA transcript was quantitated by phosphorimaging, and the amount of total P RNA synthesized, S(t), was calculated according to
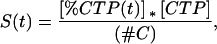 |
1 |
where [CTP] is the total concentration of CTP in the transcription mixture and #C is the number of cytidine residues in the RNA. Nonlinear synthesis of RNA over time was observed in our transcription reactions. The nonlinear behavior may be explained with a product-inhibition model in which the RNA transcripts bind to the RNAP to inhibit initiation. Assuming that the RNAP concentration is greater than the DNA template concentration and the binding of RNAP to DNA and RNA is always at equilibrium, S(t) can be described by applying equilibria equations:
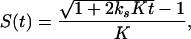 |
2 |
where ks is the synthesis rate (μM min−1) and K is the association constant (μM−1) of the RNA transcript to the RNAP in the transcription reaction. When 2 ks K t ≪ 1, Eq. 2 can be approximated as S(t) ≈ ks t.
The amount of catalytically active ribozyme was determined in two ways, depending on the synthesis rate. When ks was greater than 0.02 μM RNA/min, the amount of the folded ribozyme was measured with a molar excess of substrate (i.e., [S] > E) as described (4). When ks was less than 0.01 μM/min, the amount of the folded ribozyme was obtained by measuring the cleavage rate under single-turnover conditions (i.e., [E] ≫ [S]), where the rate of cleavage was proportional to the amount of folded ribozyme. In both cases, aliquots of the transcription reaction (typically 4.5 μl) were mixed with equal volumes of 32P-labeled substrate under high ionic conditions that terminated transcriptional initiation as determined by the identical amount of transcript at the time of substrate addition and after 20 min of incubation (data not shown). The final condition was 50 mM Tris⋅HCl, pH 8.1/100 mM MgCl2/0.6 M KCl for cleaving the pre-tRNA substrate and 50 mM Tris⋅HCl, pH 8.1/100 mM MgCl2/0.06 M KCl/1 mM spermine for cleaving the selected substrate. Aliquots (typically 2–3 μl) were taken from the cleavage reaction within 7–21 sec and quenched with an excess of EDTA and urea, and the amount of cleaved substrate was quantified by phosphorimaging.
Knowing the amount of active ribozyme present, A(t), and the amount of RNA synthesized, S(t), is sufficient to deduce the folding rate of the nascent RNA transcript. A(t) equals the sum of the amount synthesized at earlier times, S(t′), multiplied by the fraction that subsequently has folded, 1 − e−kf(t−t′)
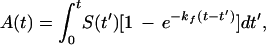 |
3 |
where kf is the folding rate (s−1) of the nascent RNA transcript. In the linear phase of synthesis [i.e., S(t) = ks t], Eq. 3 has the solution
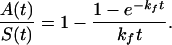 |
4 |
When the synthesis is nonlinear, the solution of Eq. 3 is an infinite series but is still well approximated by Eq. 4.
RESULTS
Folding During Transcription by T7 RNAP.
The bacterial P RNA is composed of two independently folding domains (21, 22) (Fig. 1A). One domain contains the entire active site (C domain, the catalytic domain of P RNA including nucleotides 240–409 + 1–85), whereas the other domain binds the T stem loop in a pre-tRNA substrate to confer substrate specificity (S domain, the specificity domain of P RNA including nucleotides 86–239). Cleavage of a pre-tRNA substrate requires both domains, whereas the cleavage of an in vitro-selected substrate requires only the C domain (18). In this study, we take advantage of these different cleavage specificities to identify folding intermediates in which the C domain, but not the S domain, is folded. Such an intermediate was not observed in the Mg2+-induced refolding of the wild-type P RNA, but was observed for CP RNA.
Figure 1.

(A) Schematic representation of the domain structure of wild-type P RNA and CP RNA ribozymes and requirements for different substrates cleavage: processing of the pre-tRNA substrate requires folding of both the catalytic (C) and the specificity (S) domains, whereas proper folding of the C domain is sufficient for the cleavage of the selected substrate. The C domain in the P RNA is composed of a 5′ and a 3′ part interrupted by the S domain. (B) Refolding pathway for P RNA and CP RNA (5). Completely folded domains are indicated with the shaded boxes as in A; open symbols represent not-yet-folded domains. Essentially all wild-type P RNA fold through the slow, top pathway with a kinetic intermediate in which neither domain is folded. The CP RNA can fold through upper and lower pathways. The bottom pathway has an intermediate in which only the C domain is folded. (C) Cotranscriptional folding pathway. The initial intermediate formed during transcription differs from Mg2+-initiated refolding and probably has two new misfolds. (D) NusA effect on folding during transcription by E. coli RNAP. Here, folding of the S domain does not limit formation of native CP RNA. All folding rates are in s−1 at 37°C.
Folding of P RNA during its synthesis by T7 RNAP was analyzed by measuring the rate of forming RNA structures able to catalyze cleavage of a tRNA or the selected substrate (Fig. 2). The synthesis rate of P RNA was nonlinear and increasingly slow as explained by RNA product inhibition (Eq. 2 in Materials and Methods). When assayed with a pre-tRNA substrate, folding of the nascent P RNA transcript during transcription was within a factor of 2 of the slow, Mg2+-initiated refolding rate (Fig. 2B and Table 1). Interestingly, the folding rate measured by cleavage of the selected substrate was ≈4-fold faster, indicating that the C domain folded before the S domain. Because both domains fold simultaneously in Mg2+-initiated refolding, P RNA folding during transcription is different: the C domain folds faster, even though the rate of obtaining the native structure is the same. The similarity in rate of native structure formation may be coincidental because the intermediates, and presumable the kinetic barriers, are substantially different for the two modes of folding initiation.
Figure 2.

Folding of P RNA and CP RNA during transcription by T7 RNAP. (A) The amount of P RNA synthesized [▴, S(t)] was measured by incorporation of [α-32P]CTP. The amount of active P RNA was detected by catalytic activity with a tRNA substrate [A(t), ▵] or the selected substrate [A(t), ⋄]. The synthesis of P RNA has a transcription rate (ks) of 1.3 ± 0.2 μM/min and an association constant (K) of 0.9 ± 0.2 μM−1 according to Eq. 2. (B) Catalytically active fraction [A(t)/S(t)] for P RNA folding. The folding rate determined by cleaving the selected substrate is ≈4-fold faster than that from cleaving a tRNA substrate. (C) Catalytically active fraction for CP RNA folding.
Table 1.
Folding rates of P RNA and CP RNA during transcription at 37°C
| Ribozyme | Folding process | Folding rate (s−1) by the cleavage of
|
|
|---|---|---|---|
| tRNA substrate | Selected substrate | ||
| P RNA | Refolding* | (5.2 ± 0.4) × 10−3 | (7.8 ± 0.8) × 10−3 |
| T7 RNa polymerase | (5.5 ± 0.2) × 10−3 | (20 ± 3) × 10−3 | |
| CP RNA | Refolding* | 0.075 ± 0.013† | >0.2 |
| (5.3 ± 0.4) × 10−3‡ | ND§ | ||
| T7 RNA polymerase | (4.8 ± 0.2) × 10−3 | (23 ± 2) × 10−3 | |
| E coli RNA polymerase | |||
| Wild type: core | (6.8 ± 0.4) × 10−3 | (23 ± 2) × 10−3 | |
| Wild type: (+) NusA | (23 ± 2) × 10−3 | (23 ± 2) × 10−3 | |
| Mutant: core | (3.6 ± 0.2) × 10−3 | ND | |
| Mutant: (+) NusA | (3.6 ± 0.2) × 10−3 | ND | |
ND, not determined.
Data from ref. 5.
Fast-folding population.
Slow-folding population.
Not determined at 37°C, but identical to that from cleaving a tRNA substrate at 28°C.
In our previous refolding studies, a circularly permutated P RNA with the 5′ end at nucleotide 240 was observed to fold through two pathways (Fig. 1B). When folding is initiated from low-Mg2+ concentrations or temperatures below 37°C, the pathway is similar to that of the wild-type P RNA and folding is limited by a significant interdomain misfold. However, when folding of CP RNA begins from the Mg2+-free state at 37°C, an intermediate accumulates rapidly, which has only the C domain folded. The subsequent folding of the S domain occurs on the time scale of seconds (5).
During transcription of a CP RNA template, the entire C domain (255 nt) is synthesized before the S domain (154 nt). In contrast, the wild-type P RNA is synthesized in the order of 5′ C domain (87 nt), S domain, and then 3′ C domain (168 nt) (Fig. 1A). The new synthesis order of the CP RNA might be expected to create a time window for the C domain to fold in a different way and promote rapid folding to the native structure. However, folding of the CP RNA during transcription by T7 RNAP still took minutes and was similar to that seen for the wild-type P RNA (Fig. 2C). An intermediate with a folded C domain can accumulate during both Mg2+- and transcription-initiated folding of CP RNA. Compared with refolding from the Mg2+-free state, both the intermediate and native CP RNA form more slowly (by a factor of >10) during transcription by T7 RNAP (Fig. 1 B and C).
Effect of Pausing by E. coli RNAP on Folding of CP RNA.
Transcription of E. coli genes by T7 RNAP in E. coli sometimes fails to produce functional RNAs (9, 23, 24). A likely reason is that more rapid synthesis by the phage polymerase alters RNA-folding pathways (T7 RNAP elongates in vitro at 200–400 nt/sec vs. 10–35 nt/sec for E. coli RNAP; ref. 10). This led us to ask whether synthesis of P RNA by a bacterial RNAP could promote its folding. We chose to examine transcription-initiated folding by using E. coli RNAP and only the CP RNA, which, for technical reasons, could be dissected in greater detail. We found that the folding pathway of CP RNA during synthesis with E. coli RNAP was similar to the pathway observed during synthesis with T7 RNAP (Fig. 1C and Table 1).
As suggested in the introduction, the folding pathway may be controlled by transcriptional pausing at specific sites, which is known to be modulated by elongation factors such as NusA in bacteria. To examine whether NusA-enhanced pausing could affect CP RNA folding, we tested the effect of NusA by using wild-type E. coli RNAP and an RNAP mutant known to strongly reduce hairpin-dependent pausing in vitro (14). During single-round synthesis of CP RNA at limiting GTP (conditions that enhance pausing), wild-type RNAP paused at several sites, whereas the mutant RNAP recognized only a subset of these sites and completed CP RNA synthesis faster than the wild type (Fig. 3 A and B). At 1 mM NTP (conditions used in folding assays), however, mutant RNAP transcribed slightly slower than the wild-type enzyme (Fig. 3B). Addition of NusA reduced the elongation rate of the wild-type RNAP by a factor of 2 and enhanced pausing at certain positions; the most dramatic effect was seen at position 225 of CP RNA (U55 of P RNA, mapping data not shown). Pausing at this position was detectable even at 1 mM NTP in the presence of NusA (data not shown). In contrast, NusA had little effect on the average elongation rate of the mutant RNAP (Fig. 3B) and enhanced its pausing at CP RNA 225 much less than for the wild-type enzyme both at low GTP (Fig. 3A) or 1 mM NTP (data not shown).
Figure 3.

(A) Synthesis of CP RNA by wild-type and mutant E. coli RNAP in the absence or in the presence of 90 nM NusA. The darkness of the bands reflects the dwell time at specific pausing sites. Asterisk indicates the pausing site at nucleotide U55 (225th position in the CP RNA). (B) Elongation rates of wild-type (solid bars) and mutant (shaded bars) E. coli RNAP were determined from single-round transcription assays on CP RNA template at the concentrations of NTP and NusA indicated below the bar graph. (C) Folding of CP RNA during transcription by 0.2 μM wild-type (Left) and mutant (Right) RNAP in the absence (●) and presence (□) of 0.4 μM NusA or by 0.2 μM wild-type enzyme at 0.06 mM (▵), 0.25 mM (♦), and 1 mM (curve fit) NTP. The synthesis of CP RNA by the E. coli RNAP is nonlinear and is best fit by a transcription rate (ks) of 0.046 ± 0.016 μM/min and an association constant (K) of (0.20 ± 0.08) × 10−3 μM−1 according to Eq. 2. The folding rates are determined by the cleavage of a pre-tRNA substrate.
Strikingly, NusA accelerated folding of CP RNA to the tRNA-cleavage-competent form 4-fold with wild-type RNAP, but not at all with the mutant RNAP that was defective in pausing and NusA response (Fig. 3C and Table 1). However, NusA had no effect on folding of the C domain as determined by cleavage of the selected substrate (Fig. 4 and Table 1). In fact, the NusA-stimulated folding rate was identical when assayed by either substrate, leading us to conclude that the S domain must fold fast enough to allow the C domain-folding rate of 0.02 s–1 to become rate-limiting. This requires that the S domain folds at >0.06 s−1 (Fig. 1D), which is close to the rate observed during Mg2+-initiated refolding (Fig. 1B) and >10-fold-faster than the rate observed during transcription in the absence of NusA. To confirm that the NusA effect is not caused by the slowed elongation rate in the presence of NusA, CP RNA folding was examined at comparable elongation rates in the absence of NusA by reducing the NTP concentration from 1 to 0.25 mM and 0.06 mM (Fig. 3 B and C). The folding rate remained unchanged at these low NTP concentrations. Hence, we conclude that NusA alters CP RNA folding through changes in pausing at one or more specific sites.
Figure 4.

Folding of CP RNA during transcription by 0.2 μM E. coli RNAP in the presence of 0.4 μM NusA assayed by cleavage of the selected substrate.
DISCUSSION
There are two major outcomes of this study. First, the transcription process itself changes P RNA folding though bypassing some kinetic traps and introducing new ones. Second, a component of the transcription elongation complex, NusA, significantly accelerates folding, apparently by stimulating pausing at certain sites during transcription by E. coli RNAP.
Transcription vs. Mg2+-Initiated Refolding.
Transcription by either E. coli or T7 RNAP, which differ significantly in transcription speed and recognition of pause signals (10, 25), changes the folding pathways of the wild-type P RNA and CP RNA (Fig. 1C). This change is consistent with our previous observation that tertiary RNA-folding pathways are highly malleable (5). The major difference between Mg2+- and transcription-initiated folding is the nature of the kinetic traps (Fig. 1 B and C). The major kinetic trap in the Mg2+-initiated refolding involves residues in both domains. The transcription process eliminates this major misfold and allows the C domain to fold before the S domain. However, the subsequent folding of the S domain is about 25-fold slower than for Mg2+-initiated refolding. Additionally, folding of the C domain during transcription is many times slower than that observed for Mg2+-initiated refolding. Hence, the transcription process generates misfolds different from that observed in Mg2+-initiated refolding.
The similar modulation of folding pathway during transcription by multisubunit bacterial as well as single, subunit phage RNAPs can be explained most easily by a passive model of cotranscriptional folding. By allowing the upstream regions of nascent RNA chain to fold while prohibiting long-range interactions with the yet-to-be-made downstream regions, RNA folding occurs along a different pathway with a separate set of intermediates and kinetic traps. Thus, cotranscriptional folding could lead to either overall acceleration or inhibition of the native tertiary structure formation.
NusA Effect on Folding During Transcription.
An alternative, more direct modulation of RNA folding is exemplified by the effect of the E. coli elongation factor NusA, which accelerates transcription-initiated folding of CP RNA (Fig. 1D). The large effect of NusA on folding kinetics can be explained either by its ability to reduce average transcription elongation rate (13) or by the preferential increase of the dwell time of RNAP at the specific template positions (14, 26–28). Our data are most consistent with the second possibility. First, we observe no difference in folding behavior of CP RNA during transcription by E. coli or T7 RNAP in spite of the at least 4-fold difference in the elongation rate (Table 1). Second, folding of CP RNA during transcription by E. coli RNAP is similar at 0.06, 0.25, and 1 mM NTP (Fig. 3C), even though the elongation rate at 0.25 mM NTP matches that at 1 mM NTP plus NusA, conditions that greatly accelerate folding (Fig. 3B). Third, transcription by a mutant RNAP deficient in pausing (Fig. 3A), but transcribing at the same rate as the wild-type enzyme under conditions of folding assays (1 mM NTP, Fig. 3B), reduces the rate of CP RNA folding by a factor of ≈2. Finally, folding of a double mutant of the CP RNA that eliminates the NusA-dependent pausing at the 225th nucleotide of CP RNA (asterisk, Fig. 3A) without changing its predicted secondary structure no longer depends on NusA (unpublished results), indicating that pausing at this particular site may influence significantly the folding pathway.
NusA binds elongation complexes with Ka ≈ 3 × 107 M−1 through contacts to the C-terminal domain of the α-subunit, to the nascent RNA, and to the large RNAP subunits (29, 30). NusA significantly enhances pausing at some sites preceded by RNA hairpins (26, 27, 31–33) and may stabilize the hairpins by binding to the loop and upstream part of the stem (34). The mutant RNAP we used contains two mutations in the β-subunit (see Materials and Methods), but is unlikely to prevent NusA binding because NusA slightly enhanced pausing at position 225 in CP RNA by the mutant RNAP both at low GTP concentration used to detect pausing before the addition of G226 (asterisk, Fig. 3A) and at 1 mM NTP (data not shown). This mutant RNAP probably is resistant to entering a pause-sensitive mode of transcription and, thus, responds only weakly to this pause site even in the presence of NusA.
We propose that the combined effects of transcriptional pauses and the ability of transcriptional apparatus (either RNAP itself or one of the elongation factors) to stabilize RNA structures specifically at these pause sites influences folding of newly synthesized RNA molecules. Although more work will be required to understand these effects, one possible example of such a mechanism could be reflected by the NusA-dependent S domain folding through pausing at P RNA U55 (CP RNA 225) or other sites. The NusA-dependent pause could delay RNAP long enough that the C domain assumes an altered conformation that no longer interacts with the S domain in a way that slows its folding (Fig. 1). These effects of transcription and of the transcriptional machinery on RNA folding could be species-specific, because variations in the response of different bacterial RNA polymerases to pause signals have been observed (I.A. and R.L, unpublished results).
In conclusion, our findings establish that transcription in a simple in vitro system significantly changes the folding pathway of a large ribozyme even in the absence of elongation factors. Addition of just one elongation factor resulted in drastic changes in the folding pathway through the alteration of the pausing pattern. We expect that important insights into the mechanism of both RNA folding and transcription controls can be gained by discovering how transcriptional pausing at specific sites influences the folding pathway.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM57880 to T.P. and T.R.S.; GM38660 to R.L.) and from the Cancer Research Foundation (T.R.S.).
ABBREVIATIONS
- C domain
the catalytic domain of P RNA including nucleotides 240–409 + 1–85
- CP RNA
circularly permutated P RNA with the 5′ end at nucleotide 240
- P RNA
ribozyme component of the B. subtilis ribonuclease P
- RNAP
RNA polymerase
- S domain
the specificity domain of P RNA including nucleotides 86–239
References
- 1.Netzer W J, Hartl F U. Nature (London) 1997;388:343–349. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 2.Altman S, Kirsebom L, Talbot S. FASEB J. 1993;7:7–14. doi: 10.1096/fasebj.7.1.7916700. [DOI] [PubMed] [Google Scholar]
- 3.Pace N R, Brown J W. J Bacteriol. 1995;177:1919–1928. doi: 10.1128/jb.177.8.1919-1928.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan T, Sosnick T R. Nat Struct Biol. 1997;4:931–938. doi: 10.1038/nsb1197-931. [DOI] [PubMed] [Google Scholar]
- 5.Pan T, Fang X, Sosnick T. J Mol Biol. 1999;286:721–731. doi: 10.1006/jmbi.1998.2516. [DOI] [PubMed] [Google Scholar]
- 6.Brehm S L, Cech T R. Biochemistry. 1983;22:2390–2397. doi: 10.1021/bi00279a014. [DOI] [PubMed] [Google Scholar]
- 7.Zarrinkar P P, Williamson J R. Science. 1994;265:918–924. doi: 10.1126/science.8052848. [DOI] [PubMed] [Google Scholar]
- 8.Zhang F, Ramsay E S, Woodson S A. RNA. 1995;1:284–292. [PMC free article] [PubMed] [Google Scholar]
- 9.Lewicki B T, Margus T, Remme J, Nierhaus K H. J Mol Biol. 1993;231:581–593. doi: 10.1006/jmbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- 10.Uptain S M, Kane C M, Chamberlin M J. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- 11.Landick R. Cell. 1997;88:741–744. doi: 10.1016/s0092-8674(00)81919-4. [DOI] [PubMed] [Google Scholar]
- 12.Wang D, Severinov K, Landick R. Proc Natl Acad Sci USA. 1997;94:8433–8438. doi: 10.1073/pnas.94.16.8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt M C, Chamberlin M J. Biochemistry. 1984;23:197–203. doi: 10.1021/bi00297a004. [DOI] [PubMed] [Google Scholar]
- 14.Landick R, Stewart J, Lee D N. Genes Dev. 1990;4:1623–1636. doi: 10.1101/gad.4.9.1623. [DOI] [PubMed] [Google Scholar]
- 15.Hager D A, Jin D J, Burgess R R. Biochemistry. 1990;29:7890–7894. doi: 10.1021/bi00486a016. [DOI] [PubMed] [Google Scholar]
- 16.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 17.Pan T, Zhong K. Biochemistry. 1994;33:14207–14212. doi: 10.1021/bi00251a032. [DOI] [PubMed] [Google Scholar]
- 18.Pan T, Jakacka M. EMBO J. 1996;15:2249–2255. [PMC free article] [PubMed] [Google Scholar]
- 19.Reich C, Gardiner K J, Olsen G J, Pace B, Marsh T L, Pace N R. J Biol Chem. 1986;261:7888–7893. [PubMed] [Google Scholar]
- 20.Landick R, Wang D, Chan C L. Methods Enzymol. 1996;274:334–353. doi: 10.1016/s0076-6879(96)74029-6. [DOI] [PubMed] [Google Scholar]
- 21.Pan T. Biochemistry. 1995;34:902–909. doi: 10.1021/bi00003a024. [DOI] [PubMed] [Google Scholar]
- 22.Loria A, Pan T. RNA. 1996;2:551–563. [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez P J, Iost I, Dreyfus M. Nucleic Acids Res. 1994;22:1186–1193. doi: 10.1093/nar/22.7.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao M Y, Kan M C, Lin-Chao S. Nucleic Acids Res. 1995;23:1691–1695. doi: 10.1093/nar/23.10.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyakhov D L, He B, Zhang X, Studier F W, Dunn J J, McAllister W T. J Mol Biol. 1998;280:201–213. doi: 10.1006/jmbi.1998.1854. [DOI] [PubMed] [Google Scholar]
- 26.Winkler M E, Yanofsky C. Biochemistry. 1981;20:3738–3744. doi: 10.1021/bi00516a011. [DOI] [PubMed] [Google Scholar]
- 27.Farnham P J, Greenblatt J, Platt T. Cell. 1982;29:945–951. doi: 10.1016/0092-8674(82)90457-3. [DOI] [PubMed] [Google Scholar]
- 28.Theissen G, Pardon B, Wagner R. Anal Biochem. 1990;189:254–261. doi: 10.1016/0003-2697(90)90117-r. [DOI] [PubMed] [Google Scholar]
- 29.Gill S C, Weitzel S E, von Hippel P H. J Mol Biol. 1991;220:307–324. doi: 10.1016/0022-2836(91)90015-x. [DOI] [PubMed] [Google Scholar]
- 30.Liu K, Zhang Y, Severinov K, Das A, Hanna M M. EMBO J. 1996;15:150–161. [PMC free article] [PubMed] [Google Scholar]
- 31.Farnham P J, Platt T. Nucleic Acids Res. 1981;9:563–577. doi: 10.1093/nar/9.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan C L, Landick R. J Biol Chem. 1989;264:20796–20804. [PubMed] [Google Scholar]
- 33.Sigmund C D, Morgan E A. Biochemistry. 1988;27:5628–5635. doi: 10.1021/bi00415a035. [DOI] [PubMed] [Google Scholar]
- 34.Landick R, Yanofsky C. J Mol Biol. 1987;196:363–377. doi: 10.1016/0022-2836(87)90697-8. [DOI] [PubMed] [Google Scholar]